Abstract
In silico studies were performed to assess the binding affinity of selected organophosphorus compounds toward the acetylcholinesterase enzyme (AChE). Quantum mechanical calculations, molecular docking, and molecular dynamics (MD) with molecular mechanics Generalized–Born surface area (MM/GBSA) were applied to assess quantitatively differences between the binding energies of acetylcholine (ACh; the natural agonist of AChE) and neurotoxic, synthetic correlatives (so-called “Novichoks”, and selected compounds from the G- and V-series). Several additional quantitative descriptors like root-mean-square fluctuation (RMSF) and the solvent accessible surface area (SASA) were briefly discussed to give—to the best of our knowledge—the first quantitative in silico description of AChE—Novichok non-covalent binding process and thus facilitate the search for an efficient and effective treatment for Novichok intoxication and in a broader sense—intoxication with other warfare nerve agents as well.
1. Introduction
Synthetic organophosphorus inhibitors of acetylcholinesterase (AChE) are ubiquitous in many areas of human activity from agriculture: in the form of insecticides [1,2] through medicine: anti-inflammatory drugs and potential treatments of neurological diseases [3,4] to modern warfare: nerve agents [5]. Despite their potential medical use however, the majority of such applications are related to termination rather than sustaining life: the acute toxicity and lethality of these compounds is caused by their ability to irreversible impair the enzymatic ability to operate.
Due to its high turnover number, AChE is known to quickly hydrolyze the cholinergic neurotransmitter, acetylcholine (ACh) to acetic acid and choline [6]. By doing so, it reduces ACh concentration in the synaptic cleft and, therefore, terminates neural signalling. Any permanent inhibition of the enzyme would obviously induce an over-accumulation of ACh in the cholinergic system, the cholinergic crisis [7], causing overstimulation, a subsequent irreparable detriment of nerve cells and a fatal muscular paralysis, leading in consequence to prompt death [8].
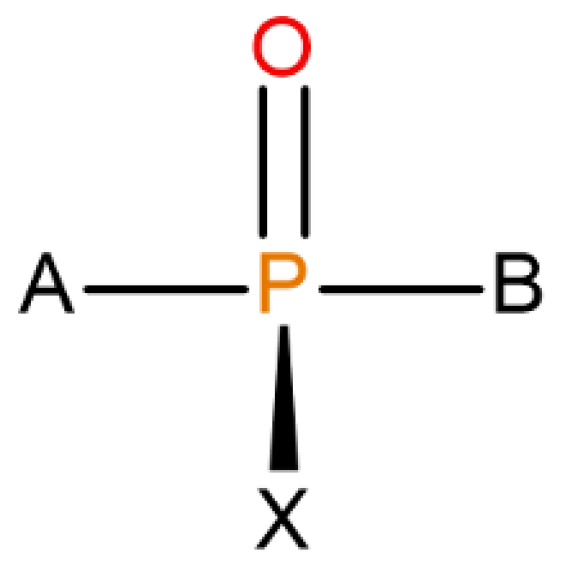
Organophosphorous compounds, especially the purpose-made, military-grade organophosphorus nerve agents (OPNAs) whose structures were assigned to halogen-containing phosphonate derivatives with their fluorinated organophosphorus core open to several modifications (see Figure 1), are believed to be able to precisely inhibit AChE by binding with one of the amino acids (serine) in the AChE catalytic triad (Ser-His-Glu) via a reactive hydroxyl group. A nucleophilic attack of Ser-OH towards the electrophilic phosphorus atom of a nerve agent in an AChE–OPNA non-covalent complex (Scheme 1) typically leads to the formation of a covalent bond between the serine and a given nerve agent (Ser-OPNA), therefore effectively impeding AChE functioning.
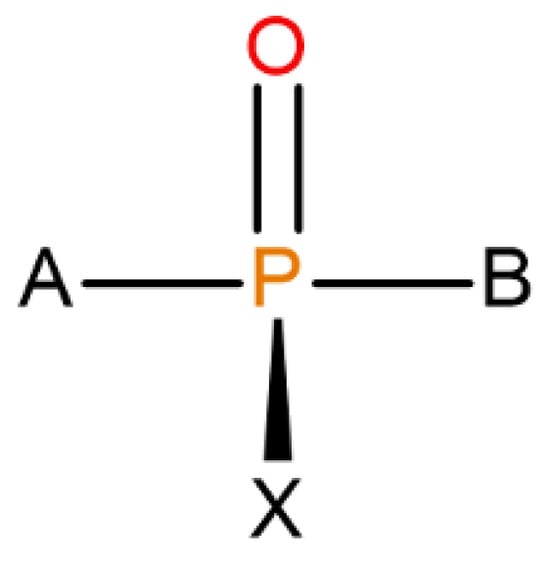
Figure 1.
The overall structure of OPNAs’ (including Novichoks) organophosphorus core: the phosphoryl bond with X—halogen or another electron-withdrawing group (usually fluorine) and other organic or heteroorganic substituents (A,B).
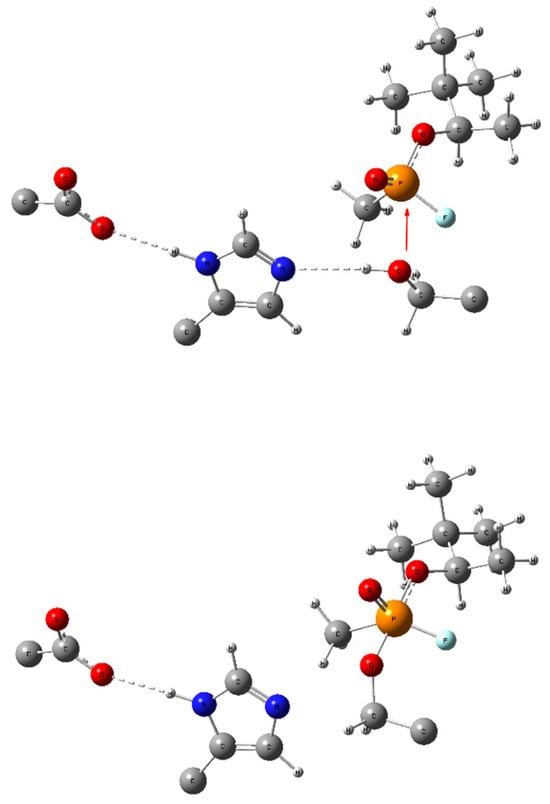
Scheme 1.
Bonding changes in the catalytic triad (GLU-HIS-SER, respectively) during the formation of a covalent bond between serine and an OPNA (here—soman). The red arrow symbolizes the nucleophilic attack of the Ser lone pair in the non-covalently bonded complex (up) leading to a pentacoordinate intermediate Ser-OPNA (low) and dashed lines symbolize hydrogen bonding. All redundant parts of the aminoacid scaffold were removed for clarity.
Numerous publications on various OPNAs’ working mechanisms have been published, e.g., [9,10] including ones that apply quantum mechanical (QM) methods [8,11] on the other hand—an efficient reactivation mechanism that could lead to a broad spectrum of possible antidotes has been explored at some depth only recently3 and the topic remains yet to be expanded.
The newest, A-generation of OPNAs, so-called “Novichoks” [12,13] which were developed shortly before the implementation of the Chemical Weapons Convention (CWC) in 1993, became anew a scientific point of interest after the infamous attempt of political poisoning of Sergei and Yulia Skripal in 2018 [10]. This renewal in scientific research on Novichoks comes arguably from both the fact that their structure and properties remain to a certain degree unknown and/or lacking particulars (as the Russian government never acknowledged developing them [11]) and the utmost threat to the public health and safety policies they should pose due to their alleged extreme toxicity (estimations of <0.1 mg lethal dose [12]).
The Novichoks’ most significant feature, distinguishing them from other, classical nerve agents, is the phosphonoamide bond, that replaced oxo- (sarin, soman, tabun) and tio- (VX) bonds between the phosphorus and heteroalkyl residues A or B (see Figure 1 and Figure 2) [14]. There are reports on the analysis of these compounds using theoretical and spectroscopic methods [9,10,13,15,16], and scarce in vitro research [17], yet there is, to the best of our knowledge, no information about any extensive in silico examinations of Novichoks’ mechanism of action and their potential reactivators. In reactivating the enzyme from OPNAs, several oxime-based compounds [18,19,20,21,22] as well as several engineered OPNA-degrading enzymes [14] are effective, and purely theoretical studies were conducted (e.g., [23,24]). Regarding Novichoks however, there is a lack of both QM and molecular mechanics (MM) investigations of Novichok–AChE interactions, including the noncovalent ones; yet, obviously, the noncovalent interplay between the ligand and the enzyme contributes to and facilitates the irreversible phosphonylation of the AChE catalytic-triad serine e.g., by adjusting the ligand electron density distribution therefore stabilizing the ligand in the enzyme active centre gorge and making it thus more susceptible to nucleophilic enzyme attack.
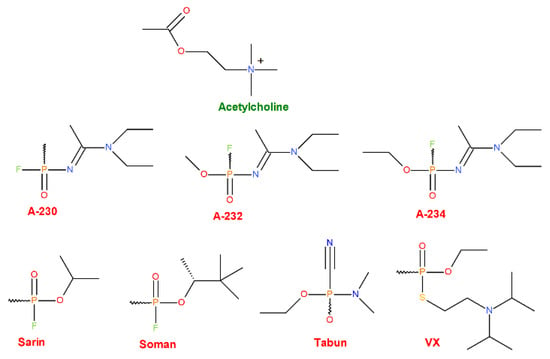
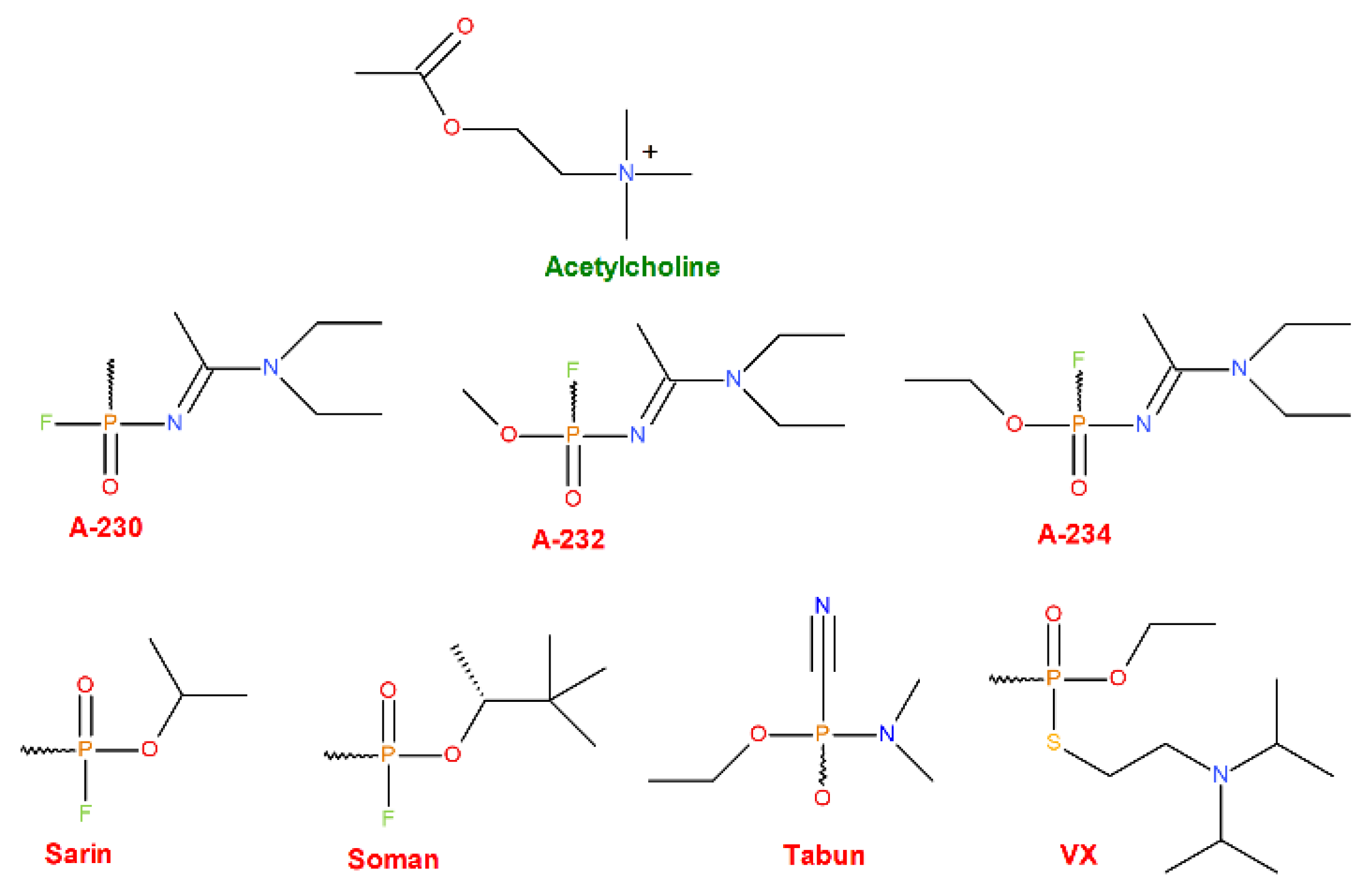
Figure 2.
Structures of the AChE natural agonist (the upper-most row, green) and various synthetic OPNAs (middle and bottom rows, red) were chosen as ligands for our research.
This work aims at in-depth investigating the human AChE–OPNAs binding affinities in comparison with the natural agonist as a starting point for further in silico research. Qualitatively and quantitatively assessed were the binding strengths of various OPNAs: the presently known, yet relatively unexplored A-series (A-230, A-232, A-234; “Novichoks”) and several arbitrarily selected, arguably the most publicly known and recognizable, examples of older OPNAs from the G- (tabun, sarin, soman) or the V-series (VX) for further comparison.
This unique assessment of the AChE binding affinity of the (officially) newest form of chemical weapon of mass destruction is in our opinion of at least twofold importance:
- (a)
- From a purely academic point of view—it allows us to assess the strength of the interaction between the AChE enzyme and the A-tier OPNAs representatives for the first time. Thereby, the amount of scientific knowledge has increased, and new ideas will certainly emerge as to how to use this knowledge for the benefit of persons affected by those compounds. This particular aspect is strictly related to the next one;
- (b)
- From the point of view of public health care and crisis management—this knowledge will contribute to the facilitation of counteracting the deadly effects of these weapons e.g., proposing new, more effective antidotes or as well as more effective pathways of removal of these substances from the human system or even blocking them from entering it in the first place.
2. Results and Discussion
Structures of the ligands selected for the research are depicted in Figure 2. whereas their average docking () and MM/GBSA binding energies () are included in Table 1. Coordinates for all G16-optimized ligand’s structures are available in Supplementary Materials.

Table 1.
An average docking score and MM/GBSA-binding energies [both in kcal/mol] for the selected ligands. An average radius of gyration (Rg) [in Å] and SASA [in Å2] for the selected ligands.
The molecular docking provides a score that mainly indicates the overall thermodynamic value for the docking (Table 1) and is regarded to be approximate [25] and always requires an indispensable refinement by an alternative method, in our case, by MM/GBSA method.
Presented data show that all the ligands are thermodynamically favourable in binding to the AChE active side. Interestingly, ACh is thermodynamically the least potent AChE ligand—as expected from a natural agonist of a high turnover number enzyme—with considerable discrepancies between the modest ACh binding energy value and the values calculated for the OPNAs. Taking into consideration the fact that all the selected OPNAs are warfare agents, designed especially to be irreversible AChE inhibitors—such results were expected and well prove the confirmation of the Novichoks’ and other OPNAs’ binding efficacy as compared to the ACh, at the first stages of the inhibition process, regardless of its possible mechanism.
The noteworthy fact is that the theoretical binding affinity for the newest (and implicitly: the most dangerous) Novichok tier is somewhat similar to the previous generation of warfare agents—at least for the first stage of the inhibition. Thus, considering as one of the first components making up the ligand potency and toxicity, this is in agreement with the reports of Carlsen [16] whose QSAR model-based study disproves the existing claims as if the Novichoks were allegedly several times more potent than the hitherto known OPNAs.
The relatively high values of MM/GBSA standard deviations may be seen as a potential source of uncertainty regarding the correctness of the simulation results or protocols but this issue arises rather due to the method itself. MM/GBSA, although overall reliable and popular due to its moderate computational cost, is known to have incorporated several approximations with the method [26] that arguably are responsible for the somewhat diminished precision of outcomes.
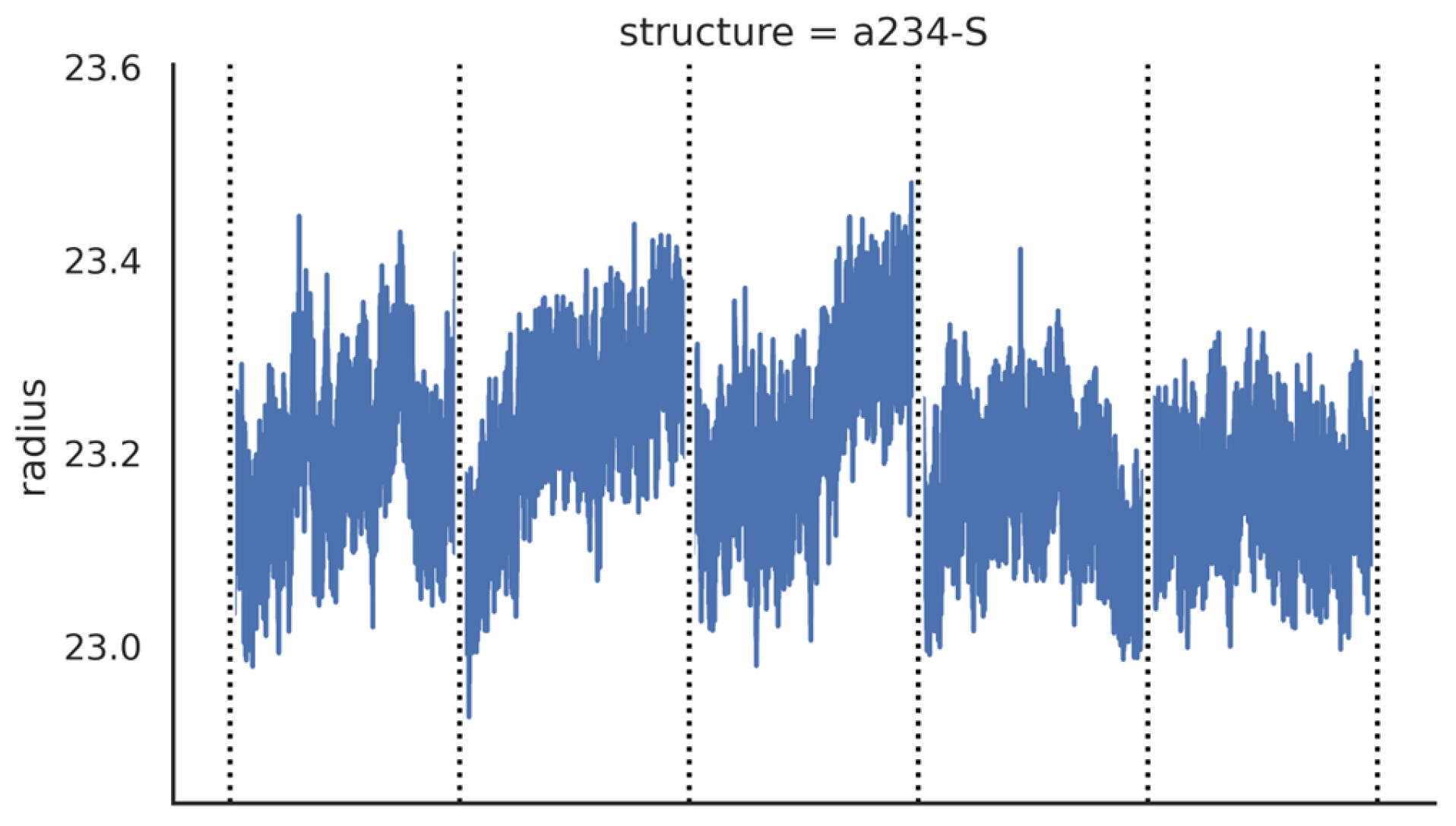
Additionally, the spatial conformation and stability of the simulated AChE–ligand system are supported by stable values of the radius of gyration (Rg) in Table 1 and Rg = Rg(t) mapping trajectory plot in Figure 3. Rg that is practically constant over the entire molecular simulation process proves that no significant conformational change is initially induced in the AChE structure by the presence of the ligands.
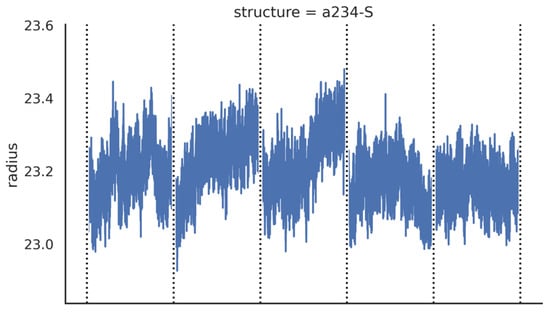
Figure 3.
An exemplary plot presenting Rg = Rg(t) dependence for a selected Novichok. Plots for all simulated systems are available free of charge as Supplementary Materials (Supplementary Figures S1–S8).
Similar ligands and complex stabilities are supported by the root-mean-square deviation per residue (RMSD) and one of the AChE Cα atoms (Figures S9–S16 and S17–S24, respectively). The Cα RMSD is in the relatively low range of values. Such results are not supportive of the induced-fit binding and support the hypothesis that Novichoks’ (and other examined OPNAs’) binding mode engages conformations resulting from preexisting conformational dynamics; the secondary mode for the AChE enzyme was advocated e.g., by Xu et al. [27]
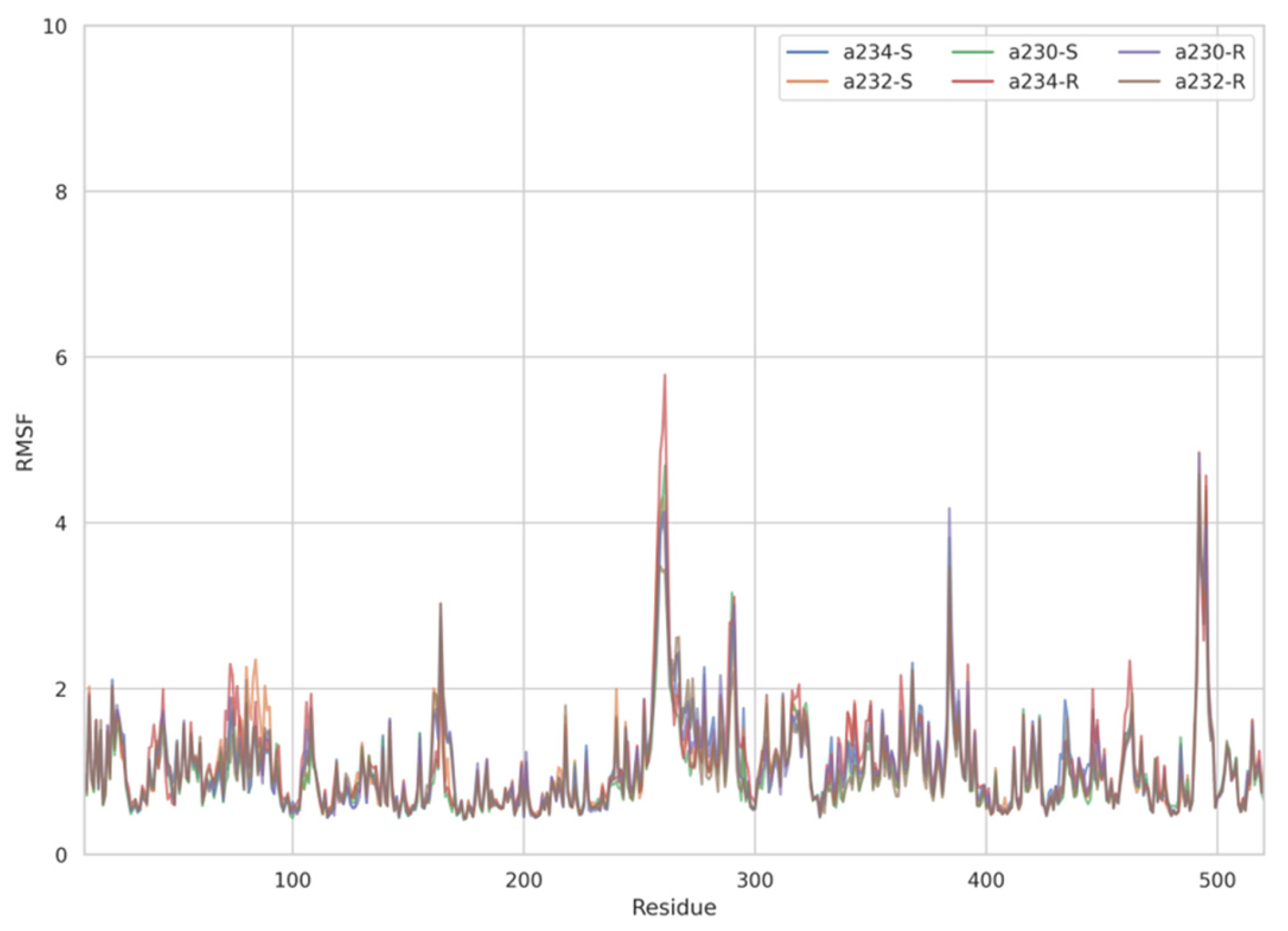
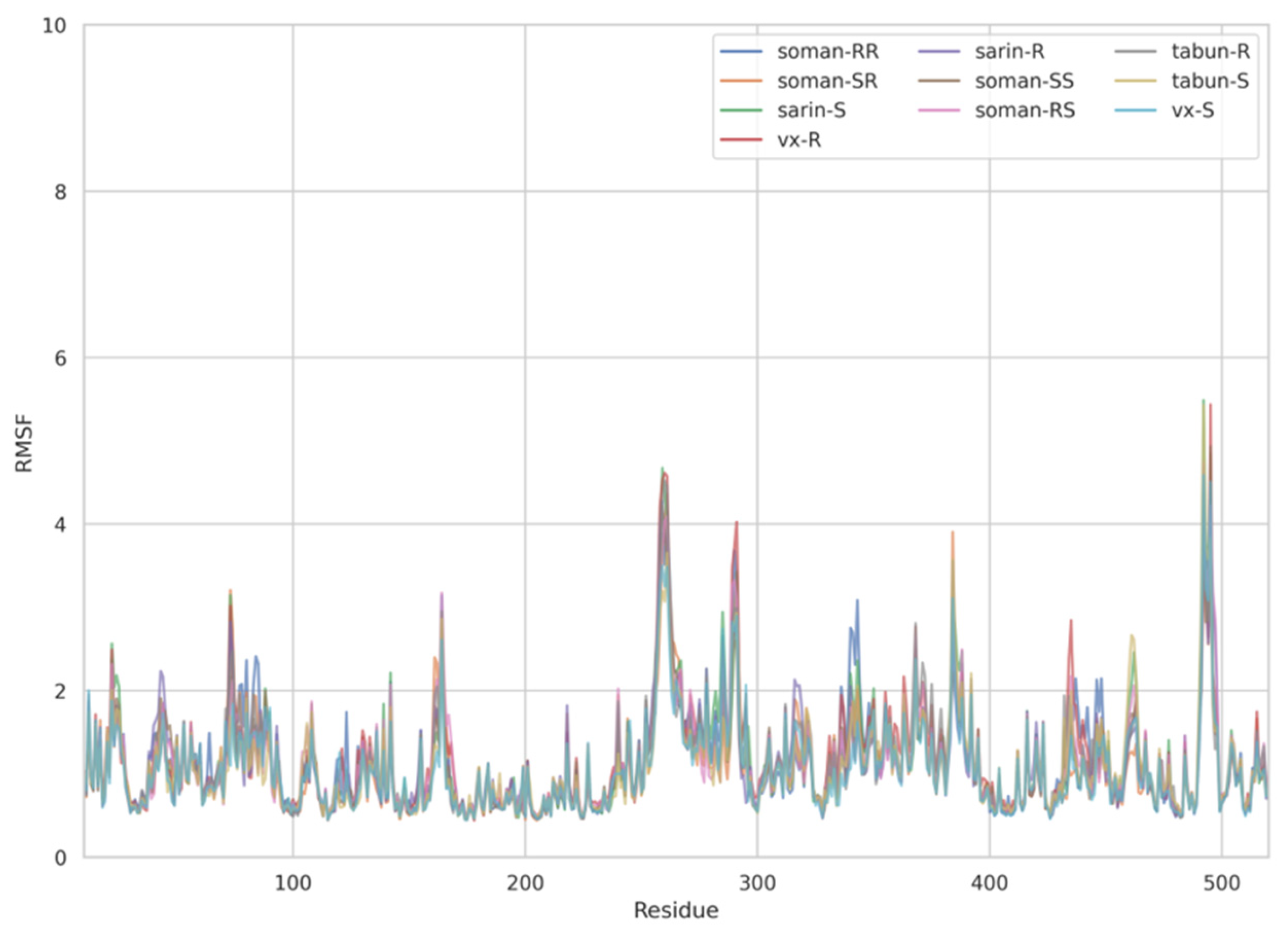
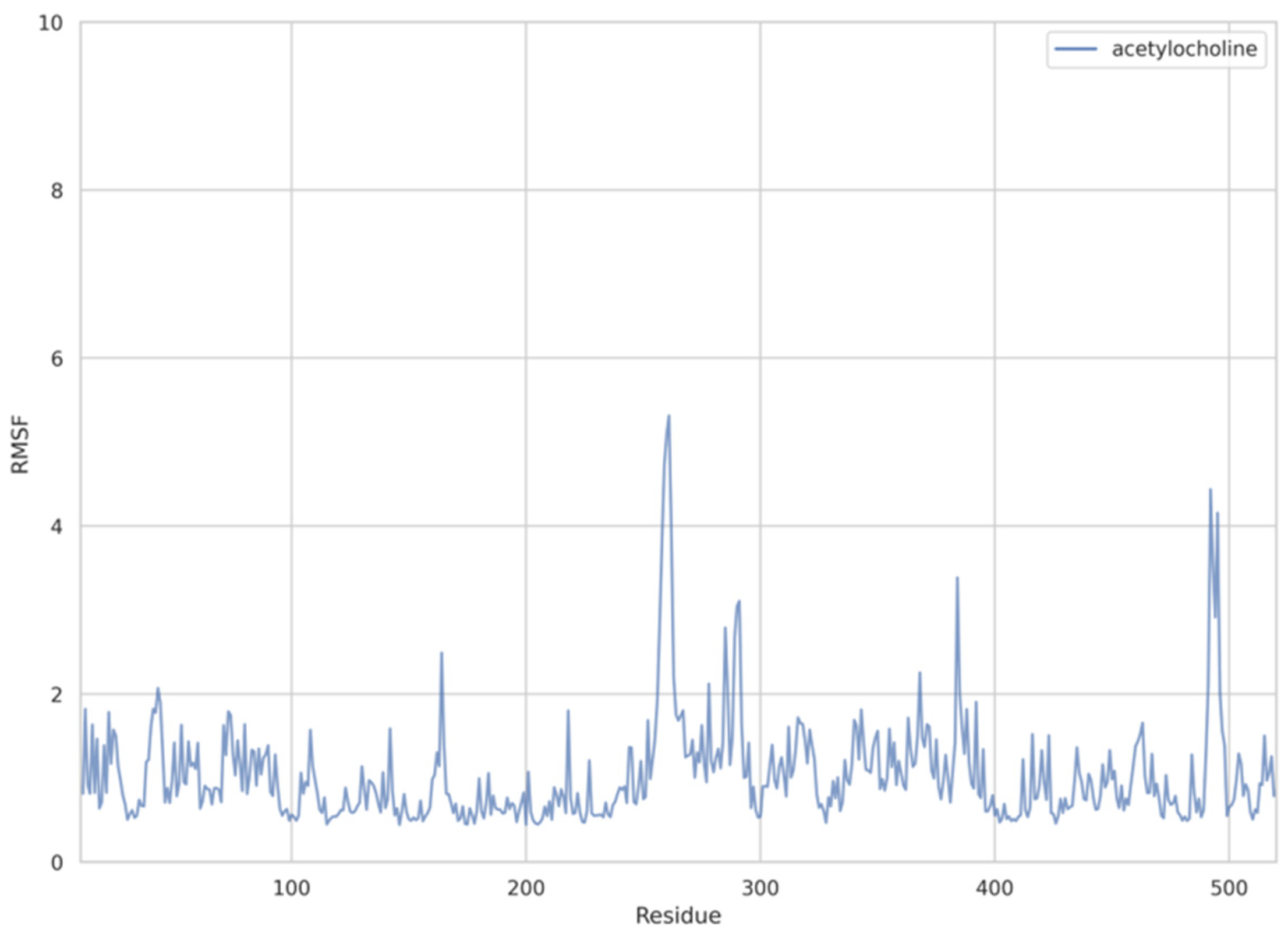
The dynamic adjustment similarity between the AChE—ACh and the AChE—OPNA systems is confirmed by RMSF calculations (Figure 4, Figure 5 and Figure 6). As can be seen, the root-mean-square fluctuations for examined systems are calculated for each residue separately (Figure 4, Figure 5 and Figure 6). The RMSF values were calculated disregarding the final five amino acids from both C- and N-termini to get rid of unnecessary fluctuations. Structural adjustments in the protein form a very similar pattern of spikes in each case and are furthermore of very similar magnitude. This allows us to presume that the protein structural folding adjusts to interact with Novichoks in the same way as with ACh and other OPNAs. This indicates that the putative Novichok working mechanism could be at least similar (if not identical) to the actual AChE—ACh mechanism.
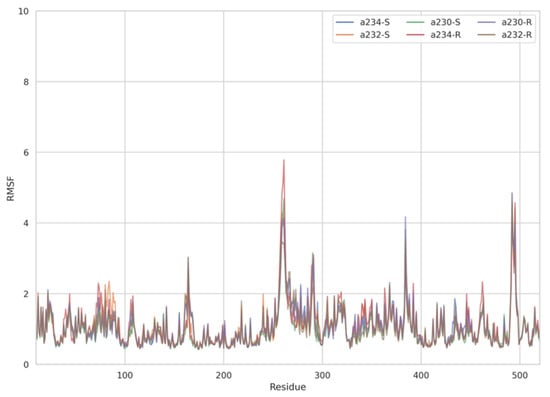
Figure 4.
Plots of RMSF per residue [in Å] for the AChE–Novichok systems.
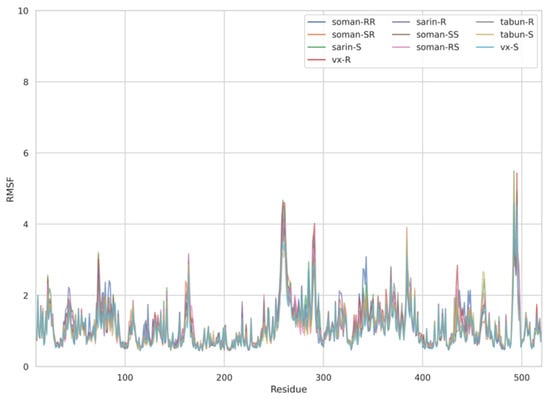
Figure 5.
Plots of RMSF per residue [in Å] for the AChE—exemplary OPNAs systems.
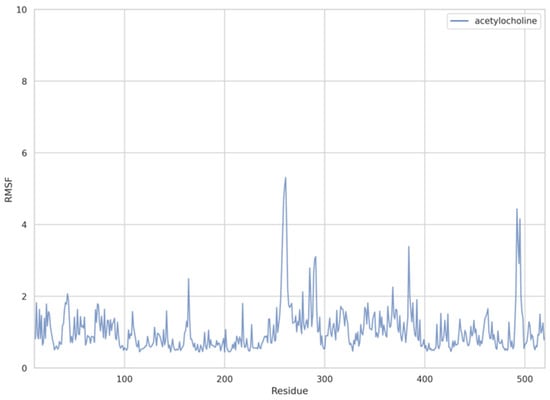
Figure 6.
Plots of RMSF per residue [in Å] for the AChE—ACh system.
The discrepancy between low RMSD values and higher RMSF most probably is due to the fact that RMSD values represent a quantitative measure of a structure divergence over time from the reference point, in this case, structures resulted from the energy minimization and the RSMF ones reveal which residues are the most mobile (fluctuate the most, i.e., diverge from the time-averaged structure).
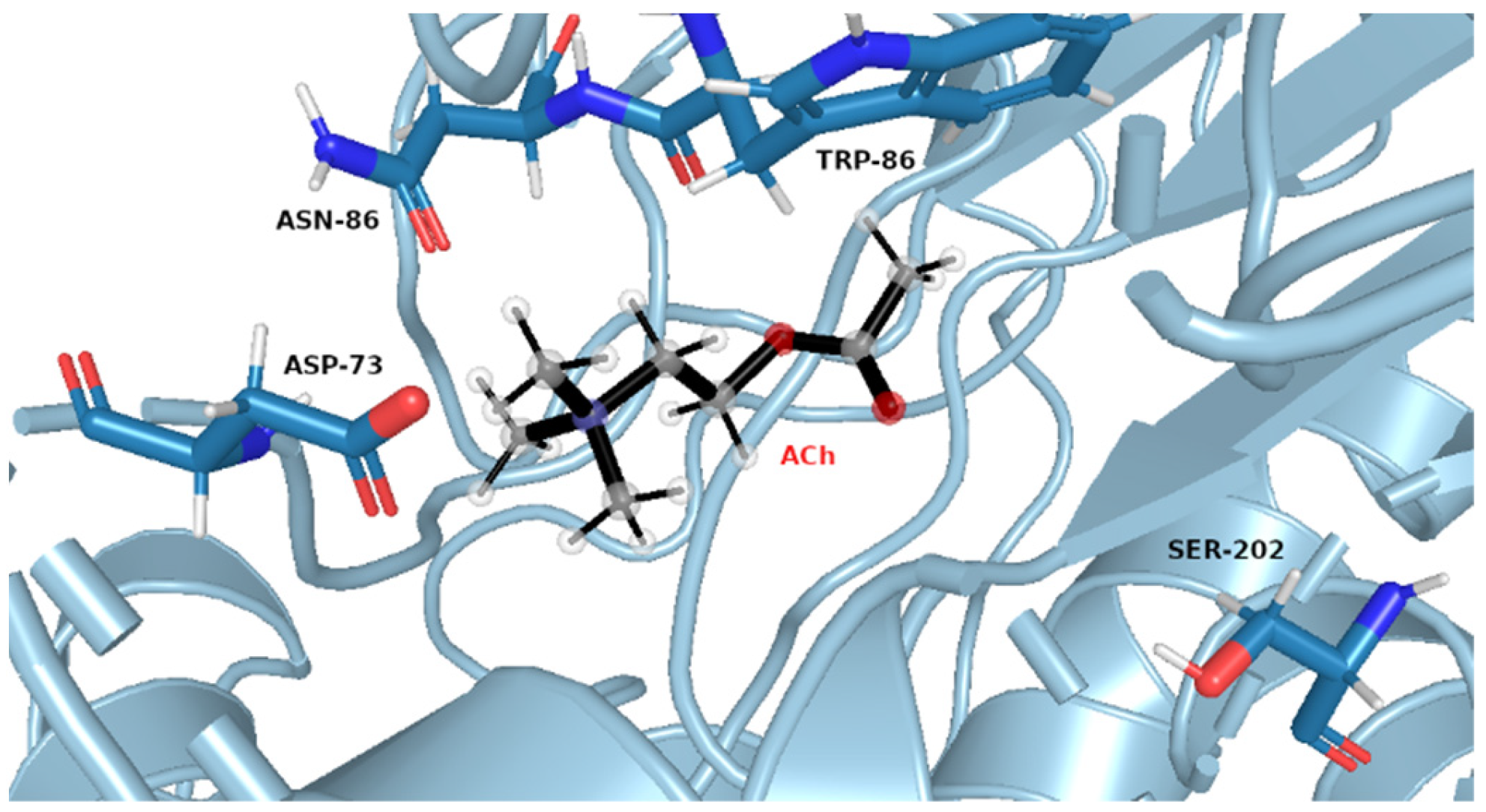
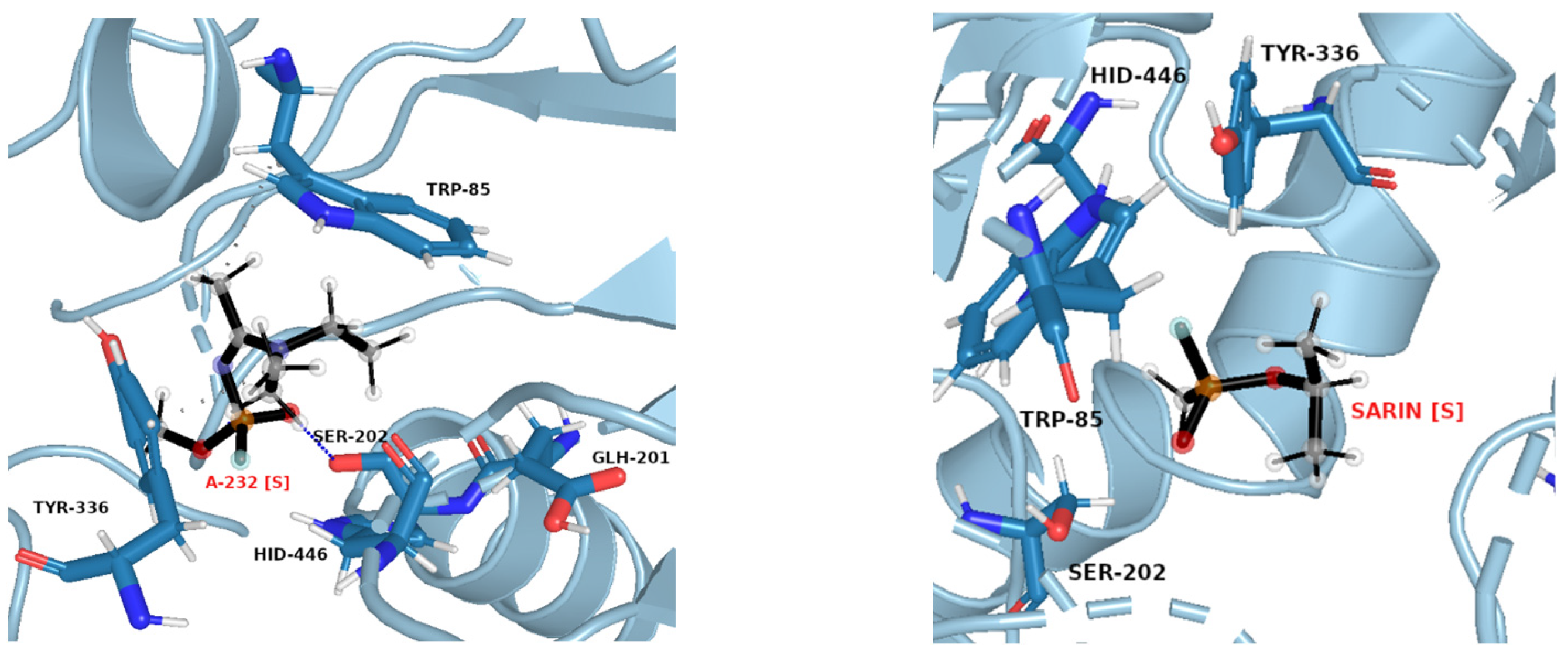
The same pattern of flexibility and rigidity emerges when considering the SASA surface for different ligands (Figures S25–S32)—molecular volume enclosed by SASA remains relatively stable and of low values for each system, except for acetylcholine. The highest SASA average value and the highest SASA standard deviation values for ACh support the view of OPNAs buried deeper into the active site and being less flexible than ACh. This native ligand may in turn operate more freely and recruit for the active site solvent molecules. Such dynamics correspond well with the AChE high turnover number concerning its natural agonist. The most frequent structures from MD simulations (see Figure 7 and Figure 8), shown together with their corresponding interacting amino acids, support the image in which Novichoks and other OPNAs are placed much more closely to Ser202 than ACh.
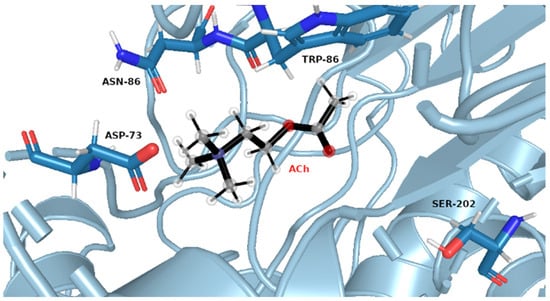
Figure 7.
Exemplary snapshot of the most frequent structure for AChE complexes with ACh. Ser202 is shown for purely illustrative purposes.
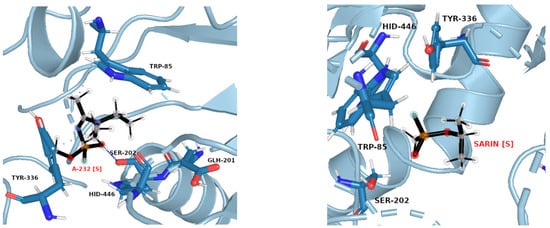
Figure 8.
Exemplary snapshots of the most frequent structures for AChE complexes with A-232 [S] (left) and sarin [S] (right). The dashed line symbolizes hydrogen bonding.
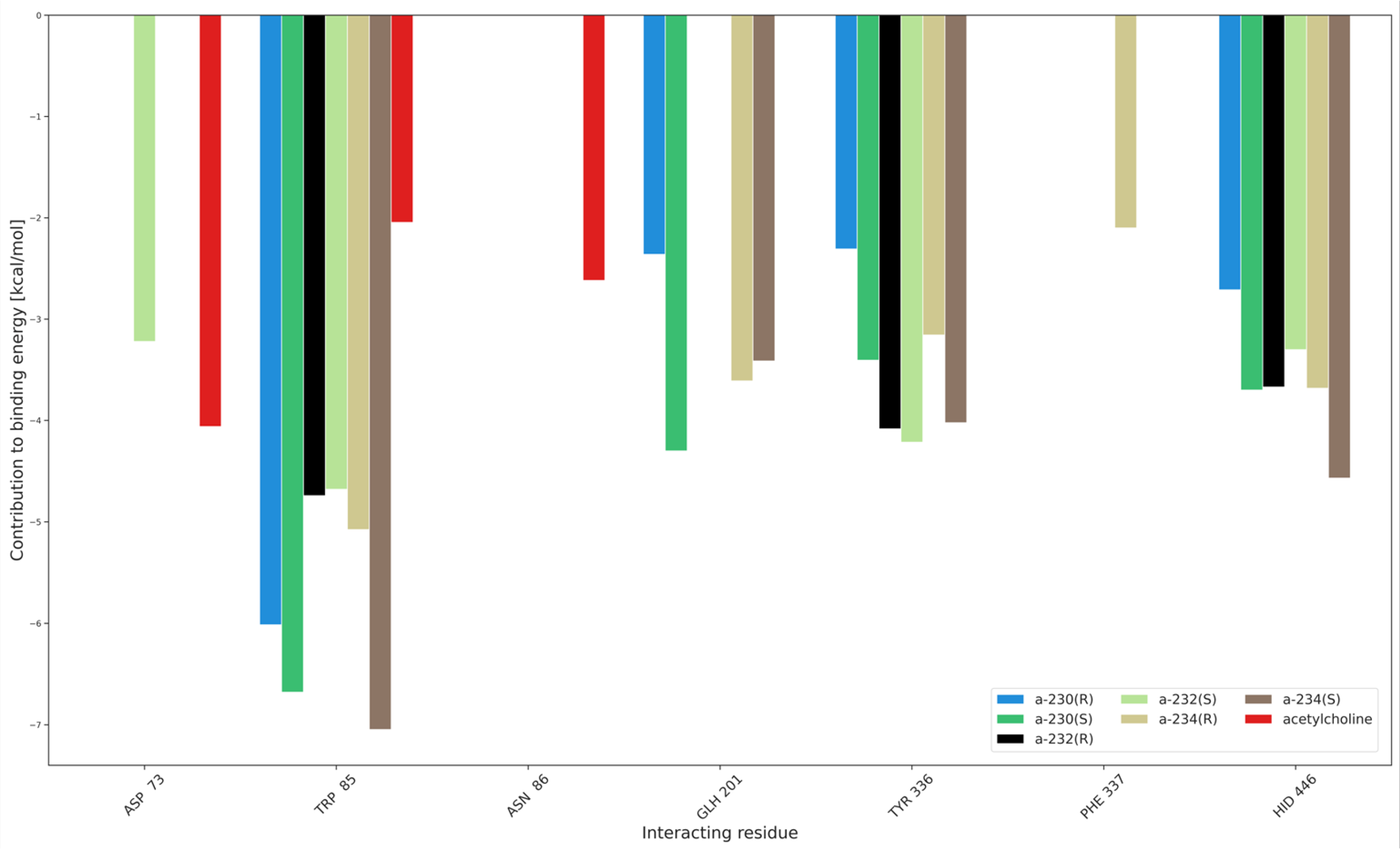
Additionally, the interaction partition analysis was performed in order to gain quantitative knowledge about how the interactions are divided between individual residues, i.e., with which amino acids the ligands interact the most. Exemplary partitioning for Novichoks is depicted in Figure 9 while Figures S33–S38 depict the amino acids and the interaction energy for the acethylcholine, Novichoks, and the remaining G- and V-series agents respectively as well as exemplary snapshots with all interacting residues common in the tier made visible.
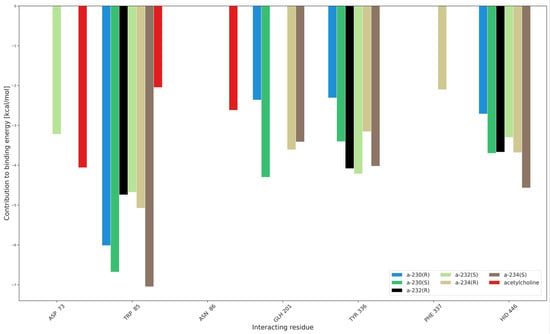
Figure 9.
Interaction partition analysis for AChE complexes with Novichoks and ACh.
As expected, our results show that the interactions within the active gorge of AChE are taking place between the ligands and amino acids that are placed in the nearest proximity of all the docked ligands. The main differences are the number of interacting residues (3 in the case of ACh, from 3 to 5 in the case of OPNAs) and their maximal strength in the tiers (about −4 kcal/mol for ACh, about −7 for A-234, slightly less than −7 for VX) and the particular residua themselves. Although there are amino acids that interact with most members of all the tiers (e.g., TRP 85), there are also ones that seem to be specific exclusively to OPNAs (TYR 336 or HID 446). This may be explained by the different sizes of the ligands and their different overall chemical construction. The results support all the previous considerations regarding the strength of ligand interactions and their flexibility.
The criteria of the final quality assessment were as follows: the best ligand was considered to be the one minimizing not only MM-GBSA energies but SASA outcomes as well because the low results of the latter reflect the ligand’s ability to remain in the binding site.
Considering all of the above factors, we conclude that the best binding OPNA turned out to be the S isomer of A-232, while the lowest affinity was shown by the RS diastereomer of soman. The worst ligand overall was acetylcholine, a fully expected outcome, considering the high turnover number of AChE.
3. Materials and Methods
3.1. Methodology
The workflow for this research consisted of the following steps:
- Preparation and optimization of the protein and all ligands, including their stereoisomers;
- Docking of each ligand to the AChE binding cavity, yielding preliminary quantitative forecasts as to whether the binding is thermodynamically favourable;
- Molecular dynamics and MM/GBSA simulations of any thermodynamically favourable dockings, giving insights into how ligands may stabilize in the active site and yield the binding affinity of their non-covalent complexes.
3.1.1. Preparation of the Protein and Ligands
The protein structure, downloaded from the RCSB PDB database [28] (PDB ID: 4m0e [29]), was reconstructed using Modeller 10.3 software to fill missing residues, then after removing ligands, it was subjected to structure validation using SAVES 6.0 server [30], passing the vast majority of tests, with high overall quality factor. The protein was protonated accordingly to its optimum pH using the Poisson-Boltzmann method [31] and prepared for molecular docking with the AMBER force field by merging non-polar hydrogens and calculating partial charges on residues.
Ligand structures (see Figure 2) were created using GaussView 6.0 [32], based on structures deposited in the PubChem database (https://pubchem.ncbi.nlm.nih.gov/, accessed on 27 October 2023), and subsequently optimized using one of the DFT methods (B3LYP-GD3/cc-pvtz level of theory) as implemented in Gaussian16 software [33]. Optimization and vibrational analysis for each ligand were performed for the gas phase. Additionally, the Merz–Kolmann population analysis and RESP partial charge [34] derivation were performed with HF/6-31G* level of theory. The latter resulted in a series of .gesp files that were subsequently used for the generation of .mol2 molecular dynamics input and library files by Antechamber 20.0 software [35] from AmberTools20 [36].
3.1.2. Molecular Docking
The protein was protonated, non-polar hydrogens were merged and calculation of partial charges on its residues was performed as mentioned above. The charges as well as the number of torsion angles were calculated using AutoDockTools 1.5.6 [37]. The flexible residues were selected based on a centroid of the active site and extracted to separate output files, also using AutoDockTools 1.5.6. The molecular docking of all selected ligands was performed using AutoDockVina 1.1.2 [38]. The docking grid was set on a centroid of the active site with flexible residues and a total volume of a maximum of 27,000 Å3.
Every docking yielded up to 20 ligand conformations and was repeated 4 times for each ligand. Then the ligands in the vicinity of Ser202 with the lowest energy and optimized spatial structure of flexible residues were merged with input protein structure using an in-house Python script. This procedure yielded initial energies of binding and spatial orientation of ligands inside the protein’s active centre and subsequently served as starting points for molecular dynamics simulations.
3.1.3. Molecular Dynamics
The Antechamber software was applied for the conversion of the ligand .gesp files to the input .mol2 ones with RESP partial charges assigned. Topology and input coordinate files were generated using tLeap, a module of the Amber20 package, using ff14SB force field [39] for protein, GAFF [40] for ligand, and TIP3P water model [41] for the solvent. The system was placed in a truncated octahedral box, solvated, and neutralized with sodium or chloride ions, depending on its total charge. MD simulation was performed with the Amber20 pmemd module, with minimizations and heating performed using multiple CPU threads, and productions using GPU. The first 10 ns of each production simulation were treated as an equilibration phase and omitted in further analysis. The trajectory was saved every 5000 MD step. Validation of the simulation was accomplished using CPPTRAJ 5.1.0 software [42], through the calculation of the radius of gyration (Rg) of the complex, root-mean-square deviation (RMSD) of the ligand atoms related to the structure after optimization, the Cα atoms of the protein, root-mean-square fluctuations (RMSF) of system residues, solvent accessible surface area (SASA) of the ligand. Additionally, an interaction energy partitioning (i.e., contributions from amino acids that interact with the given ligand the most) was calculated for every 10th trajectory frame and averaged. In order to obtain the most frequent orientation of ligands within the binding cavity, the trajectories were clustered using a hierarchical agglomeration algorithm. The relative binding energies of ligand to the enzyme: were calculated based on the MM/GBSA algorithm [26]. This yields relative binding energies with much higher accuracy than the one obtained through molecular docking and also indicates the most optimal conformation of the ligand within the binding cavity prior to covalent binding to the enzyme.
4. Conclusions
We think that the threat the possible usage of these compounds poses for the safety of public health is so significant that the current state of knowledge on this topic is far from being sufficient in order to take countermeasures effectively and efficiently at large. However, the failed Yulia and Sergei Skripal assassination attempt is apparent evidence that adequate help, when applied promptly, is able to obstruct the disastrous, fatal impact these compounds were designed to cause.
Therefore, this paper aims to slowly begin filling the yawning chasm between what is already known and what should be known regarding the Novichoks–acetylcholinesterase enzyme (AChE) interactions because, due to their putatively higher affinity to AChE, combined with recalcitrance to degradation and potential resistance to oxime’s reactivation mechanism, Novichoks is a significant threat that requires investigation. Data obtained by our research allow us to draw several conclusions about the Novichok-AChE binding affinity and provide certain clues regarding their possible working mechanism.
The selected organophosphorus nerve agents (OPNAs) bind to AChE in a thermodynamically favourable way and the natural agonist, acetylcholine (ACh), turns out to be the least potent binder with a huge discrepancy between itself and the OPNAs, which is expected from a high turnover-number enzyme. The comparable binding affinities of all the nerve agents support rather the scarce literature reports, according to which Novichoks are similar in potency to the older OPNAs instead of being much superior in toxicity.
The AChE-Novichok complexes are temporarily and spatially stable entities as indicated by the radius of gyration, root-mean-square deviation (RMSD), and Cα (alpha carbon) RMSD values. The results do not support the induced-fit binding mode; exploiting conformations from preexisting conformational dynamics offers a better explanation of the observed features.
ACh is the most flexible as indicated by e.g., solvent-accessible surface area values as well as the interaction partition analysis. Almost identical patterns of the OPNAs RMSF spikes and their magnitude, as compared to the ACh ones, prove the dynamic adjustment similarity which in turn indicates that the working mechanism may resemble the actual AChE—ACh mechanism or be identical. Although, while focusing in this work solely on the non-covalent binding as the very first step of the Novichok inhibition process, we intentionally refrain from unambiguously choosing any specific putative mechanism, we think that the results presented above allow us to surmise that the process in question should be the nucleophilic substitution at the phosphorus centre, proceeding either via pure SN2 mechanism or via the substitution-elimination (S–E) scheme, exactly as in the case of the rest of examined OPNAs.
This hypothesis however is outside the scope of the present paper and could be answered with more degree of certainty by more elaborate studies, engaging more sophisticated levels of theory applied (preferably, pure quantum mechanical (QM) or hybrid QM/MM methods with one of the ab initio approaches) allowing to follow and probe the reaction coordinate and obtain activation energy barriers. At present, we are in the process of verifying it.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29020338/s1, 1. Cartesian coordinates for all DFT-optimized ligands; 2. Figures S1–S8: Radius of gyration of protein-ligand complex as a function of simulation steps; 3. Figures S9–S16: RMSD of protein-ligand complex as a function of simulation steps; 4. Figures S17–S24: Cα RMSD of protein-ligand complex as a function of simulation steps; 5. Figures S25–S32: SASA of protein-ligand complex as a function of simulation steps; 6. Figures S33–S38: Interaction partitioning for the given ligands with exemplary snapshots.
Author Contributions
Conceptualization R.M. and B.G., investigation R.M. and B.G., methodology R.M. and B.G., supervision A.C. and M.C., validation R.M., B.G., A.C. and M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
This research was supported in part by PLGrid Infrastructure.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Todd, S.W.; Lumsden, E.W.; Aracava, Y.; Mamczarz, J.; Albuquerque, E.X.; Pereira, E.F. Gestational exposures to organophosphorus insecticides: From acute poisoning to developmental neurotoxicity. Neuropharmacology 2020, 180, 108271. [Google Scholar] [CrossRef] [PubMed]
- Dăneţ, A.F.; Bucur, B.; Cheregi, M.C.; Badea, M.; Şerban, S. Spectrophotometric determination of organophosphoric insecticides in a FIA system based on AChE inhibition. Anal. Lett. 2003, 36, 59–73. [Google Scholar] [CrossRef]
- Lushchekina, S.V.; Masson, P. Slow-binding inhibitors of acetylcholinesterase of medical interest. Neuropharmacology 2020, 177, 108236. [Google Scholar] [CrossRef] [PubMed]
- Kosińska, A.; Virieux, D.; Pirat, J.-L.; Czarnecka, K.; Girek, M.; Szymański, P.; Wojtulewski, S.; Vasudevan, S.; Chworos, A.; Rudolf, B. Synthesis and Biological Studies of Novel Aminophosphonates and Their Metal Carbonyl Complexes (Fe, Ru). Int. J. Mol. Sci. 2022, 23, 8091. [Google Scholar] [CrossRef]
- Aroniadou-Anderjaska, V.; Apland, J.P.; Figueiredo, T.H.; Furtado MD, A.; Braga, M.F. Acetylcholinesterase inhibitors (nerve agents) as weapons of mass destruction: History, mechanisms of action, and medical countermeasures. Neuropharmacology 2020, 181, 108298. [Google Scholar] [CrossRef] [PubMed]
- Anglister, L.; Stiles, J.R.; Salpetert, M.M. Acetylcholinesterase density and turnover number at frog neuromuscular junctions, with modeling of their role in synaptic function. Neuron 1994, 12, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, C.; Forsgren, N.; Hoster, N.; Akfur, C.; Artursson, E.; Edvinsson, L.; Svensson, R.; Worek, F.; Ekstrom, F.; Linusson, A. Broad-Spectrum Antidote Discovery by Untangling the Reactivation Mechanism of Nerve-Agent-Inhibited Acetylcholinesterase. Chem. Eur. J. 2022, 28, e202200678. [Google Scholar] [CrossRef] [PubMed]
- Sirin, G.S.; Zhang, Y. How is acetylcholinesterase phosphonylated by soman? An ab initio QM/MM molecular dynamics study. J. Phys. Chem. A 2014, 118, 9132–9139. [Google Scholar] [CrossRef]
- Bhakhoa, H.; Rhyman, L.; Ramasami, P. Theoretical study of the molecular aspect of the suspected novichok agent A234 of the Skripal poisoning. R. Soc. Open Sci. 2019, 6, 181831. [Google Scholar] [CrossRef]
- Imrit, Y.A.; Bhakhoa, H.; Sergeieva, T.; Danés, S.; Savoo, N.; Elzagheid, M.I.; Rhyman, L.; Andrada, D.M.; Ramasami, P. A theoretical study of the hydrolysis mechanism of A-234; the suspected novichok agent in the Skripal attack. RSC Adv. 2020, 10, 27884–27893. [Google Scholar] [CrossRef]
- Wang, J.; Gu, J.; Leszczynski, J. Molecular basis of the recognition process: Hydrogen-bonding patterns in the guanine primary recognition site of ribonuclease T1. J. Phys. Chem. B 2006, 110, 7567–7573. [Google Scholar] [CrossRef] [PubMed]
- Franca, T.C.C.; Kitagawa, D.A.S.; Cavalcante, S.F.A.; da Silva, J.A.V.; Nepovimova, E.; Kuca, K. Novichoks: The dangerous fourth generation of chemical weapons. Int. J. Mol. Sci. 2019, 20, 1222. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.A.; Almeida, J.S.F.D.; França, T.C.C.; Borges, I. Electronic and spectroscopic properties of A-series nerve agents. Comput. Theor. Chem. 2021, 1202, 113321. [Google Scholar] [CrossRef]
- Chai, P.R.; Hayes, B.D.; Erickson, T.B.; Boyer, E.W. Novichok agents: A historical, current, and toxicological perspective. Toxicol. Commun. 2018, 2, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yoon, U.H.; Ryu, T.I.; Jeong, H.J.; Kim, S.I.; Park, J.; Kye, Y.S.; Hwang, S.-R.; Kim, D.; Cho, Y.; et al. Calculation of the infrared spectra of organophosphorus compounds and prediction of new types of nerve agents. New J. Chem. 2022, 46, 8653–8661. [Google Scholar] [CrossRef]
- Carlsen, L. After Salisbury Nerve Agents Revisited. Mol. Inform. 2019, 38, e1800106. [Google Scholar] [CrossRef]
- Harvey, S.P.; McMahon, L.R.; Berg, F.J. Hydrolysis and enzymatic degradation of Novichok nerve agents. Heliyon 2020, 6, e03153. [Google Scholar] [CrossRef]
- Mercey, G.; Verdelet, T.; Renou, J.; Kliachyna, M.; Baati, R.; Nachon, F.; Jean, L.; Renard, P.Y. Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. Acc. Chem. Res. 2012, 45, 756–766. [Google Scholar] [CrossRef]
- Sharma, R.; Gupta, B.; Singh, N.; Acharya, J.R.; Musilek, K.; Kuca, K.; Ghosh, K.K. Development and structural modifications of cholinesterase reactivators against chemical warfare agents in last decade: A review. Mini Rev. Med. Chem. 2015, 15, 58–72. [Google Scholar] [CrossRef]
- Hoskovcova, M.; Halamek, E.; Kobliha, Z. Efficacy of structural homoloques and isomers of pralidoxime in reactivation of immobilised acetylcholinesterase inhibited with sarin, cyclosarin and soman. Neuro Endocrinol. Lett. 2009, 30, 152–155. [Google Scholar]
- Kuca, K.; Musilek, K.; Jun, D.; Zdarova-Karasova, J.; Nepovimova, E.; Soukup, O.; Hrabinova, M.; Mikler, J.; Franca, T.C.C.; Da Cunha, E.F.F.; et al. A newly developed oxime K203 is the most effective reactivator of tabun-inhibited acetylcholinesterase. BMC Pharmacol. Toxicol. 2018, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, P.; Remy, B.; Bross, R.P.T.; van Grol, M.; Gaucher, F.; Chabriere, E.; de Koning, M.C.; Daude, D. Enzymatic decontamination of G-type, V-type and Novichok nerve agents. Int. J. Mol. Sci. 2021, 22, 8152. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, J.S.; Guizado, T.R.C.; Guimaraes, A.P.; Ramalho, T.C.; Goncalves, A.S.; de Koning, M.C.; Franca, T.C. Docking and molecular dynamics studies of peripheral site ligand-oximes as reactivators of sarin-inhibited human acetylcholinesterase. J. Biomol. Struct. Dyn. 2016, 34, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- de Castro, A.A.; Soares, F.V.; Pereira, A.F.; Silva, T.C.; Silva, D.R.; Mancini, D.T.; Caetano, M.S.; da Cunha, E.F.F.; Ramalho, T.C. Asymmetric biodegradation of the nerve agents Sarin and VX by human dUTPase: Chemometrics, molecular docking and hybrid QM/MM calculations. J. Biomol. Struct. Dyn. 2019, 37, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, L.; Pan, X. An evaluation of combined strategies for improving the performance of molecular docking. J. Bioinform. Comput. Biol. 2021, 19, 2150003. [Google Scholar] [CrossRef]
- Xu, Y.; Colletier, J.P.; Jiang, H.; Silman, I.; Sussman, J.L.; Weik, M. Backdoor opening mechanism in acetylcholinesterase based on X-ray crystallography and molecular dynamics simulations. Protein Sci. 2008, 17, 601–605. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef]
- SAVES v6.0. Available online: https://saves.mbi.ucla.edu/ (accessed on 2 June 2022).
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).