

Thermal Conductivity Calculation in Organic Liquids: Application to Poly-α-Olefin

Abstract
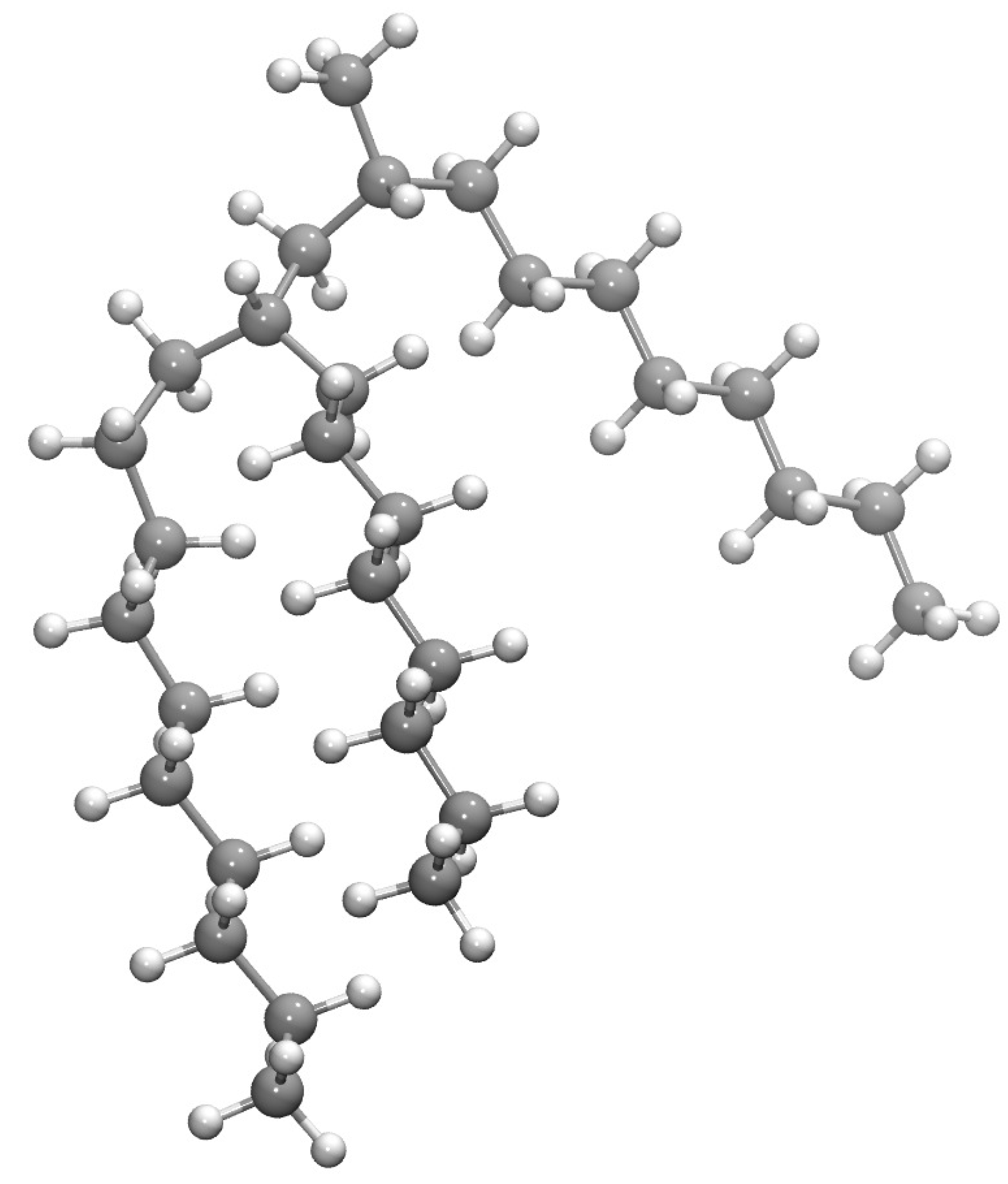
1. Introduction
2. Results and Discussion
2.1. Density Comparison between Force Fields
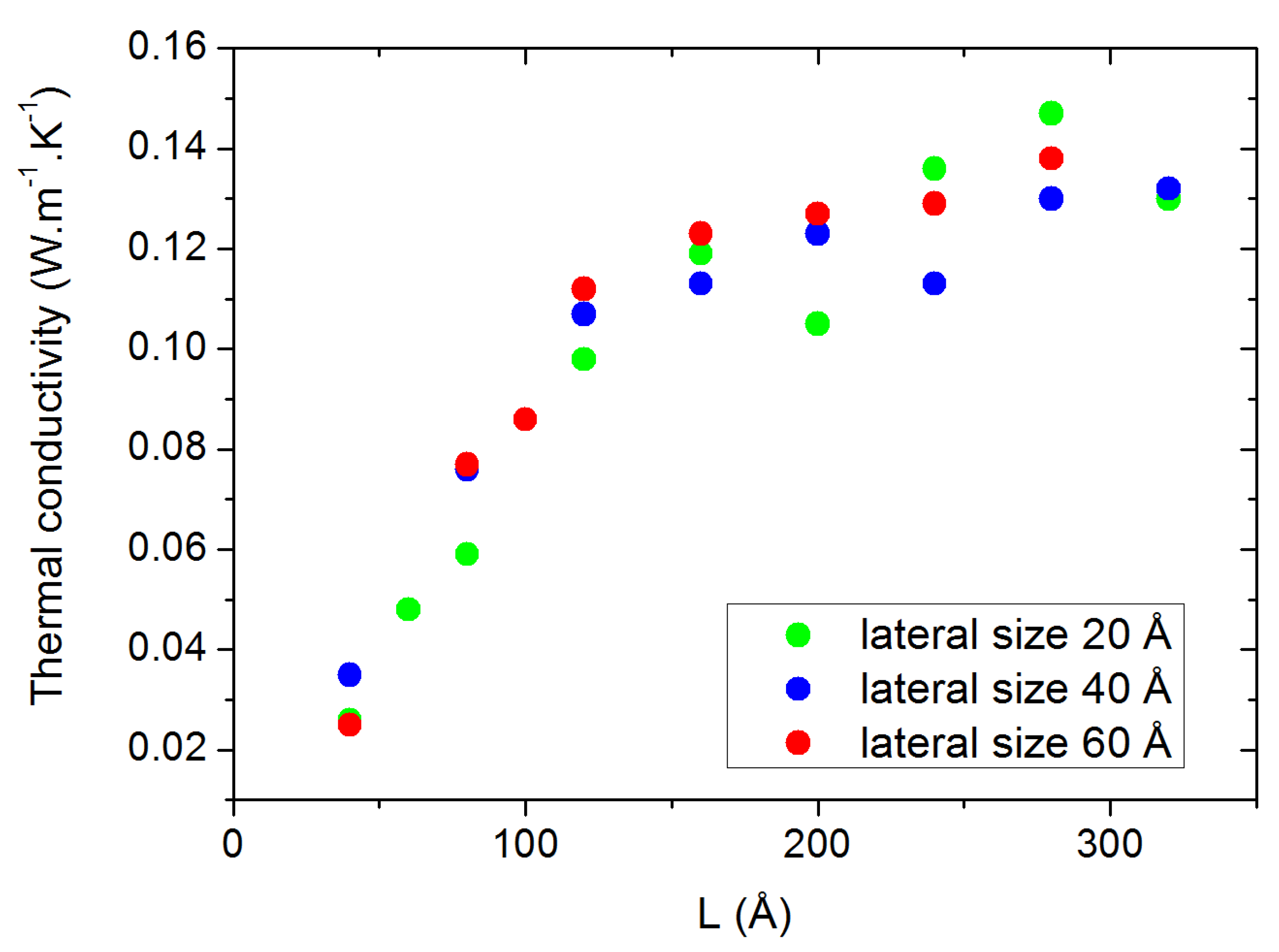
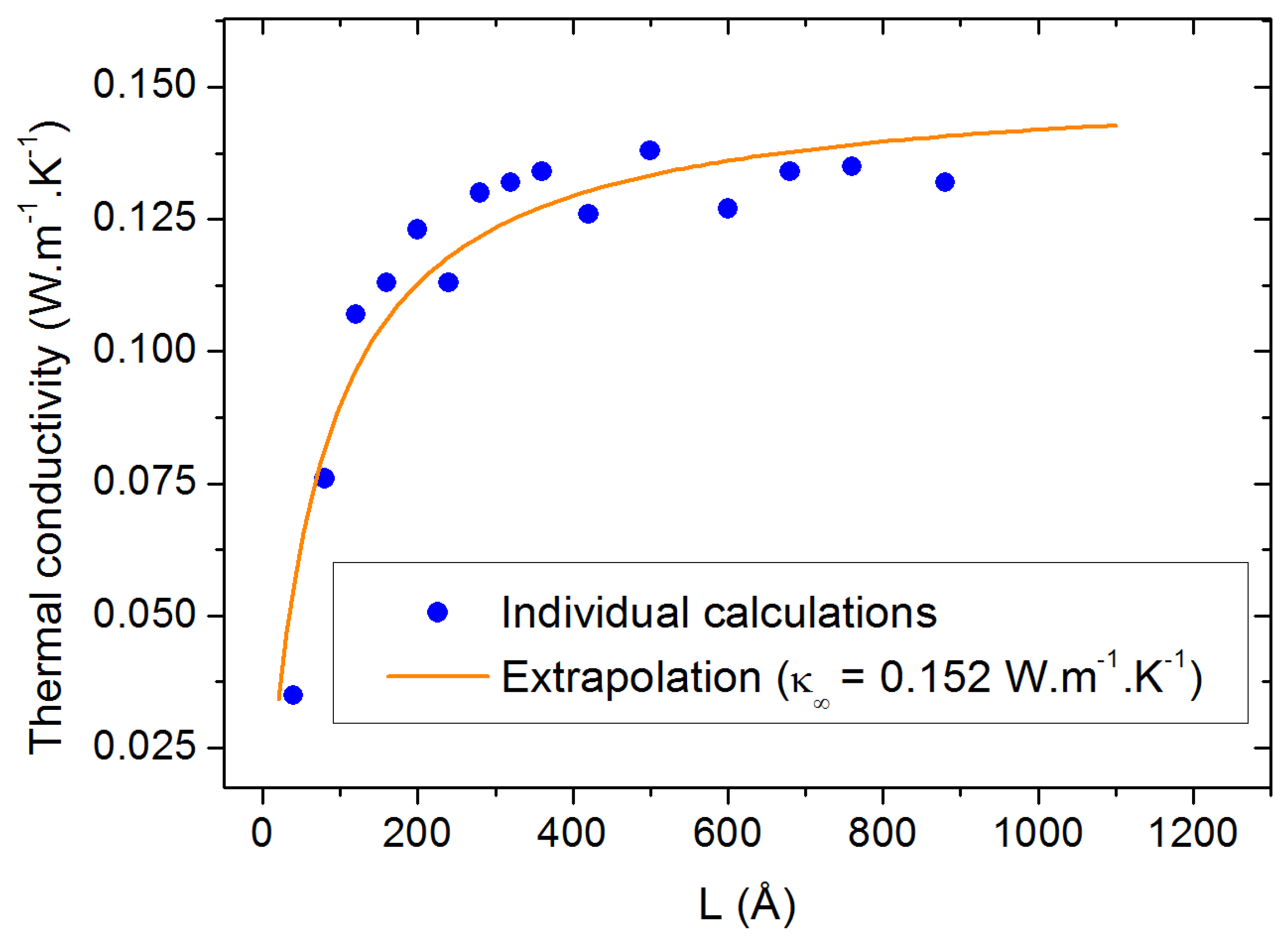
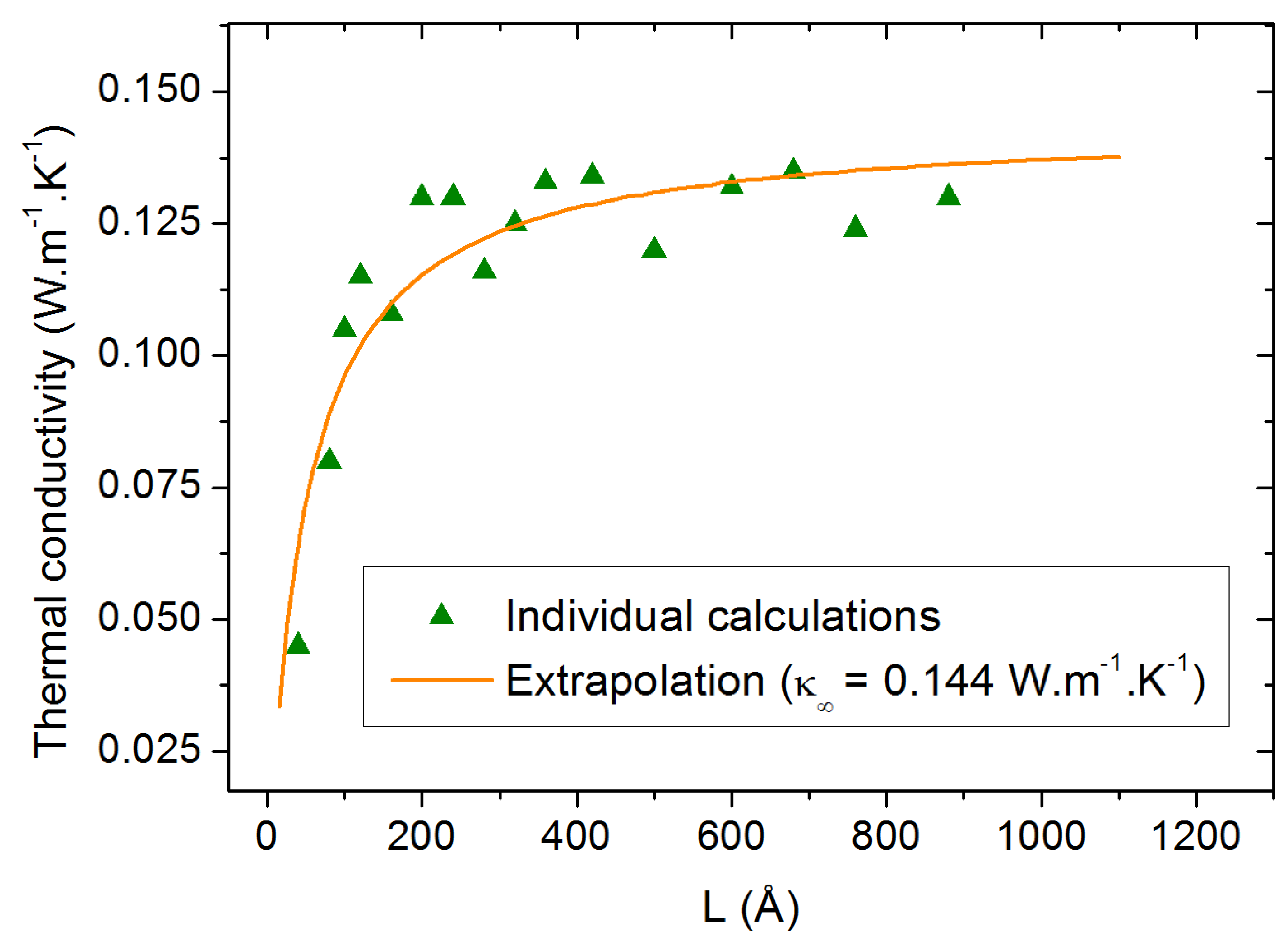
2.2. Size-Dependent Simulations
2.3. Temperature Dependence and Force Field Comparison
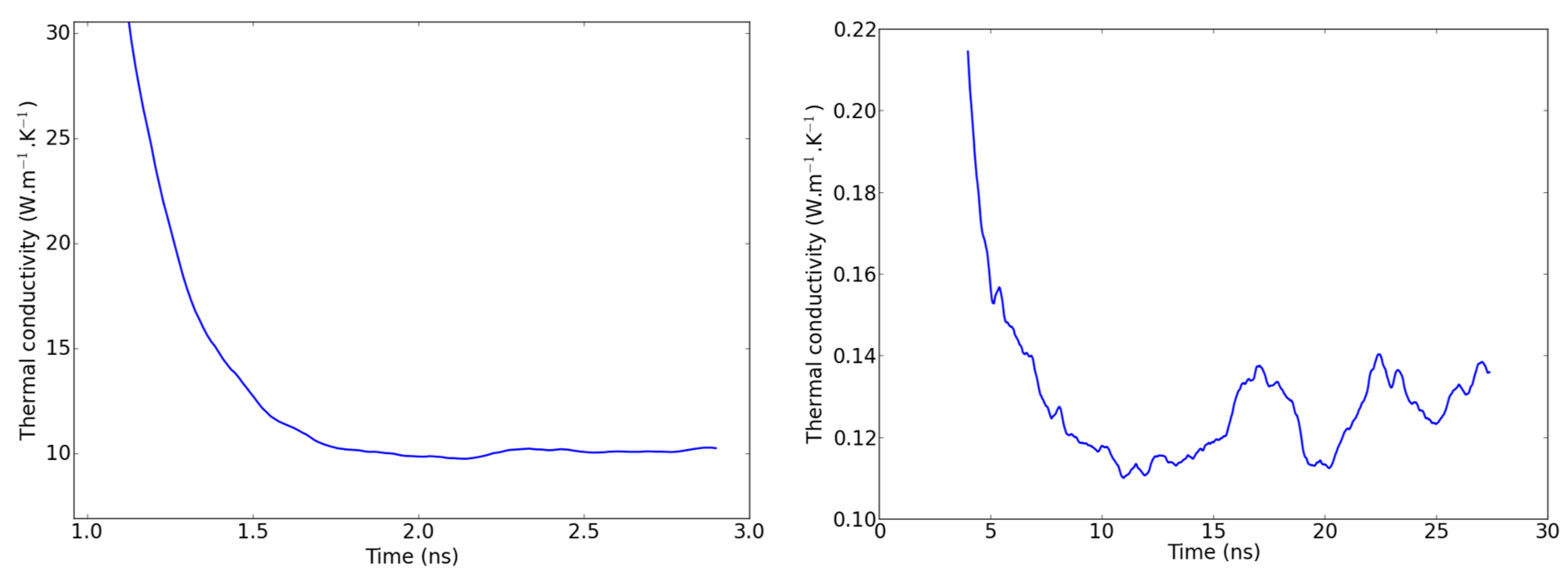
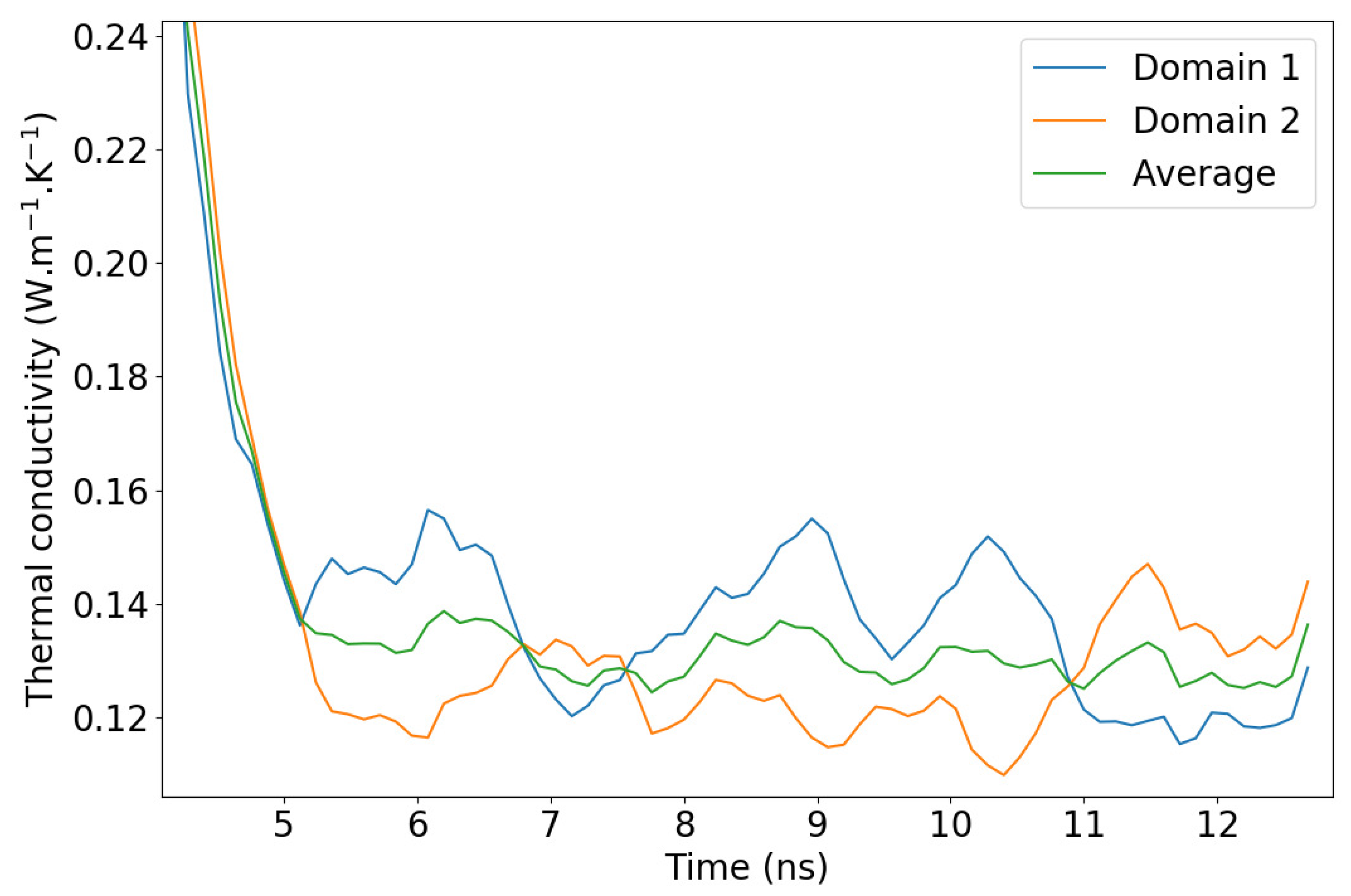
2.4. Remarks on Methodology
3. Methods
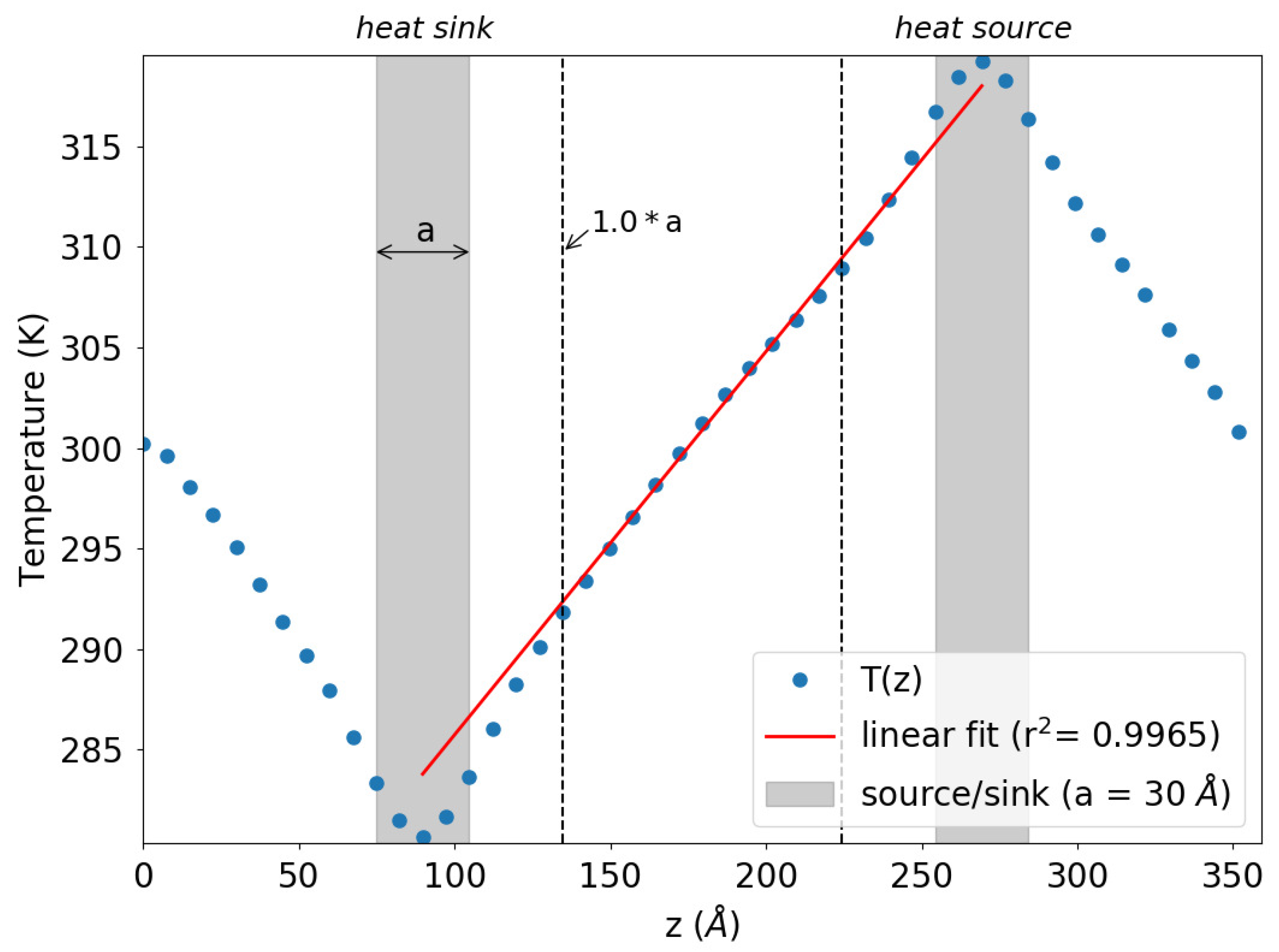
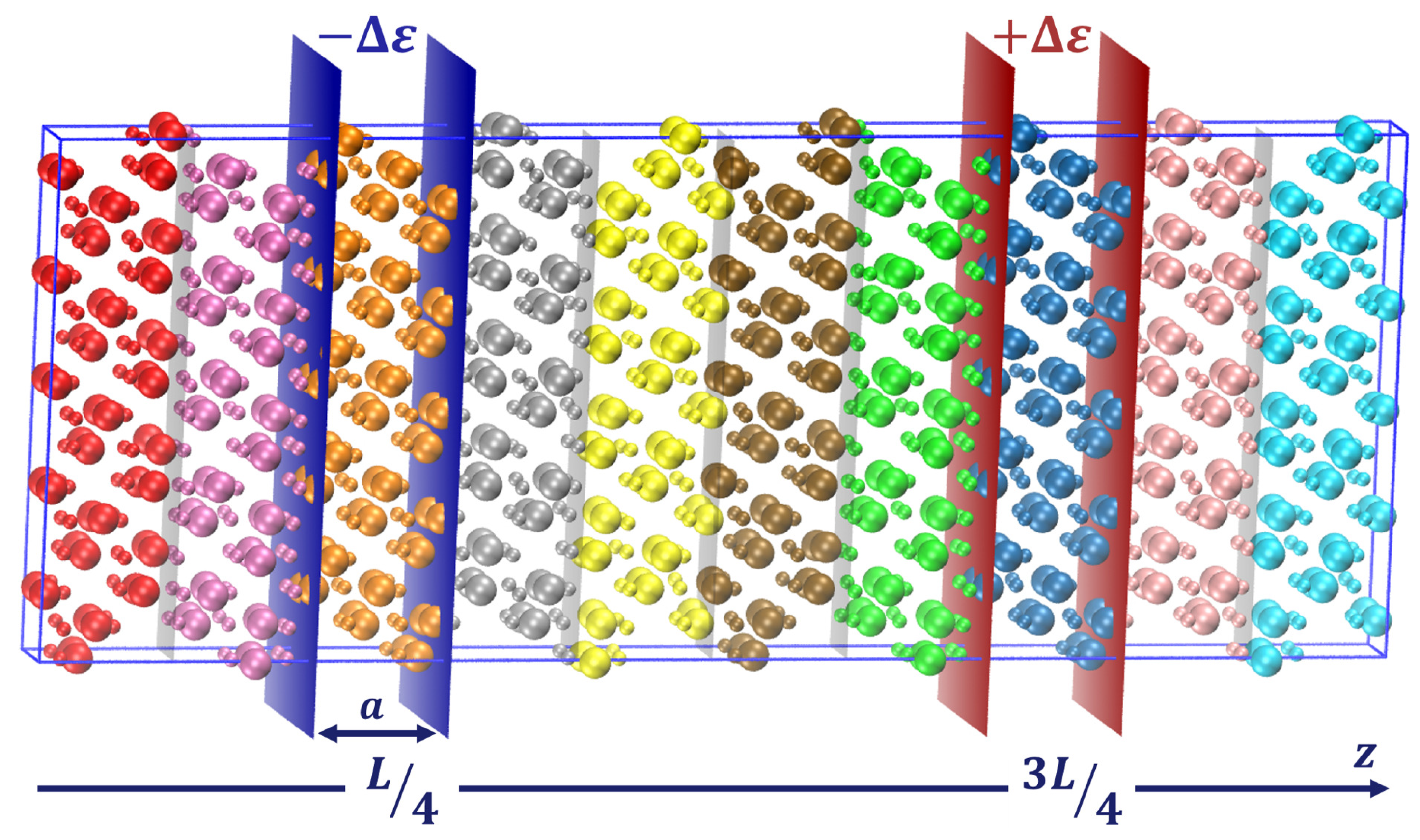
3.1. Molecular Dynamics and NEMD
3.2. Interaction Potential
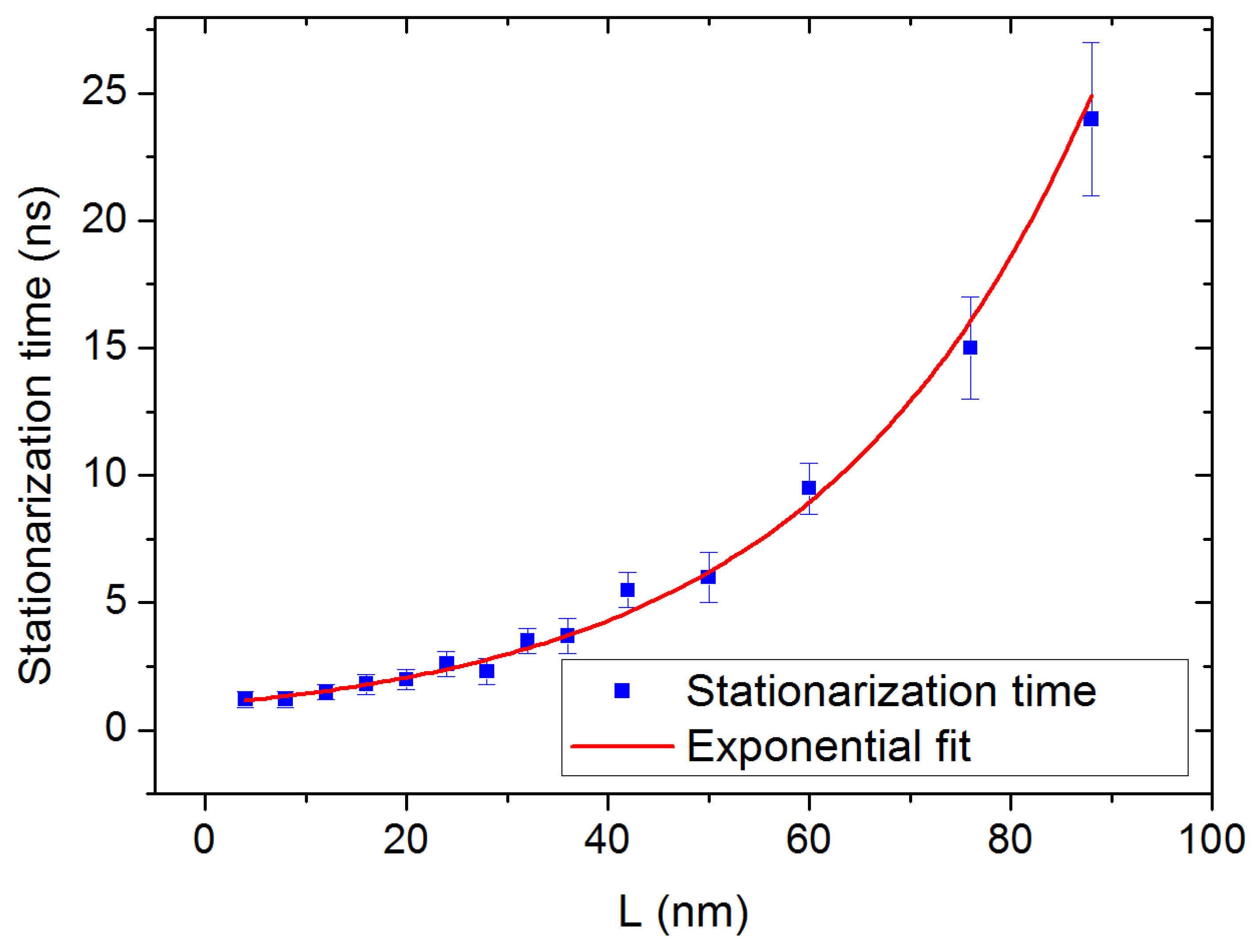
3.3. Finite Size Effects
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinha, S.; Goodson, K.E. Review: Multiscale thermal modeling in nanoelectronics. Int. J. Multiscale Comput. Eng. 2005, 3, 107–133. [Google Scholar] [CrossRef]
- Lee, E.K.; Yin, L.; Lee, Y.; Lee, J.W.; Lee, S.J.; Lee, J.; Cha, S.N.; Whang, D.; Hwang, G.S.; Hippalgaonkar, K.; et al. Large thermoelectric figure-of-merits from SiGe nanowires by simultaneously measuring electrical and thermal transport properties. Nano Lett. 2012, 12, 2918–2923. [Google Scholar] [CrossRef]
- Yang, J. Potential applications of thermoelectric waste heat recovery in the automotive industry. In Proceedings of the ICT 2005, 24th International Conference on Thermoelectrics, Clemson, SC, USA, 19–23 June 2005; IEEE: Piscataway Township, NJ, USA, 2005; pp. 170–174. [Google Scholar]
- Jelle, B.P. Traditional, state-of-the-art and future thermal building insulation materials and solutions–Properties, requirements and possibilities. Energy Build. 2011, 43, 2549–2563. [Google Scholar] [CrossRef]
- Evans, D.J. Homogeneous NEMD algorithm for thermal conductivity—Application of non-canonical linear response theory. Phys. Lett. A 1982, 91, 457–460. [Google Scholar] [CrossRef]
- Schelling, P.K.; Phillpot, S.R.; Keblinski, P. Comparison of atomic-level simulation methods for computing thermal conductivity. Phys. Rev. B 2002, 65, 144306. [Google Scholar] [CrossRef]
- Heino, P.; Ristolainen, E. Thermal conduction at the nanoscale in some metals by MD. Microelectron. J. 2003, 34, 773–777. [Google Scholar] [CrossRef]
- Müller-Plathe, F. A simple nonequilibrium molecular dynamics method for calculating the thermal conductivity. J. Chem. Phys. 1997, 106, 6082–6085. [Google Scholar] [CrossRef]
- Ikeshoji, T.; Hafskjold, B. Non-equilibrium molecular dynamics calculation of heat conduction in liquid and through liquid-gas interface. Mol. Phys. 1994, 81, 251–261. [Google Scholar] [CrossRef]
- Jund, P.; Jullien, R. Molecular-dynamics calculation of the thermal conductivity of vitreous silica. Phys. Rev. B 1999, 59, 13707. [Google Scholar] [CrossRef]
- Aubry, S.; Bammann, D.; Hoyt, J.; Jones, R.; Kimmer, C.; Klein, P.; Wagner, G.; Webb, E., III; Zimmerman, J. A Robust, Coupled Approach for Atomistic-Continuum Simulation; SAND2004-4778; Sandia National Laboratories (SNL): Albuquerque, MN, USA, 2004. [Google Scholar]
- Bracht, H.; Eon, S.; Frieling, R.; Plech, A.; Issenmann, D.; Wolf, D.; Hansen, J.L.; Larsen, A.N.; Ager, J., III; Haller, E. Thermal conductivity of isotopically controlled silicon nanostructures. New J. Phys. 2014, 16, 015021. [Google Scholar] [CrossRef]
- Ge, S.; Chen, M. Vibrational coupling and Kapitza resistance at a solid–liquid interface. Int. J. Thermophys. 2013, 34, 64–77. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, M.; Tang, D. Thermal rectification at silicon/horizontally aligned carbon nanotube interfaces. J. Appl. Phys. 2013, 113, 194307. [Google Scholar] [CrossRef]
- Zhou, Y.; Anglin, B.; Strachan, A. Phonon thermal conductivity in nanolaminated composite metals via molecular dynamics. J. Chem. Phys. 2007, 127, 184702. [Google Scholar] [CrossRef] [PubMed]
- Schelling, P.K.; Phillpot, S.R. Mechanism of Thermal Transport in Zirconia and Yttria-Stabilized Zirconia by Molecular-Dynamics Simulation. J. Am. Ceram. Soc. 2001, 84, 2997–3007. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, J. Thermal conductivity of zeolitic imidazolate framework-8: A molecular simulation study. J. Phys. Chem. C 2013, 117, 18441–18447. [Google Scholar] [CrossRef]
- Severin, J.; Jund, P. Thermal conductivity calculation in anisotropic crystals by molecular dynamics: Application to α-Fe2O3. J. Chem. Phys. 2017, 146, 054505. [Google Scholar] [CrossRef]
- Ewen, J.P.; Gattinoni, C.; Morgan, N.; Spikes, H.A.; Dini, D. Nonequilibrium Molecular Dynamics Simulations of Organic Friction Modifiers Adsorbed on Iron Oxide Surfaces. Langmuir 2016, 32, 4450–4463. [Google Scholar] [CrossRef]
- Gouron, A.; Le Mapihan, K.; Camperos, S.; Al Farra, A.; Lair, V.; Ringuedé, A.; Cassir, M.; Diawara, B. New insights in self-assembled monolayer of imidazolines on iron oxide investigated by DFT. Appl. Surf. Sci. 2018, 456, 437–444. [Google Scholar] [CrossRef]
- Souvi, S.M.O.; Badawi, M.; Paul, J.F.; Cristol, S.; Cantrel, L. A DFT study of the hematite surface state in the presence of H2, H2O and O2. Surf. Sci. 2013, 610, 7–15. [Google Scholar] [CrossRef]
- Loehle, S. Understanding of Adsorption Mechanism and Tribological Behaviors of C18 Fatty Acids on Iron-Based Surfaces: A Molecular Simulation Approach. Ph.D. Thesis, Tōhoku Daigaku, Sendai, Japon, 2014. [Google Scholar]
- Siqueira, A.L.M.; Beaumesnil, M.; Hubert-Roux, M.; Loutelier-Bourhis, C.; Afonso, C.; Pondaven, S.; Bai, Y.; Racaud, A. Characterization of polyalphaolefins using halogen anion attachment in atmospheric pressure photoionization coupled with ion mobility spectrometry-mass spectrometry. Analyst 2018, 143, 3934–3940. [Google Scholar] [CrossRef]
- Parker, W.; Jenkins, R.; Butler, C.; Abbott, G. Flash method of determining thermal diffusivity, heat capacity, and thermal conductivity. J. Appl. Phys. 1961, 32, 1679–1684. [Google Scholar] [CrossRef]
- Tada, Y.; Harada, M.; Tanigaki, M.; Eguchi, W. Laser flash method for measuring thermal conductivity of liquids—application to low thermal conductivity liquids. Rev. Sci. Instruments 1978, 49, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Mahan, G.; Pantelides, S. Dynamical simulations of nonequilibrium processes—Heat flow and the Kapitza resistance across grain boundaries. Solid State Commun. 1997, 102, 517–521. [Google Scholar] [CrossRef]
- Hu, M.; Goicochea, J.V.; Michel, B.; Poulikakos, D. Thermal rectification at water/functionalized silica interfaces. Appl. Phys. Lett. 2009, 95, 151903. [Google Scholar] [CrossRef]
- Jiang, B.; Kim, J.; Keffer, D.J.; Edwards, B. Comparison of perfluoropolyethers and n-alkanes under shear via nonequilibrium molecular dynamics simulation. Mol. Simul. 2008, 34, 231–242. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA; Materials Studio Modeling Environment: San Diego, CA, USA, 2015.
- Wirnsberger, P.; Frenkel, D.; Dellago, C. An enhanced version of the heat exchange algorithm with excellent energy conservation properties. J. Chem. Phys. 2015, 143, 124104. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Damm, W.; Frontera, A.; Tirado-Rives, J.; Jorgensen, W.L. OPLS all-atom force field for carbohydrates. J. Comput. Chem. 1997, 18, 1955–1970. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Jorgensen, W.L. OPLS all-atom model for amines: Resolution of the amine hydration problem. J. Am. Chem. Soc. 1999, 121, 4827–4836. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G. Comparison of the AMBER, CHARMM, COMPASS, GROMOS, OPLS, TraPPE and UFF force fields for prediction of vapor–liquid coexistence curves and liquid densities. Fluid Phase Equilibria 2006, 248, 50–55. [Google Scholar] [CrossRef]
- Sun, H. Ab initio calculations and force field development for computer simulation of polysilanes. Macromolecules 1995, 28, 701–712. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Dauber-Osguthorpe, P.; Roberts, V.A.; Osguthorpe, D.J.; Wolff, J.; Genest, M.; Hagler, A.T. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins Struct. Funct. Bioinform. 1988, 4, 31–47. [Google Scholar] [CrossRef]
- Sellan, D.; Landry, E.; Turney, J.; McGaughey, A.; Amon, C. Size effects in molecular dynamics thermal conductivity predictions. Phys. Rev. B 2010, 81, 214305. [Google Scholar] [CrossRef]
- Hermet, P.; Jund, P. Lattice thermal conductivity of NiTiSn half-Heusler thermoelectric materials from first-principles calculations. J. Alloy. Compd. 2016, 688, 248–252. [Google Scholar] [CrossRef]
- Hu, L.; Evans, W.; Keblinski, P. One-dimensional phonon effects in direct molecular dynamics method for thermal conductivity determination. J. Appl. Phys. 2011, 110, 113511. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temp. | OPLS | PCFF | COMPASS | In-House FF | Exp. 1 |
|---|---|---|---|---|---|
| 300 K | 788 | 742 | 789 | 865 | 806 |
| 500 K | 615 | 595 | 640 | 840 | 679 |
| OPLS | PCFF | COMPASS | In-House FF |
|---|---|---|---|
| 61 | 46 | 48 | 76 |
| Atom | D (eV) | a (1/Å) |
|---|---|---|
| C1-C2 | 3.81598 | 1.915 |
| C1-C3 | 3.81598 | 1.915 |
| C1-H | 4.70927 | 1.771 |
| C2-C2 | 3.81598 | 1.915 |
| C2-C3 | 3.81598 | 1.915 |
| C2-H | 4.70927 | 1.771 |
| C3-H | 4.70927 | 1.771 |
| Atom | z | (eV) | (Å) |
|---|---|---|---|
| C1 | −0.053 | 8.61719 × 10−4 | 4.65000 |
| C2 | −0.106 | 3.96391× 10−3 | 3.95000 |
| C3 | −0.159 | 8.44484 × 10−2 | 2.55485 |
| H | 0.053 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Severin, J.; Loehlé, S.; Jund, P. Thermal Conductivity Calculation in Organic Liquids: Application to Poly-α-Olefin. Molecules 2024, 29, 291. https://doi.org/10.3390/molecules29020291
Severin J, Loehlé S, Jund P. Thermal Conductivity Calculation in Organic Liquids: Application to Poly-α-Olefin. Molecules. 2024; 29(2):291. https://doi.org/10.3390/molecules29020291
Chicago/Turabian StyleSeverin, Jonathan, Sophie Loehlé, and Philippe Jund. 2024. "Thermal Conductivity Calculation in Organic Liquids: Application to Poly-α-Olefin" Molecules 29, no. 2: 291. https://doi.org/10.3390/molecules29020291
APA StyleSeverin, J., Loehlé, S., & Jund, P. (2024). Thermal Conductivity Calculation in Organic Liquids: Application to Poly-α-Olefin. Molecules, 29(2), 291. https://doi.org/10.3390/molecules29020291