
Base-Catalyzed Reaction of Isatins and (3-Hydroxyprop-1-yn-1-yl)phosphonates as a Tool for the Synthesis of Spiro-1,3-dioxolane Oxindoles with Anticancer and Anti-Platelet Properties

 , , , ,
, , , ,  , , , , and
, , , , and
Abstract

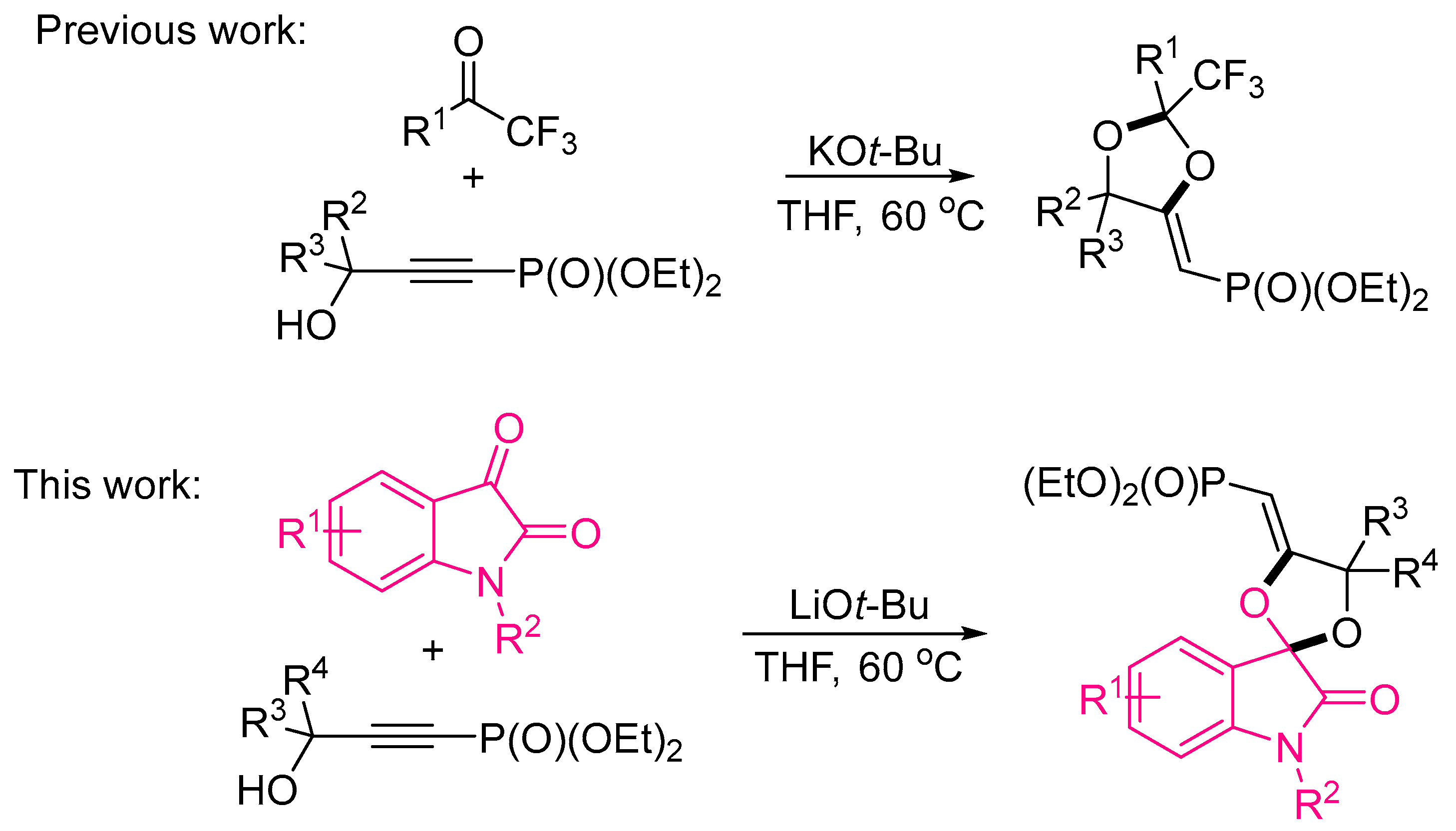
1. Introduction
2. Results
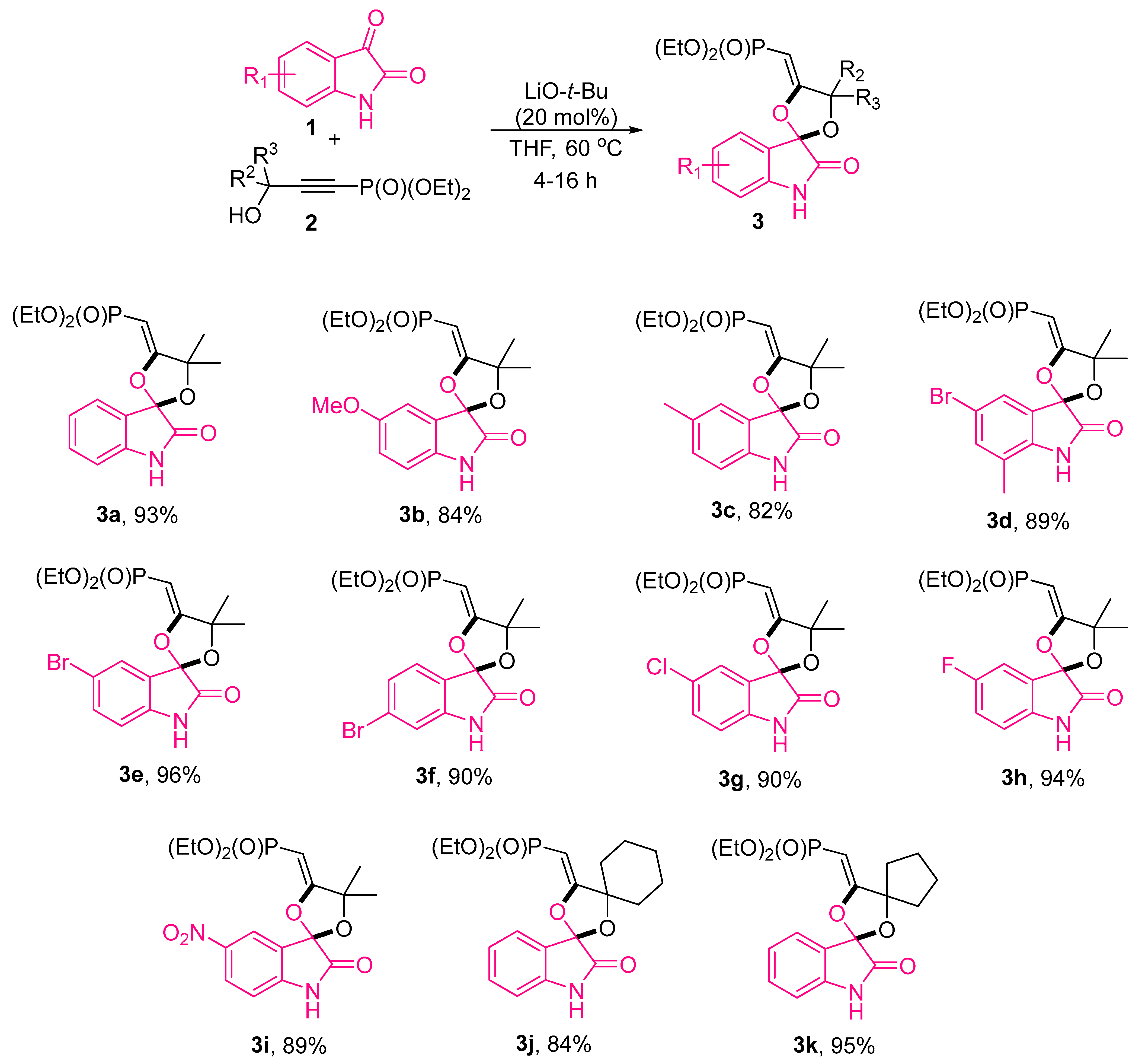
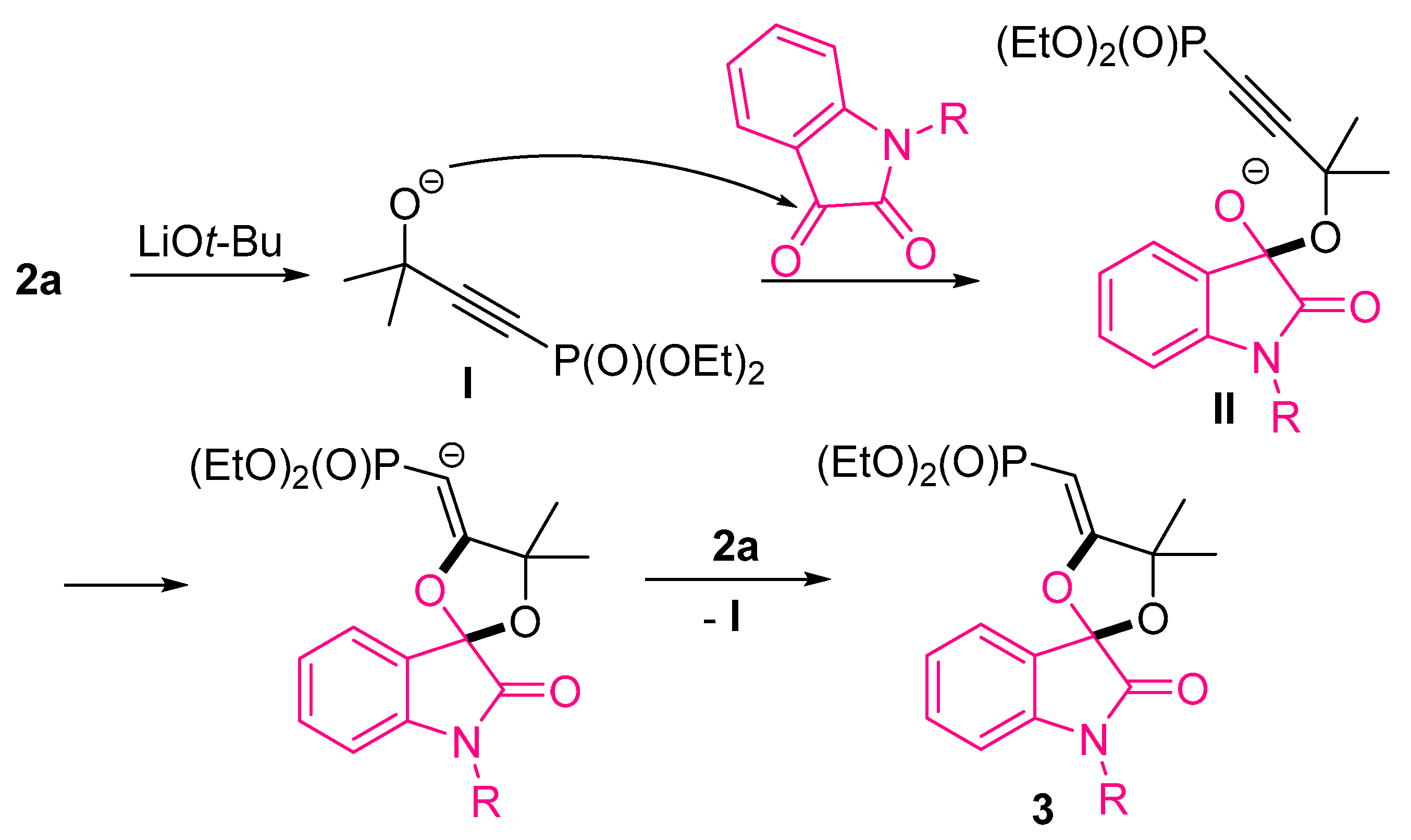
2.1. Synthesis
2.2. Bioactivity
2.2.1. Anticancer Activity
2.2.2. Anticoagulant and Antiaggregating Activities
3. Materials and Methods
3.1. Cells and Materials
3.2. Cytotoxic Assay
3.3. Anticoagulant and Antiaggregation Activities Study
3.4. Chemistry: Synthesis of 3 and 4
3.4.1. General Remarks
3.4.2. General Procedure for the Synthesis of Spiro-1,3-dioxolane Oxindoles 3 and 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Panda, S.S.; Girgis, A.S.; Aziz, M.N.; Bekheit, M.S. Spirooxindole: A Versatile Biologically Active Heterocyclic Scaffold. Molecules 2023, 28, 618. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.-W.; Cui, X.-Y.; Yu, J.-S.; Zhou, J. Spirooxindoles. In Spiro Compounds; Rios Torres, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 103–160. [Google Scholar] [CrossRef]
- Liandi, A.R.; Cahyana, A.H.; Alfariza, D.N.; Nuraini, R.; Sari, R.W.; Wendari, T.P. Spirooxindoles: Recent Report of Green Synthesis Approach. Green Synth. Catal. 2024, 5, 1–13. [Google Scholar] [CrossRef]
- Boddy, A.J.; Bull, J.A. Stereoselective Synthesis and Applications of Spirocyclic Oxindoles. Org. Chem. Front. 2021, 8, 1026–1084. [Google Scholar] [CrossRef]
- Saranya, P.V.; Neetha, M.; Aneeja, T.; Anilkumar, G. Transition Metal-Catalyzed Synthesis of Spirooxindoles. RSC Adv. 2021, 11, 7146–7179. [Google Scholar] [CrossRef]
- Kim, S.Y.; Roh, H.J.; Seo, D.Y.; Ryu, J.Y.; Lee, J.; Kim, J.N. Base-Catalyzed One-Pot Synthesis of Dispiro-1,3-Dioxolane Bisoxindoles from N-Methylisatin and Methyl Propiolate. Tetrahedron Lett. 2017, 58, 914–918. [Google Scholar] [CrossRef]
- Dos Santos, E.L.; Gomes Jr, W.A.; Ribeiro, N.M.; Andrade, H.M.C. Ecofriendly Ketalization of Isatin Using Heteropolycompounds as Catalysts. J. Mol. Catal. A Chem. 2008, 295, 18–23. [Google Scholar] [CrossRef]
- Ribeiro, N.M.; da Cunha Pinto, A.; da Silva, B.V.; de Almeida Violante, F.; Dias, M.O. A Fast, Efficient and Eco-Friendly Procedure to Prepare Isatin Ketals. Catal. Commun. 2007, 8, 2130–2136. [Google Scholar] [CrossRef]
- Khorshidi, A.; Shariati, S.; Rahimi, P. Magnetite Nanoparticles Catalyzed Preparation of Isatin Ketals under Solvent Free Conditions Promoted by Ultrasound Irradiation. Arab. J. Chem. 2019, 12, 2470–2475. [Google Scholar] [CrossRef]
- Rekhter, M.A.; Radul, O.M.; Bukhanyuk, S.M. Synthesis of N-(2-Chloroethyl)Isatins and Their Β-Ethyleneacetals and Β-Thiosemicarbazones. Chem. Heterocycl. Compd. 1999, 35, 792–794. [Google Scholar] [CrossRef]
- Zhunghietu, G. Isatin Β-Ethylene Ketals as Psychotropics. Rev. Roum. Chim. 2002, 46, 517–520. [Google Scholar]
- Rajopadhye, M.; Popp, F.D. Potential Anticonvulsants. 11. Synthesis and Anticonvulsant Activity of Spiro[1,3-Dioxolane-2,3′-Indolin]-2′-Ones and Structural Analogs. J. Med. Chem. 1988, 31, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Geronikaki, A.; Babaev, E.; Dearden, J.; Dehaen, W.; Filimonov, D.; Galaeva, I.; Krajneva, V.; Lagunin, A.; Macaev, F.; Molodavkin, G.; et al. Design, Synthesis, Computational and Biological Evaluation of New Anxiolytics. Bioorg. Med. Chem. 2004, 12, 6559–6568. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Sudo, G.; Pontes, L.B.; Gabriel, D.; Mendes, T.C.F.; Ribeiro, N.M.; Pinto, A.C.; Trachez, M.M.; Sudo, R.T. Sedative–Hypnotic Profile of Novel Isatin Ketals. Pharmacol. Biochem. Behav. 2007, 86, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Melis, C.; Meleddu, R.; Angeli, A.; Distinto, S.; Bianco, G.; Capasso, C.; Cottiglia, F.; Angius, R.; Supuran, C.T.; Maccioni, E. Isatin: A privileged scaffold for the design of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 68–73. [Google Scholar] [CrossRef]
- Singh, G.S.; Desta, Z.Y. Isatins As Privileged Molecules in Design and Synthesis of Spiro-Fused Cyclic Frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef]
- Moradi, R.; Ziarani, G.M.; Lashgari, N. Recent applications of isatin in the synthesis of organic compounds. Arkivoc 2017, 12, 148. [Google Scholar] [CrossRef]
- Borad, M.A.; Bhoi, M.N.; Prajapati, N.P.; Patel, H.D. Review of Synthesis of Spiro Heterocyclic Compounds from Isatin. Synth. Commun. 2014, 44, 897. [Google Scholar] [CrossRef]
- Zhungietu, G.I.; Rekhter, M.A. Izatin i ego proizvodnye (Isatin and Its Derivatives); Shtiintsa: Kishinev, Moldova, 1977; pp. 1–229. [Google Scholar]
- Bogdanov, A.V.; Mironov, V.F. Synthesis of isatoic anhydride derivatives. Chem. Heterocycl. Compd. 2016, 52, 90. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Musin, L.I.; Mironov, V.F. Advances in the synthesis and application of isoindigo derivatives. Arkivoc 2015, 6, 362. [Google Scholar] [CrossRef]
- Medvedev, A.; Buneeva, O.; Glover, V. Iological Targets for Isatin and Its Analogues: Implication for Therapy. Biol. Targets Ther. 2007, 1, 151. [Google Scholar]
- Pal, M.; Sharma, N.K.; Priyanka, J.K.K. Synthetic and Biological Multiplicity of Isatin: A Review. J. Adv. Sci. Res. 2011, 2, 35. [Google Scholar]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Ranson, M.; Skropeta, D. Cytotoxic and Anticancer Activities of Isatin and Its Derivatives: A Comprehensive Review from 2000–2008. Anti-Cancer Agents Med. Chem. 2009, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Darji, N.; Pillai, J.; Patel, B. Recent advance in anti-cancer activity of indole derivatives. Int. J. Drug Res. Tech. 2012, 2, 225–230. [Google Scholar]
- Sriram, D.; Yogeeswari, P. Towards the design and development of agents with broad spectrum chemotherapeutic properties for the effective treatment of HIV/AIDS. Curr. Med. Chem. 2003, 10, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Skropeta, D. Recent highlights in the development of isatin-based anticancer agents. Adv. Anticancer Agents Med. Chem. 2013, 2, 254–312. [Google Scholar] [CrossRef]
- Rane, R.A.; Karunanidhi, S.; Jain, K.; Shaikh, M.; Hampannavar, G.; Karpoormath, R. A recent perspective on discovery and development of diverse therapeutic agents inspired from isatin alkaloids. Curr. Top. Med. Chem. 2016, 16, 1262. [Google Scholar] [CrossRef]
- Hou, J.; Jin, K.; Li, J.; Jiang, Y.; Li, X.; Wang, X.; Huang, Y.; Zhang, Y.; Xu, W. LJNK, an indoline-2,3-dione-based aminopeptidase N inhibitor with promising antitumor potency. Anti-Cancer Drugs 2016, 27, 496. [Google Scholar] [CrossRef]
- Pape, V.F.S.; Tóth, S.; Füredi, A.; Szebényi, K.; Lovrics, A.; Szabó, P.; Wiese, M.; Szakács, G. Design, synthesis and biological evaluation of thiosemicarbazones, hydrazinobenzothiazoles and arylhydrazones as anticancer agents with a potential to overcome multidrug resistance. Eur. J. Med. Chem. 2016, 117, 335. [Google Scholar] [CrossRef]
- Chen, G.; Ning, Y.; Zhao, W.; Zhang, Y.; Zhang, Y.; Hao, X.; Wang, Y.; Mu, S. Synthesis, Neuro-protection and Anti-cancer Activities of Simple Isatin Mannich and Schiff Bases. Lett. Drug Des. Discov. 2016, 13, 395. [Google Scholar] [CrossRef]
- Rana, S.; Blowers, E.C.; Tebbe, C.; Contreras, J.I.; Radhakrishnan, P.; Kizhake, S.; Zhou, T.; Rajule, R.N.; Arnst, J.L.; Munkarah, A.R.; et al. Isatin Derived Spirocyclic Analogues with α-Methylene-γ-butyrolactone as Anticancer Agents: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 5121. [Google Scholar] [CrossRef]
- Premanathan, M.; Radhakrishnan, S.; Kulangiappar, K.; Singaravelu, G.; Thirumalaiarasu, V.; Sivakumar, T.; Kathiresan, K. Antioxidant & anticancer activities of isatin (1H-indole-2,3-dione), isolated from the flowers of Couroupita guianensis Aubl. Indian J. Med. Res. 2012, 136, 822. [Google Scholar] [PubMed]
- Corona, A.; Meleddu, R.; Esposito, F.; Distinto, S.; Bianco, G.; Masaoka, T.; Maccioni, E.; Menйndez-Arias, L.; Alcaro, S.; Le Grice, S.F.J.; et al. Ribonuclease H/DNA Polymerase HIV-1 Reverse Transcriptase Dual Inhibitor: Mechanistic Studies on the Allosteric Mode of Action of Isatin-Based Compound RMNC6. PLoS ONE 2016, 11, e0147225. [Google Scholar] [CrossRef]
- Meleddu, R.; Distinto, S.; Corona, A.; Bianco, G.; Cannas, V.; Esposito, F.; Artese, A.; Alcaro, S.; Matyus, P.; Bogdan, D.; et al. (3Z)-3-(2-[4-(aryl)-1,3-thiazol-2-yl]hydrazin-1-ylidene)-2,3-dihydro-1H-indol-2-one derivatives as dual inhibitors of HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2015, 93, 452. [Google Scholar] [CrossRef]
- Tavari, M.; Malan, S.F.; Joubert, J. Design, synthesis, biological evaluation and docking studies of sulfonyl isatin derivatives as monoamine oxidase and caspase-3 inhibitors. Med. Chem. Commun. 2016, 7, 1628. [Google Scholar] [CrossRef]
- Akdemir, A.; Guzel-Akdemir, O.; Karali, N.; Supuran, C.T. Isatin analogs as novel inhibitors of Candida spp. β-carbonic anhydrase enzymes. Bioorg. Med. Chem. 2016, 24, 1648. [Google Scholar] [CrossRef] [PubMed]
- Chundawat, T.S.; Kumari, P.; Sharma, N.; Bhagat, S. Strategic synthesis and in vitro antimicrobial evaluation of novel difluoromethylated 1-(1, 3-diphenyl-1H-pyrazol-4-yl)-3, 3-difluoro-1, 3-dihydro-indol-2-ones. Med. Chem. Res. 2016, 25, 2335. [Google Scholar] [CrossRef]
- Sahoo, S.; Mahendrakumar, C.B.; Setty, C.M. Synthesis, antiinflammatory and antibacterial activities of some substituted isatin and isatin fused with 3-substituted 4-amino-5-mercapto-1, 2, 4-triazoles. Int. J. Chem. Sci. 2015, 13, 613. [Google Scholar]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Synthesis of new 4-thiazolidinone-, pyrazoline-, and isatin-based conjugates with promising antitumor activity. J. Med. Chem. 2012, 55, 8630. [Google Scholar] [CrossRef]
- Chiyanzu, I.; Clarkson, C.; Smith, P.J.; Lehman, J.; Gut, J.; Rosenthal, P.J.; Chibale, K. Design, synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg. Med. Chem. 2005, 13, 3249. [Google Scholar] [CrossRef]
- Raj, R.; Biot, C.; Carrere-Kremer, S.; Kremer, L.; Guerardel, Y.; Gut, J.; Rosenthal, P.J.; Forge, D.; Kumar, V. 7-chloroquinoline-isatin conjugates: Antimalarial, antitubercular, and cytotoxic evaluation. Chem. Biol. Drug Des. 2014, 83, 622. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, S.; Gao, C.; Fan, J.; Zhao, F.; Lv, Z.-S.; Feng, L.-S. Isatin hybrids and their anti-tuberculosis activity. Chin. Chem. Lett. 2017, 28, 159. [Google Scholar] [CrossRef]
- Musin, L.I.; Bogdanov, A.V.; Mironov, V.F. Isatin Derivatives in Reactions with Phosphorus(Iii–V) Compounds. Chem. Heterocycl. Compd. 2015, 51, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Brandao, P.; Marques, C.S.; Carreiro, E.P.; Pineiro, M.; Burke, A.J. Engaging Isatins in Multicomponent Reactions (MCRs)—Easy Access to Structural Diversity. Chem. Rec. 2021, 21, 1–115. [Google Scholar] [CrossRef]
- Mayorquín-Torres, M.C.; Simoens, A.; Bonneure, E.; Stevens, C.V. Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity: An Update 2004–2024. Chem. Rev. 2024, 124, 7907–7975. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.-Y.; Zou, Y.-X. Recent Advances in the Construction of Phosphorus-Substituted Heterocycles, 2009–2019. Adv. Synth. Catal. 2020, 362, 1724–1818. [Google Scholar] [CrossRef]
- Gasperi, T.; Loreto, M.A.; Migliorini, A.; Ventura, C. Synthesis of Aziridine- and Oxirane-2-Phosphonates Spiro-Fused with Oxindoles. Eur. J. Org. Chem. 2011, 2, 385–391. [Google Scholar] [CrossRef]
- Miceli, M.; Mazziotta, A.; Palumbo, C.; Roma, E.; Tosi, E.; Longhi, G.; Abbate, S.; Lupattelli, P.; Mazzeo, G.; Gasperi, T. Asymmetric Synthesis of Spirooxindoles Via Nucleophilic Epoxidation Promoted by Bifunctional Organocatalysts. Molecules 2018, 23, 438. [Google Scholar] [CrossRef]
- Huang, N.; Zou, L.; Peng, Y. Enantioselective 1,3-Dipolar Cycloaddition of Methyleneindolinones with A-Diazomethylphosphonate to Access Chiral Spiro-Phosphonylpyrazoline-Oxindoles Catalyzed by Tertiary Amine Thiourea and 1,5-Diazabicyclo[4.3.0]Non-5-Ene. Org. Lett. 2017, 19, 5806–5809. [Google Scholar] [CrossRef]
- Du, T.; Du, F.; Ning, Y.; Peng, Y. Organocatalytic Enantioselective 1,3-Dipolar Cycloadditions between Seyferth–Gilbert Reagent and Isatylidene Malononitriles: Synthesis of Chiral Spiro-Phosphonylpyrazoline-Oxindoles. Org. Lett. 2015, 17, 1308–1311. [Google Scholar] [CrossRef]
- Zhou, Q.-Q.; Yuan, X.; Xiao, Y.-C.; Dong, L.; Chen, Y.-C. Aminocatalytic Asymmetric Diels–Alder Reaction of Phosphorus Dienophiles and 2,4-Dienals. Tetrahedron 2013, 69, 10369–10374. [Google Scholar] [CrossRef]
- Zhou, Q.-Q.; Xiao, Y.-C.; Yuan, X.; Chen, Y.-C. Asymmetric Diels-Alder Reactions of 2,4,6-Trienals via Tetraenamine Catalysis. Asian J. Org. Chem. 2014, 3, 545–549. [Google Scholar] [CrossRef]
- Mitrofanov, A.Y.; Nefedov, S.E.; Beletskaya, I.P. Base-Promoted Synthesis of Trifluoromethylated (1,3-Dioxolan-4-Ylidene)Methylphosphonates from Trifluoromethylketones and Ethynylphosphonates. Asian J. Org. Chem. 2021, 10, 2611–2617. [Google Scholar] [CrossRef]
- Salah Ayoup, M.; Wahby, Y.; Abdel-Hamid, H.; Ramadan, E.S.; Teleb, M.; Abu-Serie, M.M.; Noby, A. Design, synthesis and biological evaluation of novel α-acyloxy carboxamides via Passerini reaction as caspase 3/7 activators. Eur. J. Med. Chem. 2019, 168, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Mackman, N.; Falanga, A.; Pabinger, I.; Noble, S.; Ageno, W.; Moik, F.; Lee, A.Y.Y. Cancer-associated venous thromboembolism. Nat. Rev. Dis. Primer. 2022, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Mulder, F.I.; Horváth-Puhó, E.; van Es, N.; van Laarhoven, H.W.M.; Pedersen, L.; Moik, F.; Ay, C.; Büller, H.R.; Sørensen, H.T. Venous thromboembolism in cancer patients: A population-based cohort study. Blood 2021, 137, 1959–1969. [Google Scholar] [CrossRef]
- Moik, F.; Ay, C.; Pabinger, I. Risk prediction for cancer-associated thrombosis in ambulatory patients with cancer: Past, present and future. Thromb. Res. 2020, 191, S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Moik, F.; van Es, N.; Posch, F.; Di Nisio, M.; Fuereder, T.; Preusser, M.; Pabinger, I.; Ay, C. Gemcitabine and Platinum-Based Agents for the Prediction of Cancer-Associated Venous Thromboembolism: Results from the Vienna Cancer and Thrombosis Study. Cancers 2020, 12, 2493. [Google Scholar] [CrossRef]
- Moik, F.; Ay, C. Hemostasis and cancer: Impact of haemostatic biomarkers for the prediction of clinical outcomes in patients with cancer. J. Thromb. Haemost. JTH 2022, 20, 2733–2745. [Google Scholar] [CrossRef]
- Born, G. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- Morozova, J.E.; Gilmullina, Z.R.; Voloshina, A.D.; Lyubina, A.P.; Amerhanova, S.K.; Syakaev, V.V.; Babaeva, O.B.; Ziganshina, A.Y.; Mukhametzyanov, T.A.; Samorodov, A.V.; et al. Calix[4]Resorcinarene Carboxybetaines and Carboxybetaine Esters: Synthesis, Investigation of In Vitro Toxicity, Anti-Platelet Effects, Anticoagulant Activity, and BSA Binding Affinities. Int. J. Mol. Sci. 2022, 23, 15298. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Base (mol%) | Solvent | T, °C | t, hours | Yield (Conversion), % | Z/E |
| 1 | t-BuOK (20) | THF | 60 | 16 | 100 (100) | 93/7 |
| 2 | t-BuOK (20) | THF | 25 | 16 | 5 (5) | - |
| 3 | t-BuOK (20) | toluene | 60 | 16 | 100 (100) | 84/16 |
| 4 | t-BuOK (20) | dioxane | 60 | 16 | 85 (85) | 91/9 |
| 5 | t-BuOK (20) | MTBE | 60 | 16 | 42 (42) | 92/8 |
| 6 | t-BuOK (20) | EtOH | 60 | 16 | 95 (100) | 87/13 |
| 7 | Cs2CO3 (20) | THF | 60 | 16 | 100 (100) | 86/14 |
| 8 | K2CO3 (20) | THF | 60 | 24 | 93 (93) | 91/9 |
| 9 | DBU (20) | THF | 60 | 24 | 88 (88) | 91/9 |
| 10 | TEA (20) | THF | 60 | 24 | 0 | - |
| 11 | t-BuONa (20) | THF | 60 | 16 | 78 (78) | 96/4 |
| 12 | t-BuOLi (20) | THF | 60 | 4 | 100 (100) | >99/1 |
| Cmpd | Cancer Cell Line | Normal Cell Line | |||
|---|---|---|---|---|---|
| M-HeLa | HuTu 80 | Chang Liver | |||
| IC50 | SI | IC50 | SI | ||
| 3b | 91.0 ± 7.3 | 1.2 | 118 ± 9.3 | ns | 108 ± 8.4 |
| 3c | 102.6 ± 8.2 | ns | 180 ± 14.4 | ns | 84.5 ± 6.8 |
| 3d | 81.0 ± 6.5 | 2.5 | 119 ± 9.5 | 1.7 | 200 ± 16 |
| 3e | 86.1 ± 6.9 | 2.1 | 68.6 ± 5.5 | 2.7 | 185 ± 14.8 |
| 3f | 76.0 ± 6.1 | 1.4 | 131 ± 10 | ns | 103 ± 8.2 |
| 3g | 92.7 ± 7.4 | 1.3 | 113.3 ± 9 | 1.0 | 118 ± 9.3 |
| 3h | 107.0 ± 8.7 | 1.6 | 110.7 ± 8.9 | 1.5 | 167 ± 13.3 |
| 3i | 99.3 ± 7.8 | 1.3 | 136.2 ± 11 | ns | 125 ± 9.9 |
| 3j | 84.5 ± 6.8 | 2.5 | 124 ± 10 | 1.7 | 208 ± 16.4 |
| 3k | 92.8 ± 7.4 | ns | 107.3 ± 8.6 | ns | 61.2 ± 4.9 |
| 4a | 104.0 ± 8.1 | 1.9 | 129.0 ± 10 | 1.5 | 195 ± 15 |
| 4b | 29.0 ± 2.3 | 1.3 | 31.4 ± 2.5 | 1.2 | 39.0 ± 2.8 |
| 4c | 23.4 ± 5.0 | 1.1 | 15.4 ± 1.3 | 1.6 | 25 ± 2 |
| 4d | 30.5 ± 3.6 | 1.3 | 14.3 ± 0.4 | 3.0 | 40.0 ± 3.1 |
| 4e | 23.5 ± 1.8 | 1.1 | 23.3 ± 1.7 | 1.1 | 26 ± 2.1 |
| 3a | na | - | na | - | nd |
| 3j | na | - | na | - | nd |
| 5-fluorouracil | 75.4 ± 5.9 | 1.1 | 65.2 ± 5.6 | 1.3 | 83.3 ± 6.7 |
| Cmpd | Latent Period, % of Control | Maximum Amplitude (MA), % of Control | Aggregation Rate, % of Control | Time to MA, % of Control | APTT $, % of Control |
|---|---|---|---|---|---|
| 3b | −2.4 (1.7–3.5) | −3.8 (3.1–4.2) # | −11.4 (10.5–12.7) * | −18.5 (16.2–19.7) *,# | +5.6 (4.9–7.2) *,† |
| 3c | −3.7 (3.1–4.2) | −2.4 (2.1–3.5) # | −13.7 (12.5–16.3) *,# | +8.4 (7.5–9.6) *,# | +7.7 (6.5–8.4) *,† |
| 3d | +4.8 (3.7–5.6) # | −15.4 (13.3–16.7) * | −7.6 (7.1–10.5) * | +14.8 (13.5–16.7) * | +4.7 (3.8–5.8) † |
| 3e | +6.5 (4.7–7.6) *,# | −12.1 (10.5–14.2) * | −31.7 (30.6–34.2) *,# | −18.2 (17.4–21.3) *,# | +4.9 (4.1–8.3) *,† |
| 3f | +2.3 (1.7–3.5) # | −9.5 (8.2–10.9) *,# | −16.2 (15.7–17.1) *,# | −11.9 (10.2–13.6) *,# | +3.8 (2.4–4.3) † |
| 3g | +20.3 (19.7–21.4) *,# | −20.1 (18.7–23.5) *,# | −18.4 (17.4–20.5) *,# | −21.4 (17.4–22.9) *,# | +8.3 (7.2–10.1) *,† |
| 3h | +13.4 (11.7–15.2) *,# | −15.3 (14.8–18.2) * | −13.6 (12.4–14.9) * | −22.5 (21.7–23.9) *,# | +8.7 (6.8–10.2) *,† |
| 3i | +5.1 (4.7–5.4) # | −5.3 (4.7–7.5) *,# | +2.7 (2.5–3.9) # | −16.4 (16.1–19.5) *,# | +5.7 (4.1–7.6) *,† |
| 3j | +3.1 (2.9–4.2) # | −4.2 (2.6–4.8) # | −10.4 (9.3–12.7) * | +12.6 (10.3–15.7) * | +7.3 (5.9–8.2) † |
| 3k | +7.3 (6.2–8.5) *,# | −11.5 (9.2–13.1) * | −27.4 (26.3–29.5) *,# | −5.1 (3.7–6.4) *,# | +9.7 (8.8–10.5) *,† |
| 4a | +25.7 (24.8–27.5) *,# | −14.2 (13.1–15.7) * | −12.4 (10.2–14.3) * | +12.3 (11.7–13.5) * | +6.1 (4.5–7.9) † |
| 4b | −3.1 (2.9–4.1) | −2.4 (1.7–2.6) # | −4.1 (3.9–5.6) # | −9.7 (8.1–11.4) *,# | +7.4 (6.3–9.2) *,† |
| 4c | +5.2 (4.7–6.1) *,# | −6.8 (5.7–7.3) *,# | −8.2 (7.1–10.4) * | +11.8 (10.4–13.7) * | +2.5 (1.7–3.2) † |
| 4d | +4.3 (3.8–5.7) # | −6.7 (5.4–8.3) *,# | −7.9 (7.1–10.4) *,# | +10.2 (9.1–13.4) * | +2.7 (1.7–2.8) † |
| 4e | +2.6 (2.1–3.8) # | −7.2 (5.4–9.2) *,# | +3.1 (2.9–4.2) # | −11.4 (9.4–12.3) *,# | +2.4 (1.8–3.7) † |
| Acetylsalicylic acid | −2.1 (1.1–2.6) | −13.7 (10.8–16.4) * | −10.5 (7.6–12.3) * | +10.5 (8.7–13.4) * | - |
| Heparin sodium | - | - | - | - | +20.3 (19.7–21.4) * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murashkina, A.V.; Bogdanov, A.V.; Voloshina, A.D.; Lyubina, A.P.; Samorodov, A.V.; Mitrofanov, A.Y.; Beletskaya, I.P.; Smolyarchuk, E.A.; Zavadich, K.A.; Valiullina, Z.A.; et al. Base-Catalyzed Reaction of Isatins and (3-Hydroxyprop-1-yn-1-yl)phosphonates as a Tool for the Synthesis of Spiro-1,3-dioxolane Oxindoles with Anticancer and Anti-Platelet Properties. Molecules 2024, 29, 4764. https://doi.org/10.3390/molecules29194764
Murashkina AV, Bogdanov AV, Voloshina AD, Lyubina AP, Samorodov AV, Mitrofanov AY, Beletskaya IP, Smolyarchuk EA, Zavadich KA, Valiullina ZA, et al. Base-Catalyzed Reaction of Isatins and (3-Hydroxyprop-1-yn-1-yl)phosphonates as a Tool for the Synthesis of Spiro-1,3-dioxolane Oxindoles with Anticancer and Anti-Platelet Properties. Molecules. 2024; 29(19):4764. https://doi.org/10.3390/molecules29194764
Chicago/Turabian StyleMurashkina, Arina V., Andrei V. Bogdanov, Alexandra D. Voloshina, Anna P. Lyubina, Alexandr V. Samorodov, Alexander Y. Mitrofanov, Irina P. Beletskaya, Elena A. Smolyarchuk, Kseniya A. Zavadich, Zulfiya A. Valiullina, and et al. 2024. "Base-Catalyzed Reaction of Isatins and (3-Hydroxyprop-1-yn-1-yl)phosphonates as a Tool for the Synthesis of Spiro-1,3-dioxolane Oxindoles with Anticancer and Anti-Platelet Properties" Molecules 29, no. 19: 4764. https://doi.org/10.3390/molecules29194764
APA StyleMurashkina, A. V., Bogdanov, A. V., Voloshina, A. D., Lyubina, A. P., Samorodov, A. V., Mitrofanov, A. Y., Beletskaya, I. P., Smolyarchuk, E. A., Zavadich, K. A., Valiullina, Z. A., Nazmieva, K. A., Korunas, V. I., & Krylova, I. D. (2024). Base-Catalyzed Reaction of Isatins and (3-Hydroxyprop-1-yn-1-yl)phosphonates as a Tool for the Synthesis of Spiro-1,3-dioxolane Oxindoles with Anticancer and Anti-Platelet Properties. Molecules, 29(19), 4764. https://doi.org/10.3390/molecules29194764