
Investigation of Structural and Spectral Peculiarities of Fusarium sp. Indicator Pigment Bostrycoidin

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Theoretical Approach
3. Results and Discussions
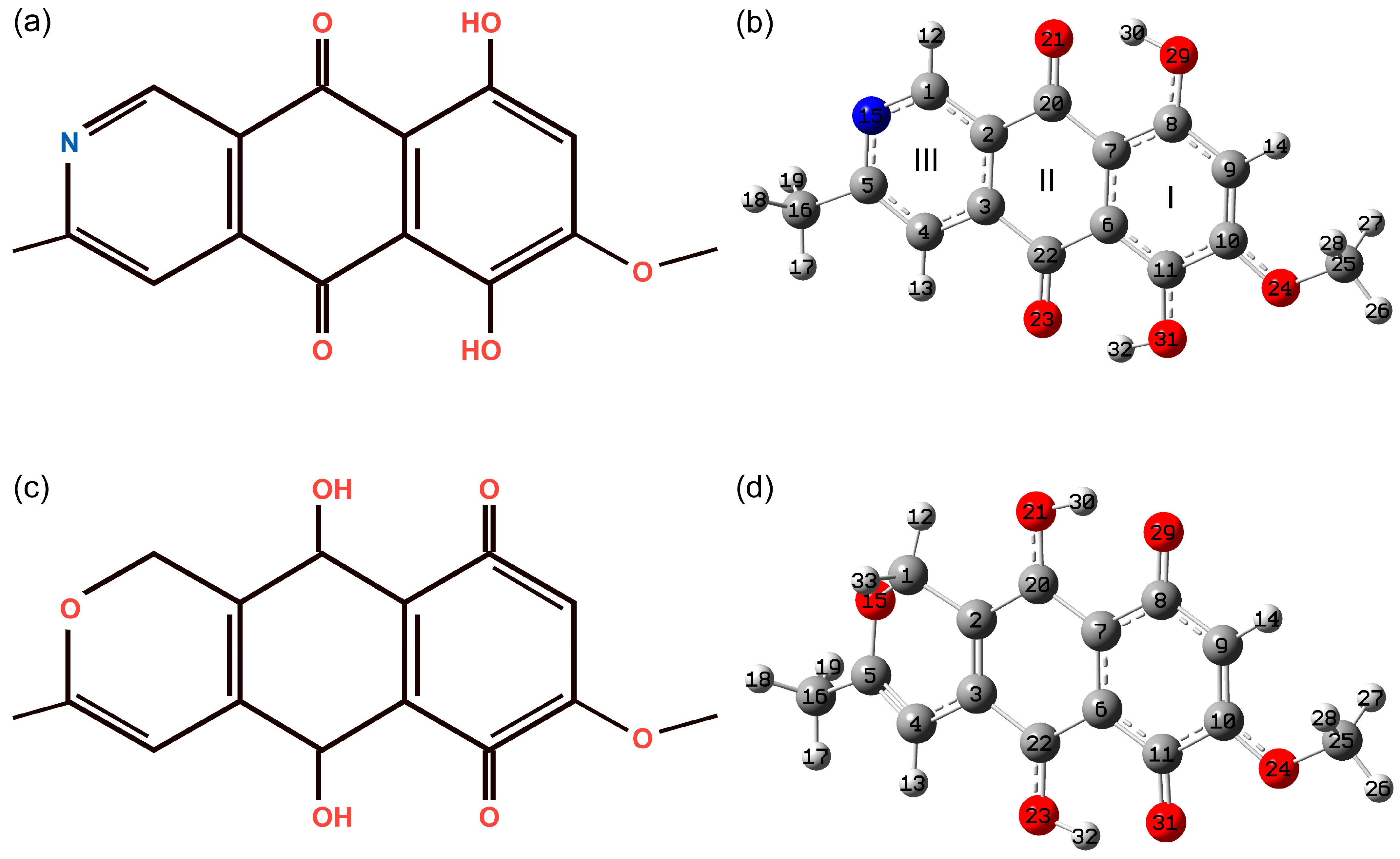
3.1. Structural Peculiarities
3.1.1. The Most Stable State
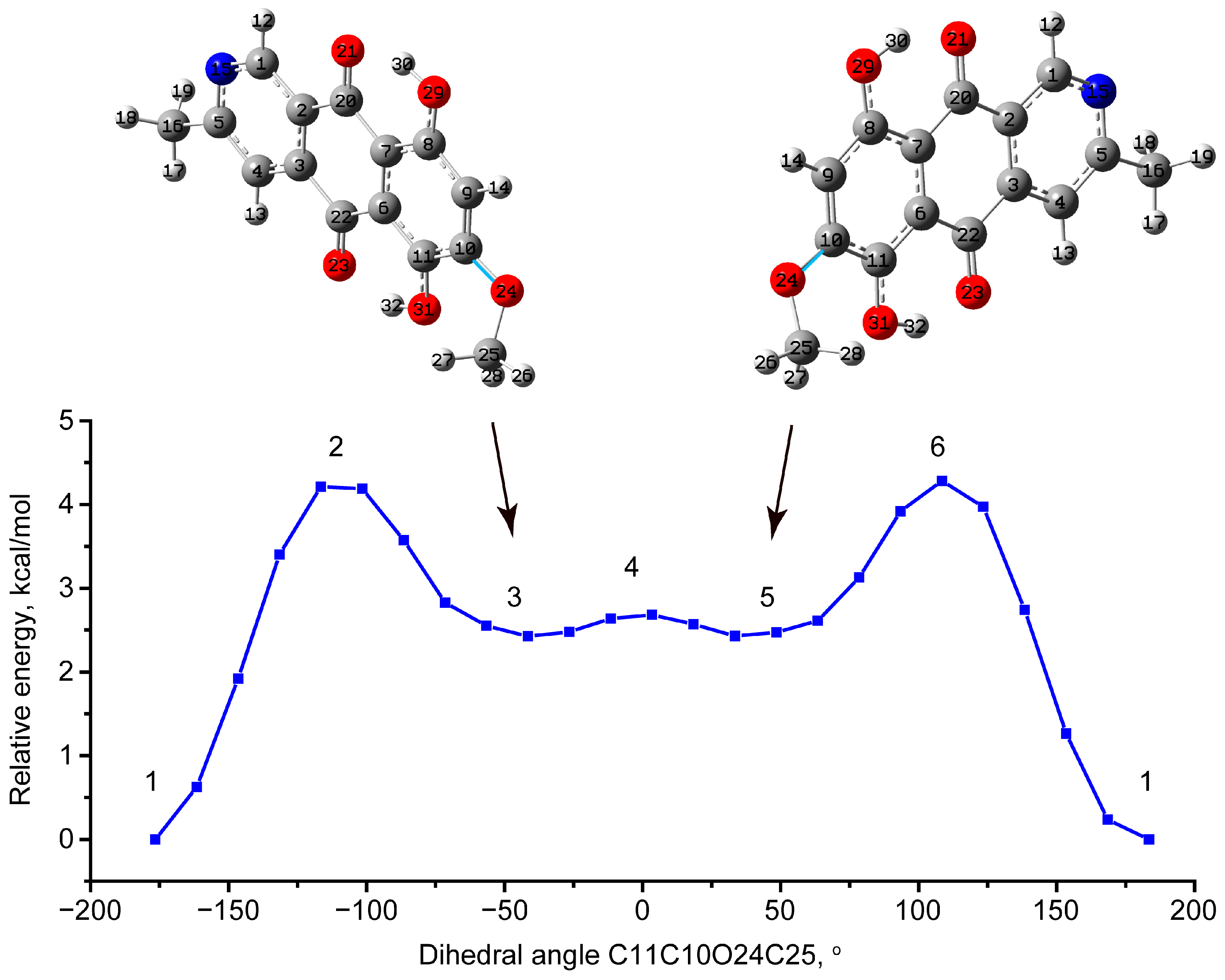
3.1.2. Conformational Enantiomers of Bostrycoidin
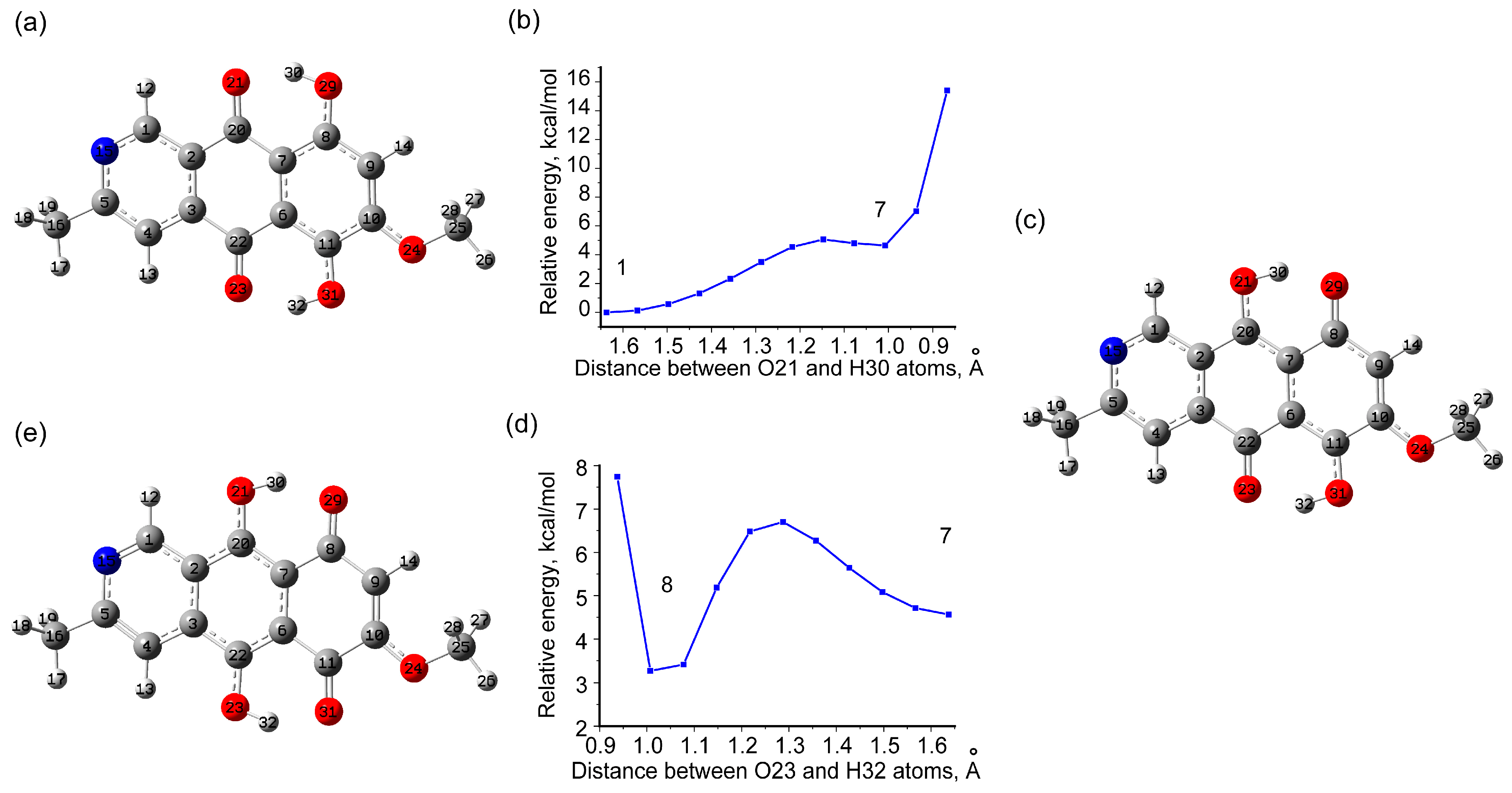
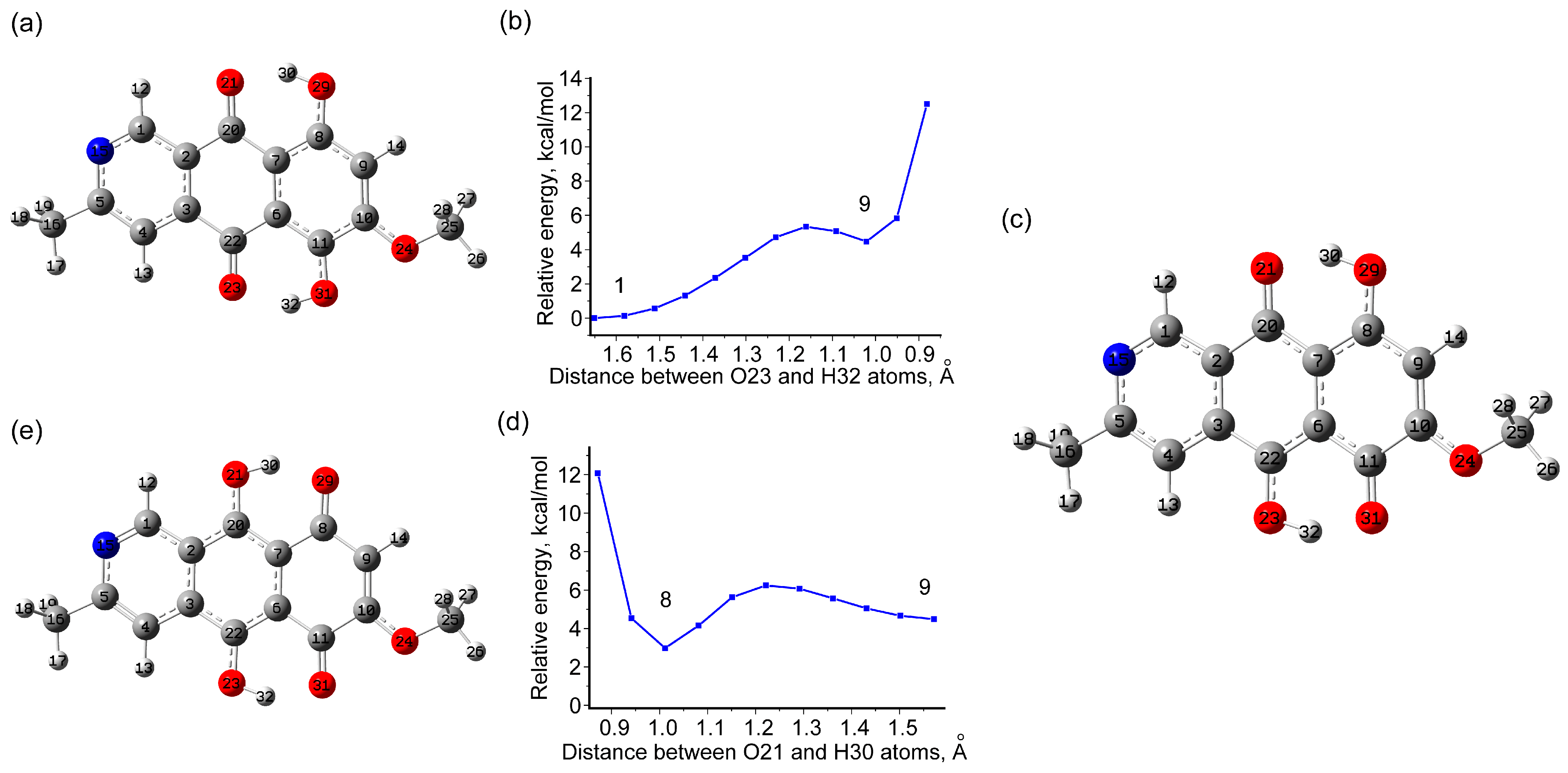
3.1.3. Tautomeric Forms of Bostrycoidin
3.2. Optical Properties
Vibrational Spectroscopy
3.3. UV–Vis Absorbance Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bycroft, B.W.; Payne, D.J. (Eds.) Dictionary of Antibiotics and Related Substances; CRC Press: Boca Raton, FL, USA, 2013; ISBN 9781482282153. [Google Scholar]
- Cajori, F.A.; Otani, T.T.; Hamilton, M.A. The Isolation and Some Properties of an Antibiotic from Fusarium Bostrycoides. J. Biol. Chem. 1954, 208, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cordell, G.A. Chemistry and Biology; Elsevier Science: Amsterdam, The Netherlands, 1999; ISBN 9780080865775. [Google Scholar]
- Mapari, S.A.S. Chemotaxonomic Exploration of Fungal Biodiversity for Polyketide Natural Food Colorants: Discovery & Evaluation of Cell Factories, and Characterization of Pigments. Ph.D. Thesis, Technical University of Denmark, Kongens Lyngby, Denmark, 2009. [Google Scholar]
- Lagashetti, A.C.; Dufossé, L.; Singh, S.K.; Singh, P.N. Fungal Pigments and Their Prospects in Different Industries. Microorganisms 2019, 7, 604. [Google Scholar] [CrossRef] [PubMed]
- Menezes, B.S.; Solidade, L.S.; Conceição, A.A.; Santos Junior, M.N.; Leal, P.L.; de Brito, E.S.; Canuto, K.M.; Mendonça, S.; de Siqueira, F.G.; Marques, L.M. Pigment Production by Fusarium solani BRM054066 and Determination of Antioxidant and Anti-Inflammatory Properties. AMB Express 2020, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.R.; Holzwarth, A.K.R.; Brew, E.; Chrapkova, N.; Kaniki, S.E.K.; Kastaniegaard, K.; Sørensen, T.; Westphal, K.R.; Wimmer, R.; Sondergaard, T.E.; et al. A New Vector System for Targeted Integration and Overexpression of Genes in the Crop Pathogen Fusarium solani. Fungal Biol. Biotechnol. 2019, 6, 25. [Google Scholar] [CrossRef]
- Dame, Z.T.; Silima, B.; Gryzenhout, M.; van Ree, T. Bioactive Compounds from the Endophytic Fungus Fusarium proliferatum. Nat. Prod. Res. 2016, 30, 1301–1304. [Google Scholar] [CrossRef]
- Kesteleyn, B.; De Kimpe, N. Synthesis of Two Naphthoquinone Antibiotics, Dehydroherbarin and 6-Deoxybostrycoidin. J. Org. Chem. 2000, 65, 640–644. [Google Scholar] [CrossRef]
- Arsenault, G.P. The Structure of Bostrycoidin, a β-Aza-Anthraquinone from D2 Purple. Tetrahedron Lett. 1965, 6, 4033–4037. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kinoshita, Y.; Ran Thor, G.; Hasumi, M.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Yoshimura, I. Isofuranonaphthoquinone Derivatives from Cultures of the Lichen Arthonia Cinnabarina (DC). Wallr. Phytochem. 2002, 60, 741–745. [Google Scholar] [CrossRef]
- Bejenari, M.; Spedtsberg, E.M.L.; Mathiesen, J.; Jeppesen, A.C.; Cernat, L.; Toussaint, A.; Apostol, C.; Stoianov, V.; Pedersen, T.B.; Nielsen, M.R.; et al. First-Class—Biosynthesis of 6-MSA and Bostrycoidin Type I Polyketides in Yarrowia lipolytica. Front. Fungal Biol. 2024, 5, 1327777. [Google Scholar] [CrossRef]
- Ekwomadu, T.I.; Mwanza, M. Fusarium Fungi Pathogens, Identification, Adverse Effects, Disease Management, and Global Food Security: A Review of the Latest Research. Agriculture 2023, 13, 1810. [Google Scholar] [CrossRef]
- World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Smith, J.S.; Fotso, J.; Leslie, J.F.; Wu, X.; Vandervelde, D.; Thakur, R.A. Characterization of Bostrycoidin: An Analytical Analog of Zearalenone. J. Food Sci. 2004, 69, FCT227–FCT232. [Google Scholar] [CrossRef]
- Busman, M. Utilization of High Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry for Characterization of 8-O-Methylbostrycoidin Production by Species of the Fungus Fusarium. J. Fungi 2017, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Kenoyer, J.M.; Cameron, A.; Bukhchuluun, D.; Amartuvshin, C.; Byambatseren, B.; Honeychurch, W.; Dussubieux, L.; Law, R. Carnelian Beads in Mongolia: New Perspectives on Technology and Trade. Archaeol. Anthr. Sci. 2022, 14, 6. [Google Scholar] [CrossRef]
- Khan, N.; Afroz, F.; Begum, M.N.; Roy Rony, S.; Sharmin, S.; Moni, F.; Mahmood Hasan, C.; Shaha, K.; Sohrab, M.H. Endophytic Fusarium Solani: A Rich Source of Cytotoxic and Antimicrobial Napthaquinone and Aza-Anthraquinone Derivatives. Toxicol. Rep. 2018, 5, 970–976. [Google Scholar] [CrossRef]
- Nielsen, K. Fungal Metabolite Screening: Database of 474 Mycotoxins and Fungal Metabolites for Dereplication by Standardised Liquid Chromatography–UV–Mass Spectrometry Methodology. J. Chromatogr. A 2003, 1002, 111–136. [Google Scholar] [CrossRef]
- Egging, V.; Nguyen, J.; Kurouski, D. Detection and Identification of Fungal Infections in Intact Wheat and Sorghum Grain Using a Hand-Held Raman Spectrometer. Anal. Chem. 2018, 90, 8616–8621. [Google Scholar] [CrossRef]
- Salman, A.; Lapidot, I.; Pomerantz, A.; Tsror, L.; Hammody, Z.; Moreh, R.; Huleihel, M.; Mordechai, S. Detection of Fusarium oxysporum Fungal Isolates Using ATR Spectroscopy. J. Spectrosc. 2012, 27, 551–556. [Google Scholar] [CrossRef]
- Farber, C.; Kurouski, D. Detection and Identification of Plant Pathogens on Maize Kernels with a Hand-Held Raman Spectrometer. Anal. Chem. 2018, 90, 3009–3012. [Google Scholar] [CrossRef]
- Pankin, D.; Povolotckaia, A.; Kalinichev, A.; Povolotskiy, A.; Borisov, E.; Moskovskiy, M.; Gulyaev, A.; Lavrov, A.; Izmailov, A. Complex Spectroscopic Study for Fusarium Genus Fungi Infection Diagnostics of “Zalp” Cultivar Oat. Agronomy 2021, 11, 2402. [Google Scholar] [CrossRef]
- Pankin, D.; Povolotckaia, A.; Borisov, E.; Povolotskiy, A.; Borzenko, S.; Gulyaev, A.; Gerasimenko, S.; Dorochov, A.; Khamuev, V.; Moskovskiy, M. Investigation of Spectroscopic Peculiarities of Ergot-Infected Winter Wheat Grains. Foods 2023, 12, 3426. [Google Scholar] [CrossRef]
- Long, Y.; Huang, W.; Wang, Q.; Fan, S.; Tian, X. Integration of Textural and Spectral Features of Raman Hyperspectral Imaging for Quantitative Determination of a Single Maize Kernel Mildew Coupled with Chemometrics. Food Chem. 2022, 372, 131246. [Google Scholar] [CrossRef] [PubMed]
- Almoujahed, M.B.; Rangarajan, A.K.; Whetton, R.L.; Vincke, D.; Eylenbosch, D.; Vermeulen, P.; Mouazen, A.M. Non-Destructive Detection of Fusarium Head Blight in Wheat Kernels and Flour Using Visible near-Infrared and Mid-Infrared Spectroscopy. Chemom. Intell. Lab. Syst. 2024, 245, 105050. [Google Scholar] [CrossRef]
- Schlücker, S.; Szeghalmi, A.; Schmitt, M.; Popp, J.; Kiefer, W. Density Functional and Vibrational Spectroscopic Analysis of Β-carotene. J. Raman Spectrosc. 2003, 34, 413–419. [Google Scholar] [CrossRef]
- Martin, D.; Amado, A.M.; Gonzálvez, A.G.; Marques, M.P.M.; Batista de Carvalho, L.A.E.; Ureña, Á.G. FTIR Spectroscopy and DFT Calculations to Probe the Kinetics of β-Carotene Thermal Degradation. J. Phys. Chem. A 2019, 123, 5266–5273. [Google Scholar] [CrossRef]
- Novikov, V.S.; Kuzmin, V.V.; Darvin, M.E.; Lademann, J.; Sagitova, E.A.; Prokhorov, K.A.; Ustynyuk, L.Y.; Nikolaeva, G.Y. Relations between the Raman Spectra and Molecular Structure of Selected Carotenoids: DFT Study of α-Carotene, β-Carotene, γ-Carotene and Lycopene. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 270, 120755. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.M.; Lyon, J.P.; Lima, A.; Codognoto, L.; Machado, A.E.D.H.; Tiago, F.D.S.; Araújo, D.M.S.; Silva, E.L.; Hioka, N.; Rodrigues, M.R.; et al. Quinquangulin and Rubrofusarin: A Spectroscopy Study. Orbital Electron. J. Chem. 2017, 9, 290–298. [Google Scholar] [CrossRef]
- Pankin, D.; Povolotckaia, A.; Borisov, E.; Belyakov, M.; Borzenko, S.; Gulyaev, A.; Moskovskiy, M. Theoretical Modelling of Structure, Vibrational and UV–Vis Absorbance Spectra of Rubrofusarin Molecule. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2023, 293, 122469. [Google Scholar] [CrossRef]
- Pankin, D.; Povolotckaia, A.; Smirnov, M.; Borisov, E.; Gulyaev, A.; Dorochov, A.; Novikov, V.; Kuznetsov, S.; Noy, O.; Belousov, S.; et al. Theoretical Investigation of Anhydrofusarubin: Structural and Optical Properties. Crystals 2023, 13, 1556. [Google Scholar] [CrossRef]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.; Cannarile, L.; Thuy, T.; Sung, T.; Sohrab, H.; Hasan, C.; Ayroldi, E.; et al. Fusarubin and Anhydrofusarubin Isolated from A Cladosporium Species Inhibit Cell Growth in Human Cancer Cell Lines. Toxins 2019, 11, 503. [Google Scholar] [CrossRef]
- Shao, C.-L.; Wang, C.-Y.; Deng, D.-S.; She, Z.-G.; Gu, Y.-C.; Lin, Y.-C. Crystal Structure of a Marine Natural Compound, Anhydrofusarubin. Chin. J. Struct. Chem. 2008, 27, 824–828. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z. =11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Pankin, D.; Smirnov, M.; Povolotckaia, A.; Povolotskiy, A.; Borisov, E.; Moskovskiy, M.; Gulyaev, A.; Gerasimenko, S.; Aksenov, A.; Litvinov, M.; et al. DFT Modelling of Molecular Structure, Vibrational and UV-Vis Absorption Spectra of T-2 Toxin and 3-Deacetylcalonectrin. Materials 2022, 15, 649. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A New Definition of Cavities for the Computation of Solvation Free Energies by the Polarizable Continuum Model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General Formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Jamróz, M.H. Vibrational Energy Distribution Analysis (VEDA): Scopes and Limitations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 114, 220–230. [Google Scholar] [CrossRef]
- Jamróz, M.H. Vibrational Energy Distribution Analysis: VEDA 4. Program, Warsaw. Available online: https://smmg.pl/software/veda/ (accessed on 7 October 2024).
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01. Available online: https://gaussian.com/glossary/g09/ (accessed on 7 October 2024).
- Zahedi Tabrizi, M.; Tayyari, S.F.; Tayyari, F.; Behforouz, M. Fourier Transform Infrared and Raman Spectra, Vibrational Assignment and Density Functional Theory Calculations of Naphthazarin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2004, 60, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.O.; Schutte, C.J.H. Infrared Spectra of the Polymorphs of Naphthazarin: Microscopy and Low Temperature Studies. Mikrochim. Acta 1988, 94, 171–173. [Google Scholar] [CrossRef]
- Mayo, D.W.; Miller, F.A.; Hannah, R.W. Course Notes on the Interpretation of Infrared and Raman Spectra; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; Wiley: Hoboken, NJ, USA, 2004; ISBN 9780470093078. [Google Scholar]
- Paul, S.O.; Schutte, C.J.H.; Hendra, P.J. The Fourier Transform Raman and Infrared Spectra of Naphthazarin. Spectrochim. Acta A 1990, 46, 323–329. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Bond Length, Å | ||
|---|---|---|---|
| Bostrycoidin | Anhydrofusarubin | ||
| Ring I | C7C8 | 1.401 | 1.401 |
| C8C9 | 1.410 | 1.407 | |
| C9C10 | 1.376 | 1.386 | |
| C10C11 | 1.434 | 1.424 | |
| C11C6 | 1.396 | 1.401 | |
| C8O29 | 1.333 | 1.339 | |
| O29H30 | 0.997 | - | |
| C11O31 | 1.330 | 1.336 | |
| O31H32 | 0.994 | - | |
| C10O24 | 1.343 | 1.335 | |
| O24C25 | 1.424 | 1.426 | |
| C9H14 | 1.080 | - | |
| C25H26 | 1.087 | - | |
| C25H27 | 1.093 | - | |
| C25H28 | 1.093 | - | |
| Ring II | C2C20 | 1.477 | 1.458 |
| C20C7 | 1.449 | 1.455 | |
| C7C6 | 1.430 | 1.420 | |
| C6C22 | 1.457 | 1.454 | |
| C22C3 | 1.486 | 1.498 | |
| C20O21 | 1.245 | 1.248 | |
| C22O23 | 1.240 | 1.236 | |
| Ring III | N15C1 | 1.326 | - |
| C1C2 | 1.398 | - | |
| C2C3 | 1.398 | - | |
| C3C4 | 1.390 | - | |
| C4C5 | 1.393 | - | |
| C5N15 | 1.346 | - | |
| C1H12 | 1.084 | - | |
| C4H13 | 1.082 | - | |
| C5C16 | 1.502 | 1.491 | |
| C16H17 | 1.090 | - | |
| C16H18 | 1.093 | - | |
| C16H19 | 1.093 | - | |
| Angle | Angle, ° | ||
| Ring I | C7C8C9 | 120.3 | 119.6 |
| C8C9C10 | 120.9 | 121.4 | |
| C9C10C11 | 120.1 | 119.5 | |
| C10C11C6 | 119.2 | 119.7 | |
| C11C6C7 | 120.6 | 120.2 | |
| C6C11O31 | 123.4 | 122.4 | |
| C7C8O29 | 122.3 | 122.0 | |
| C11O31H32 | 105.7 | 106.2 | |
| C8O29H30 | 105.7 | 105.5 | |
| C11C10O24 | 114.5 | 112.2 | |
| C8C9H14 | 117.3 | - | |
| Ring II | C2C20C7 | 117.6 | 118.5 |
| C20C7C6 | 121.5 | 120.4 | |
| C7C6C22 | 120.8 | 120.7 | |
| C6C22C3 | 117.7 | 117.4 | |
| C22C3C2 | 120.9 | 121.2 | |
| C2C20O21 | 120.3 | 119.7 | |
| C3C22O23 | 120.0 | 119.6 | |
| C20C7C8 | 119.5 | - | |
| C22C6C11 | 118.6 | - | |
| Ring III | N15C1C2 | 123.7 | - |
| C1C2C3 | 117.7 | - | |
| C2C3C4 | 118.8 | - | |
| C3C4C5 | 119.4 | - | |
| C5N15C1 | 122.0 | - | |
| C4C5C16 | 121.8 | - | |
| C5C4H13 | 121.5 | - | |
| N15C1H12 | 117.3 | - | |
| Theory | Experiment | |||||
|---|---|---|---|---|---|---|
| [2] | [19] | |||||
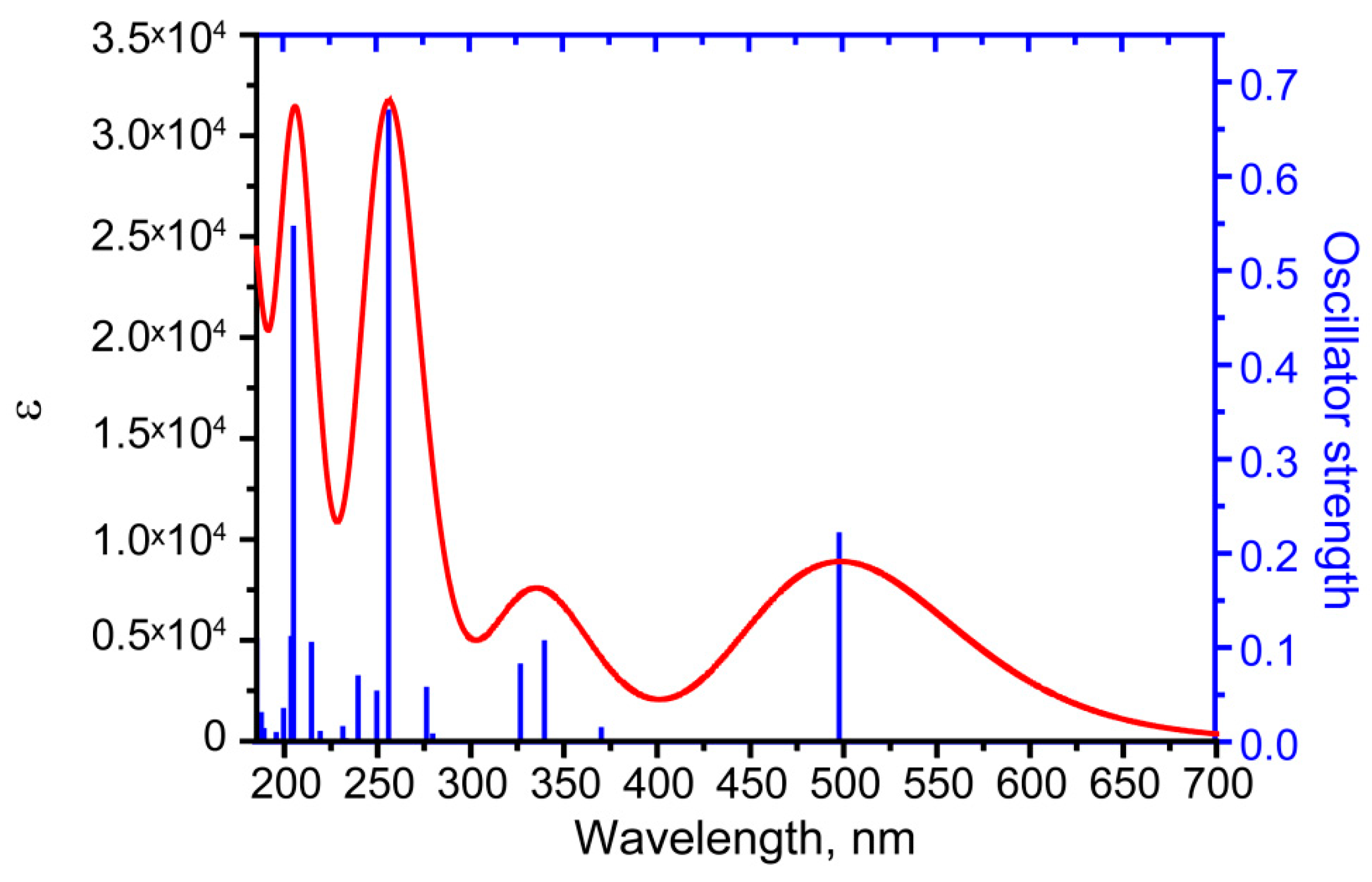
| Peak № | Excited State | Orbitals with >10% Contribution (Percent) | Oscillator Strength | Wavelength, nm (Energy, eV) | Wavelength, nm | |
| 1 | 1 | 74 -> 75 (99) | 0.2196 | 498.43 (2.4875) | 497 (475sh * and 525sh) * | 492 (472sh * and 526sh *) |
| 2 | 5 | 74 -> 76 (85) | 0.1050 | 340.43 (3.6420) | ||
| and | 320 | 324 | ||||
| 327.46 | ||||||
| 7 | 71 -> 75 (85) | 0.804 | (3.7863) | |||
| 3 | 11 | 73 -> 76 (85) | 0.6683 | 256.73 | 251 | 252 |
| (4.8293) | ||||||
| 4 | 22 | 65 ->75 (37) | 0.5449 | 205.74 | N.O. ** | 204 |
| 68 -> 76 (27) | (6.0263) | |||||
| 71 -> 77 (19) | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Povolotckaia, A.; Pankin, D.; Novikov, V.; Borisov, E.; Kuznetsov, S.; Dorokhov, A.; Gulyaev, A.; Zavyalova, E.; Alieva, R.; Akulov, S.; et al. Investigation of Structural and Spectral Peculiarities of Fusarium sp. Indicator Pigment Bostrycoidin. Molecules 2024, 29, 4765. https://doi.org/10.3390/molecules29194765
Povolotckaia A, Pankin D, Novikov V, Borisov E, Kuznetsov S, Dorokhov A, Gulyaev A, Zavyalova E, Alieva R, Akulov S, et al. Investigation of Structural and Spectral Peculiarities of Fusarium sp. Indicator Pigment Bostrycoidin. Molecules. 2024; 29(19):4765. https://doi.org/10.3390/molecules29194765
Chicago/Turabian StylePovolotckaia, Anastasia, Dmitrii Pankin, Vasiliy Novikov, Evgenii Borisov, Sergey Kuznetsov, Alexey Dorokhov, Anatoly Gulyaev, Elena Zavyalova, Rugiya Alieva, Sergey Akulov, and et al. 2024. "Investigation of Structural and Spectral Peculiarities of Fusarium sp. Indicator Pigment Bostrycoidin" Molecules 29, no. 19: 4765. https://doi.org/10.3390/molecules29194765
APA StylePovolotckaia, A., Pankin, D., Novikov, V., Borisov, E., Kuznetsov, S., Dorokhov, A., Gulyaev, A., Zavyalova, E., Alieva, R., Akulov, S., Belousov, S., & Moskovskiy, M. (2024). Investigation of Structural and Spectral Peculiarities of Fusarium sp. Indicator Pigment Bostrycoidin. Molecules, 29(19), 4765. https://doi.org/10.3390/molecules29194765