
Optimized Method for the Synthesis of Alkyne-Modified 2′-Deoxynucleoside Triphosphates

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
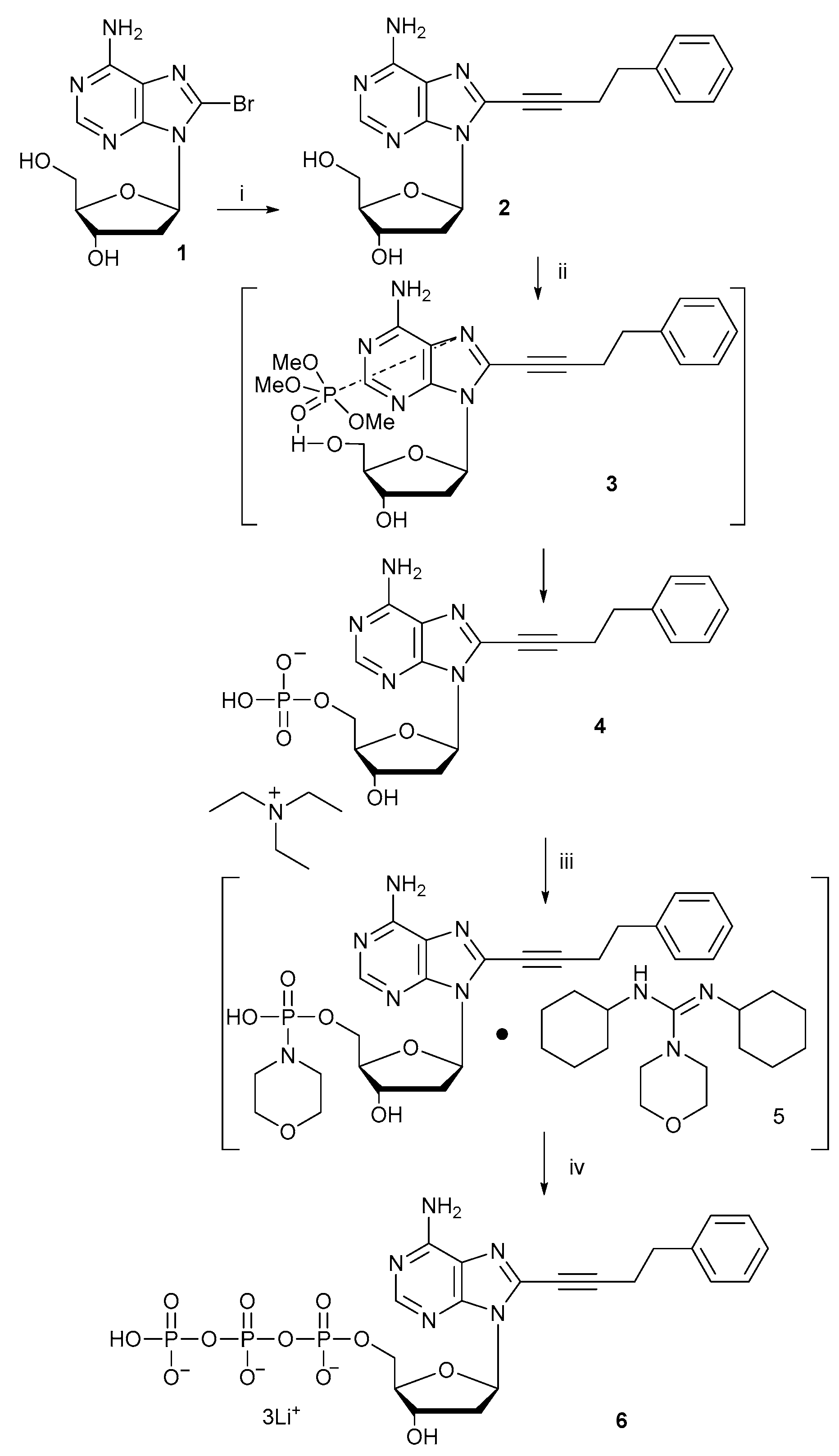
2.1. Synthesis of 8-Alkyne-Modified 2′-Deoxyadenosine Triphosphate
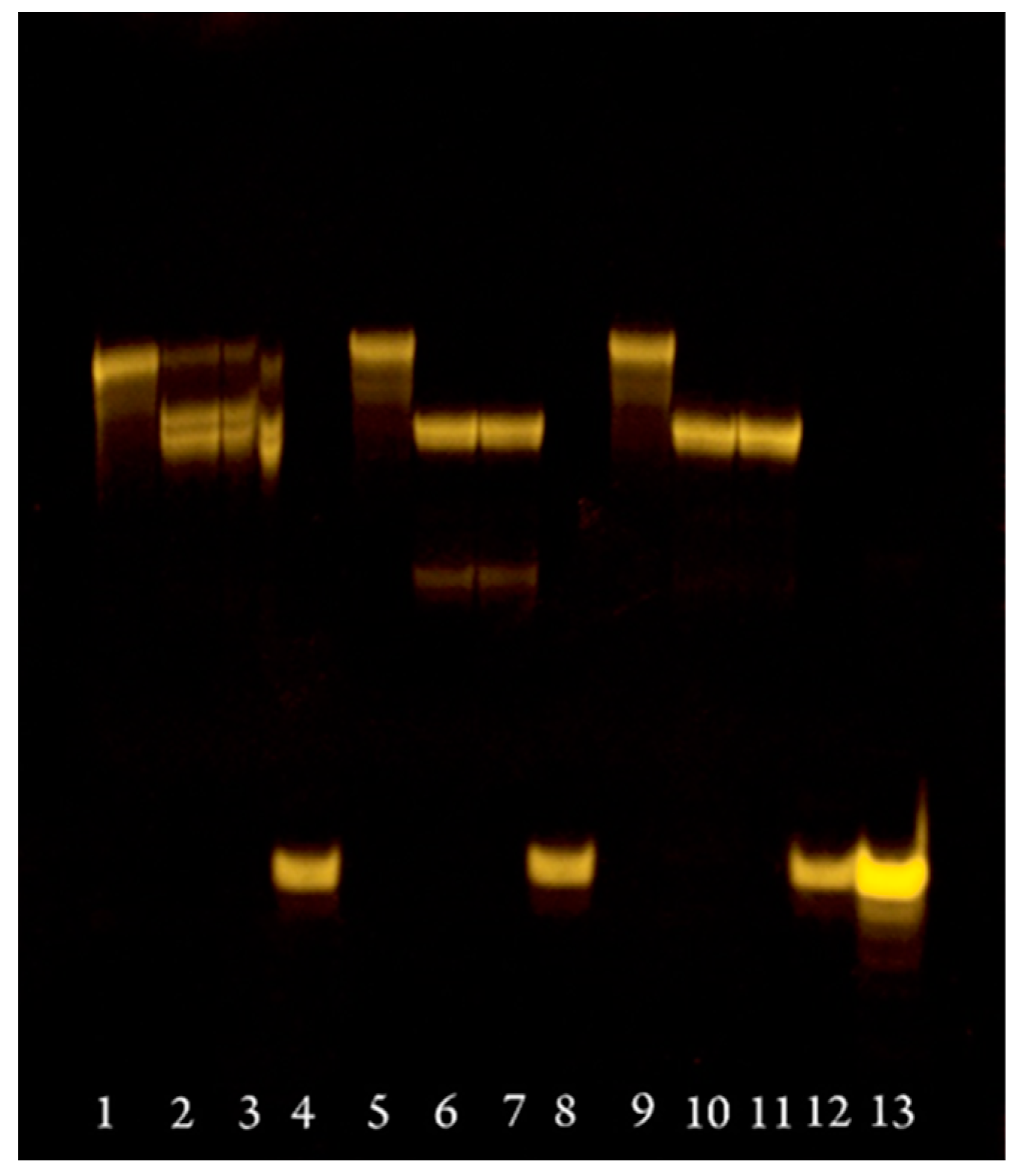
2.2. Enzymatic Incorporation of 8-Alkyne-Modified 2′-Deoxyadenosine Triphosphate during Primer Extensions
- T1: (T)20-TTG-TCA-CTC-AGA-CCA-ACT-CCC-T,
- T2.AGCAGCACAGAGGTCAGATGCCGCCAGGCCACCCATACACCAACAACCCCTATGCGTGCTACCGT
3. Materials and Methods
3.1. General Information
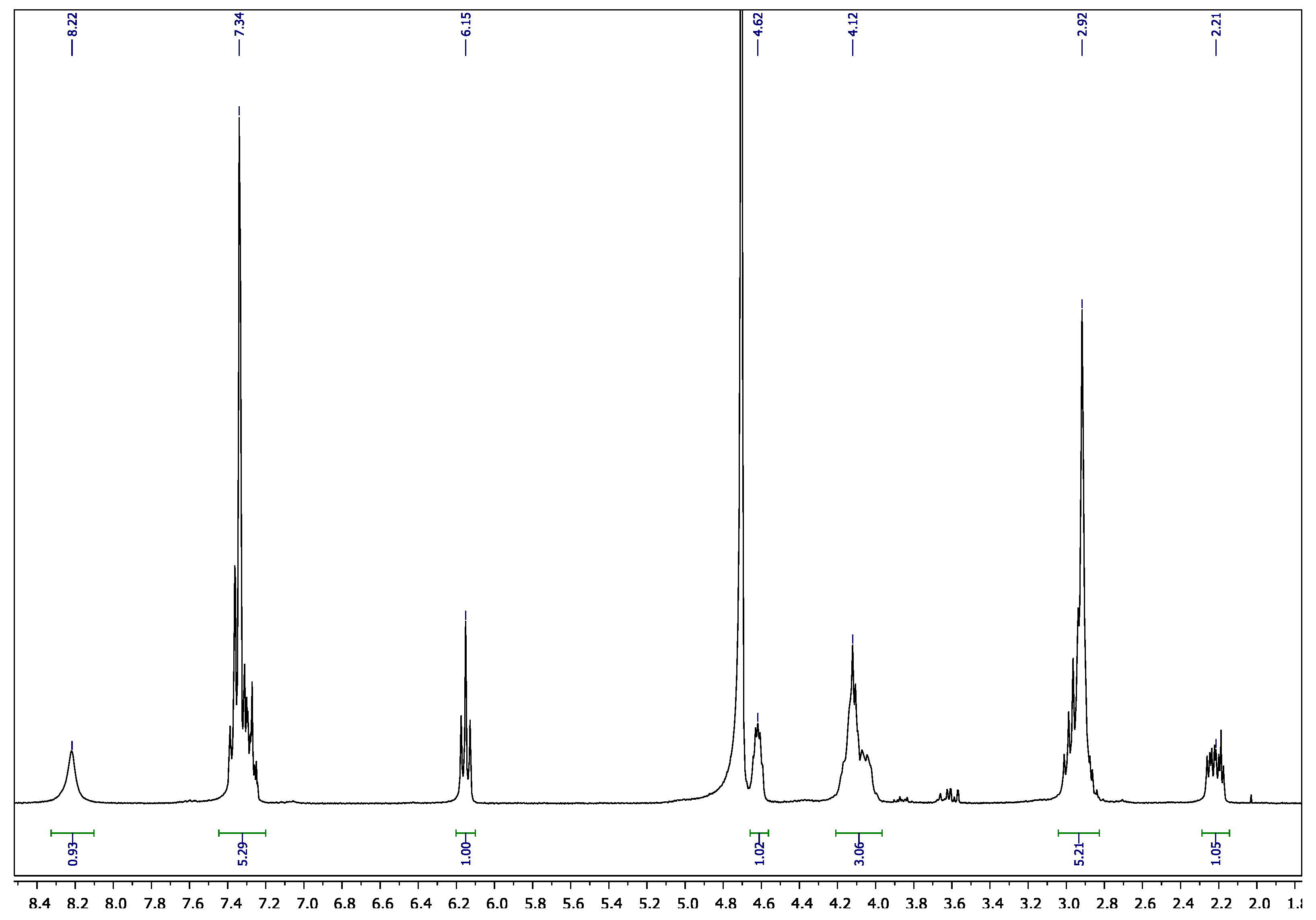
3.2. Synthesis of 8-Alkyne-Modified 2′-Deoxyadenosine Triphosphate (6)
3.3. Enzymatic Incorporation of Modified dATP 6 during Primer Extensions
- T1: (T)20-TTG-TCA-CTC-AGA-CCA-ACT-CCC-T;
- Primer1: Fitc-A-GGG-AGT-TGG-TCT-GAG-TGA-CAA;
- T2: AGCAGCACAGAGGTCAGATGCCGCCAGGCCACCCATACACCAACAACCCCTATGCGTGCTACCGT;
- Primer2: Cy3-ACG-GTA-GCA-CGC-ATA-GG.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kanda, T.; Jackson, M.J.; Smith, L.A.; Pearce, R.K.B.; Nakamura, J.; Kase, H.; Kuwana, Y.; Jenner, P. Adenosine A2A Antagonist: A Novel Antiparkinsonian Agent that does not Provoke Dyskinesia in Parkinsonian Monkeys. Ann. Neurol. 1998, 43, 507–513. [Google Scholar] [CrossRef] [PubMed]
- DeClercq, E. A 40-year Journey in Search of Selective Antiviral Chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, F.; Huecas, S.; Ruiz-Avila, L.B.; Canada, F.J.; Perona, A.; Poveda, A.; Martin-Santamaria, S.; Morreale, A.; Jimenez-Barbero, J.; Andreu, J.M. Interactions of Bacterial Cell Division Protein FtsZ with C8-Substituted Guanine Nucleotide Inhibitors. A combined NMR, Biochemical and Molecular Modeling Perspective. J. Am. Chem. Soc. 2013, 135, 16418–16428. [Google Scholar] [CrossRef]
- Jager, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A Versatile Toolbox for Variable DNA Functionalization at High Density. J. Am. Chem. Soc. 2005, 127, 15071–15082. [Google Scholar] [CrossRef]
- Lam, C.; Hipolito, C.; Perrin, D.M. Synthesis and Enzymatic Incorporation of Modified Deoxyadenosine Triphosphates. European J. Org. Chem. 2008, 29, 4915–4923. [Google Scholar] [CrossRef]
- Jonathan, D.; Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; et al. Selection of DNA Aptamers with two Modified Bases. Expanding the Chemistry of DNA for in vitro Selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar]
- Percze, K.; Meszaros, D.T. Analysis of Modified Nucleotide Aptamer Library Generated by Thermophilic DNA Polymerases. Chembiochem 2020, 21, 2939–2944. [Google Scholar] [CrossRef]
- Anderson, J.P.; Angerer, B.; Loeb, L.A. Incorporation of Reporter-Labeled Nucleotides by DNA Polymerases. Biotechniques 2005, 38, 257–264. [Google Scholar] [CrossRef]
- Kore, A.R.; Senthilvelan, A.; Shanmugasundaram, M. Highly Regioselective C-5 Iodination of Pyrimidine Nucleotides and Subsequent Chemoselective Sonogashira Coupling with Propargylamine. Nucleosides Nucleotides Nucleic Acids 2015, 34, 92–102. [Google Scholar] [CrossRef]
- Herve, G.; Len, C. Heck and Sonogashira Couplings in Aqueous Media—Application to Unprotected Nucleosides and Nucleotides. Sustain. Chem. Process. 2015, 3, 3. [Google Scholar] [CrossRef]
- Zasedateleva, O.A.; Vasiliskov, V.A.; Surzhikov, S.A.; Kuznetsova, V.E.; Shershov, V.E.; Guseinov, T.O. dUTPs Conjugated with Zwitterionic Cy3 or Cy5 Fluorophore Analogues are Effective Substrates for DNA Amplification and Labelling by Taq Polymerase. Nucleic Acids Res. 2018, 46, e73. [Google Scholar] [CrossRef] [PubMed]
- Hocek, M.; Silhar, P.; Shih, I.; Mabery, E.; Mackman, R. Cytostatic and Antiviral 6-Arylpurine Ribonucleosides. Part 7: Synthesis and Evaluation of 6-Substituted Purine l-Ribonucleosides. Bioorg. Med. Chem. Lett. 2006, 16, 5290–5293. [Google Scholar] [CrossRef]
- Ashton, T.D.; Aumann, K.M.; Baker, S.P.; Schiesser, C.H.; Scammells, P.J. Structure—Activity Relationships of Adenosines with Heterocyclic N6-Substituents. Bioorg. Med. Chem. Lett. 2007, 17, 6779–6784. [Google Scholar] [CrossRef] [PubMed]
- Voller, J.; Zatloukal, M.; Lenobel, R.; Dolezal, K.; Beres, T.; Krystof, V.; Spichal, L.; Niemann, P.; Dzubak, P.; Hajduch, M.; et al. Anticancer Activity of Natural Cytokinins: A Structure—Activity Relationship Study. Phytochemistry 2010, 71, 1350–1359. [Google Scholar] [CrossRef]
- Collier, A.; Wagner, G. A Facile Two-Step Synthesis of 8-Arylated Guanosine Mono- and Triphosphates (8-Aryl GXPs). Org. Biomol. Chem. 2006, 4, 4526–4532. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Shaughnessy, K.H. Aqueous-Phase Sonogashira Alkynylation to Synthesize 5-Substituted Pyrimidine and 8-Substituted Purine Nucleosides. Curr. Protoc. Nucleic Acid Chem. 2012, 49, 1.27.1–1.27.10. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, Q.; Wei, S.; Fu, B.; Yang, Q.; Ming, X. Synthesis of 8-Substituted 2′-Deoxyisoguanosines via Unprotected 8-Brominated 2-Amino-2′-Deoxyadenosine. Chem. Biodivers 2018, 15, e1700335. [Google Scholar] [CrossRef]
- Firth, A.G.; Fairlamb, I.J.S.; Darleyc, K.; Baumannb, C.G. Sonogashira Alkynylation of Unprotected 8-Brominated Adenosines and Guanosines: Fluorescence Properties of Compact Conjugated Acetylenes Containing a Purine Ring. Tetrahedron Lett. 2006, 47, 3529–3533. [Google Scholar] [CrossRef]
- Ikonen, S.; Macickova-Cahova, H.; Pohl, R.; Sanda, M.; Hocek, M. Synthesis of Nucleoside and Nucleotide Conjugates of Bile Acids, and Polymerase Construction of Bile Acid-Functionalized DNA. Org. Biomol. Chem. 2010, 8, 1194–1201. [Google Scholar] [CrossRef]
- Kuznetsova, V.E.; Shershov, V.E.; Guseinov, T.O.; Miftakhov, R.A.; Solyev, P.N.; Novikov, R.A.; Levashova, A.I.; Zasedatelev, A.S.; Lapa, S.A.; Chudinov, A.V. Synthesis of Cy5-Labelled C5-Alkynyl-Modified Cytidine Triphosphates via Sonogashira Coupling for DNA Labelling. Bioorg. Chem. 2023, 131, 106315–106330. [Google Scholar] [CrossRef]
- Vrabel, M.; Pohl, R.; Klepetarova, B.; Votruba, I.; Hocek, M. Synthesis of 2′-Deoxyadenosine Nucleosides Bearing Bipyridine-Type Ligands and their Ru-Complexes in Position 8 through Cross-Coupling Reactions. Org. Biomol. Chem. 2007, 5, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Kölmel, D.K.; Barandun, L.J.; Kool, E.T. Efficient Synthesis of Fluorescent Alkynyl C-Nucleosides via Sonogashira Coupling for the Preparation of DNA-Based Polyfluorophores. Org. Biomol. Chem. 2016, 14, 6407–6412. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, A.; Wang, Y.H.; Ho, T.I. Sonogashira Coupling Reaction with Diminished Homocoupling. Org. Lett. 2003, 5, 1841–1844. [Google Scholar] [CrossRef] [PubMed]
- Leophairatana, P.; Samanta, S.; De Silva, C.C.; Koberstein, J.T. Preventing Alkyne–Alkyne (i.e., Glaser) Coupling Associated with the ATRP Synthesis of Alkyne-Functional Polymers/Macromonomers and for Alkynes under Click (i.e., CuAAC) Reaction Conditions. J. Am. Chem. Soc. 2017, 139, 3756–3766. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Patel, A.D.; Hampton, A. Formation of Ribonucleotide 2′, 3′-Cyclic Carbonates During Conversion of Ribonucleoside 5′-Phosphates to Diphosphates and Triphosphates by the Phosphorimidazolidate Procedure. Nucleic Acids Res. 1977, 4, 2843–2853. [Google Scholar] [CrossRef]
- Ludwig, J. A New Route to Nucleoside 5′-Triphosphates. Biochim Biophys. Acad. Sci. Hung. 1981, 16, 131–133. [Google Scholar]
- Simon, E.S.; Grabowski, S.; Whitesides, G.M. Convenient Syntheses of Cytidine 5′-Triphosphate, Guanosine 5′-Triphosphate, and Uridine 5′-Triphosphate and Their Use in the Preparation of UDP-Glucose, UDP-Glucuronic Acid, and GDP-Mannose. J. Org. Chem. 1990, 55, 1834–1841. [Google Scholar] [CrossRef]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef]
- Xu, Z. A Review on the Chemical Synthesis of Pyrophosphate Bonds in Bioactive Nucleoside Diphosphate Analogs. Med. Chem. Lett. 2015, 25, 3777–3783. [Google Scholar] [CrossRef]
- Rachwalak, M.; Romanowska, J.; Sobkowski, M.; Stawinski, J. Nucleoside Di- and Triphosphates as a New Generation of Anti-Hiv Pronucleotides. Chemical and Biological Aspects. Appl. Sci. 2021, 11, 2248–2276. [Google Scholar] [CrossRef]
- Kozlov, M.; Bergendahl, V.; Burgess, R.; Goldfarb, A.; Mustaev, A. Homogeneous Fluorescent Assay for RNA Polymerase. Anal. Biochem. 2005, 342, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Kore, A.R.; Xiao, Z.; Senthilvelan, A.; Charles, I.; Snanmugasundaram, M.; Mukundarajan, S.; Srinivasan, B. An Efficient Synthesis of Pyrimidine Specific 2′-Deoxynucleoside-5′-Tetraphosphates. Nucleosides Nucleotides Nucleic Acids 2012, 31, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.G.; Khorana, H.G. Nucleoside Polyphosphates. X.1 The Synthesis and Some Reactions of Nucleoside-5′ Phosphoromorpholidates and Related Compounds. Improved Methods for the Preparation of Nucleoside-5′ Polyphosphates. J. Am. Chem. Soc. 1961, 83, 649–658. [Google Scholar] [CrossRef]
- Moffatt, J.G. A General Synthesis of Nucleoside 5′-Triphosphates. Can. J. Chem. 1964, 42, 599–604. [Google Scholar] [CrossRef]
- Xia, R.; Sunb, L.-P.; Chena, L.-S. Improved Synthesis of Cytidine Diphosphate Choline (CDP-Choline) via Selective Phosphorylation. J. Chem. Res. 2016, 40, 358–360. [Google Scholar] [CrossRef]
- Gillerman, I.; Fischer, B. An Improved One-Pot Synthesis of Nucleoside 5′-Triphosphate Analogues. Nucleosides Nucleotides Nucleic Acids 2010, 29, 245–256. [Google Scholar] [CrossRef]
- Kore, A.R.; Shanmugasundaram, M.; Senthilvelan, A.; Srinivasan, B. Gram-Scale Chemical Synthesis of 2-Deoxynucleoside-5-O-Triphosphates. Curr. Protoc. Nucleic Acid Chem. 2012, 49, 13.10.1–13.10.12. [Google Scholar] [CrossRef] [PubMed]
- Matyasovskya, J.; Hocek, M. 2-Substituted 2′-deoxyinosine 5′-Triphosphates as Substrates for Polymerase Synthesis of Minor-Groove-Modified DNA and Effects on Restriction Endonuclease Cleavage. Org. Biomol. Chem. 2020, 18, 255–262. [Google Scholar] [CrossRef]
- Ondrus, M.; Sykorov, V.; Bednarov, L.; Pohl, R.; Hocek, M. Enzymatic Synthesis of Hypermodified DNA Polymers for Sequence-Specific Display of Four Different Hydrophobic Groups. Nucleic Acids Res. 2020, 48, 11982–11993. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. A Novel Method for Phosphorylation of Nucleosides to 5′-Nucleotides. Tetrahedron Lett. 1967, 50, 5065–5068. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. Studies of Phosphorylation. III. Selective Phosphorylation of Unprotected Nucleosides. Bull. Chem. Soc. Jpn. 1969, 42, 3505–3508. [Google Scholar] [CrossRef]
- Ikemoto, T.; Haze, A.; Hatano, H.; Kitamoto, Y.; Ishida, M.; Nara, K. Phosphorylation of Nucleosides with Phosphorus Oxychloride in Trialkyl Phosphate. Chem. Pharm. Bull. 1995, 43, 210–215. [Google Scholar] [CrossRef]
- Kowalska, J.; Lewdorowicz, M.; Darzynkiewicz, E.; Jemielity, J. A Simple and Rapid Synthesis of Nucleotide Analogues Containing a Phosphorothioate Moiety at the Terminal Position of the Phosphate Chain. Tetrahedron Lett. 2007, 48, 5475–5479. [Google Scholar] [CrossRef]
- Kadokura, M.; Wada, T.; Urashima, C.; Sekine, M. Efficient Synthesis of γ-Methyl-Capped Guanosine 5′-Triphosphate as a 5′-Terminal Unique Structure of U6 RNA via a New Triphosphate Bond Formation Involving Activation of Methyl Phosphorimidazolidate using ZnCl2 as a Catalyst in DMF under Anhydrous Conditions. Tetrahedron Lett. 1997, 38, 8359–8362. [Google Scholar]
- Tam, C.P.; Zhou, L.; Fahrenbach, A.C.; Zhang, W.; Walton, T.; Szostak, J.W. Synthesis of a Nonhydrolyzable Nucleotide Phosphoroimidazolide Analogue that Catalyzes Nonenzymatic RNA Primer Extension. J. Am. Chem. Soc. 2018, 140, 783–792. [Google Scholar] [CrossRef]
- Appy, L.; Chardet, C.; Peyrottes, S.; Roy, B. Synthetic Strategies for Dinucleotides Synthesis. Molecules 2019, 24, 4334–4361. [Google Scholar] [CrossRef]
- Wittmann, V.; Wong, C.W. 1H-Tetrazole as Catalyst in Phosphomorpholidate Coupling Reactions: Efficient Synthesis of GDP-Fucose, GDP-Mannose, and UDP-Galactose. J. Org. Chem. 1997, 62, 2144–2147. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, S.; Sun, J.; Liu, S.; Xiao, Q.; Pu, S. A P(V)−N Activation Strategy for the Synthesis of Nucleoside Polyphosphates. J. Org. Chem. 2013, 78, 8417–8426. [Google Scholar] [CrossRef]
- Kodr, D.; Kuzmova, E.; Pohl, R.; Tand, K.; Hocek, M. Lipid-Linked Nucleoside Triphosphates for Enzymatic Synthesis of Hydrophobic Oligonucleotides with Enhanced Membrane Anchoring Efficiency. Chem. Sci. 2023, 14, 4059–4069. [Google Scholar] [CrossRef]
- Hollenstein, M. Enzymatic Synthesis of Base-Modified Nucleic Acids. Handbook of Chemical Biology of Nucleic Acids; Springer Nature: Berlin/Heidelberg, Germany, 2023; pp. 1–39. [Google Scholar]
- Chardet, C.; Serres, S.; Payrastre, C.; Escudier, J.-M.; Gerland, B. Functionalized Oligonucleotides, Synthetic Catalysts as Enzyme Mimics. Comptes Rendus Chim. 2023, 26, 1–13. [Google Scholar] [CrossRef]
- Xie, R.; Li, W.; Ge, Y.; Zhou, Y.; Xiao, G.; Zhao, Q.; Han, Y.; Li, Y.; Chen, G. Late-stage guanine C8–H alkylation of nucleosides, nucleotides, and oligonucleotides via photo-mediated Minisci reaction. Nat. Commun. 2024, 15, 2549–2563. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activator, eqv. | (nBu4N)3HP2O7, eqv. | Solvent | t, °C | Time | Yield | |
|---|---|---|---|---|---|---|
| - | 3 | DMF, DMSO | 20 | 7 days | traces | |
| 40 | 2 days | mixture of dAMP, dADP and dATP | ||||
| 1H-tetrazole | 4 | 2 | DMF, DMSO, HMP | 20 | 7 days | 10% |
| 50 | 1 day | mixture of dAMP, dADP and dATP | ||||
| 4,5-dicyanoimidazole | 4 | 1 | DMF | 20 | 6 h | 19% |
| 40 | mixture of dAMP, dADP and dATP | |||||
| 20 | 10 | HMP | 30 | 25 h | 51% | |
| 35 | 64% | |||||
| 40 | 7 h | dAMP, dADP, dATP, dinucleoside 5′,5′-polyphosphate | ||||
| 50 | 6 h | |||||
| DMSO | 40 | 6 h | dATP/dADP, 1:1 ratio | |||
| 15 | 5 | HMP | 35 | 25 h | 25% | |
| 10 | 5 | 19% | ||||
| 5 | 10 | 13% | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsova, V.E.; Shershov, V.E.; Shtylev, G.F.; Shishkin, I.Y.; Butvilovskaya, V.I.; Stomakhin, A.A.; Grechishnikova, I.V.; Zasedateleva, O.A.; Chudinov, A.V. Optimized Method for the Synthesis of Alkyne-Modified 2′-Deoxynucleoside Triphosphates. Molecules 2024, 29, 4747. https://doi.org/10.3390/molecules29194747
Kuznetsova VE, Shershov VE, Shtylev GF, Shishkin IY, Butvilovskaya VI, Stomakhin AA, Grechishnikova IV, Zasedateleva OA, Chudinov AV. Optimized Method for the Synthesis of Alkyne-Modified 2′-Deoxynucleoside Triphosphates. Molecules. 2024; 29(19):4747. https://doi.org/10.3390/molecules29194747
Chicago/Turabian StyleKuznetsova, Viktoriya E., Valeriy E. Shershov, Georgiy F. Shtylev, Ivan Yu. Shishkin, Veronika I. Butvilovskaya, Andrey A. Stomakhin, Irina V. Grechishnikova, Olga A. Zasedateleva, and Alexander V. Chudinov. 2024. "Optimized Method for the Synthesis of Alkyne-Modified 2′-Deoxynucleoside Triphosphates" Molecules 29, no. 19: 4747. https://doi.org/10.3390/molecules29194747
APA StyleKuznetsova, V. E., Shershov, V. E., Shtylev, G. F., Shishkin, I. Y., Butvilovskaya, V. I., Stomakhin, A. A., Grechishnikova, I. V., Zasedateleva, O. A., & Chudinov, A. V. (2024). Optimized Method for the Synthesis of Alkyne-Modified 2′-Deoxynucleoside Triphosphates. Molecules, 29(19), 4747. https://doi.org/10.3390/molecules29194747