

Synthesis of Carborane–Thiazole Conjugates as Tyrosinase and 11β-Hydroxysteroid Dehydrogenase Inhibitors: Antiproliferative Activity and Molecular Docking Studies

 ,
,  ,
,  , , , , ,
, , , , ,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
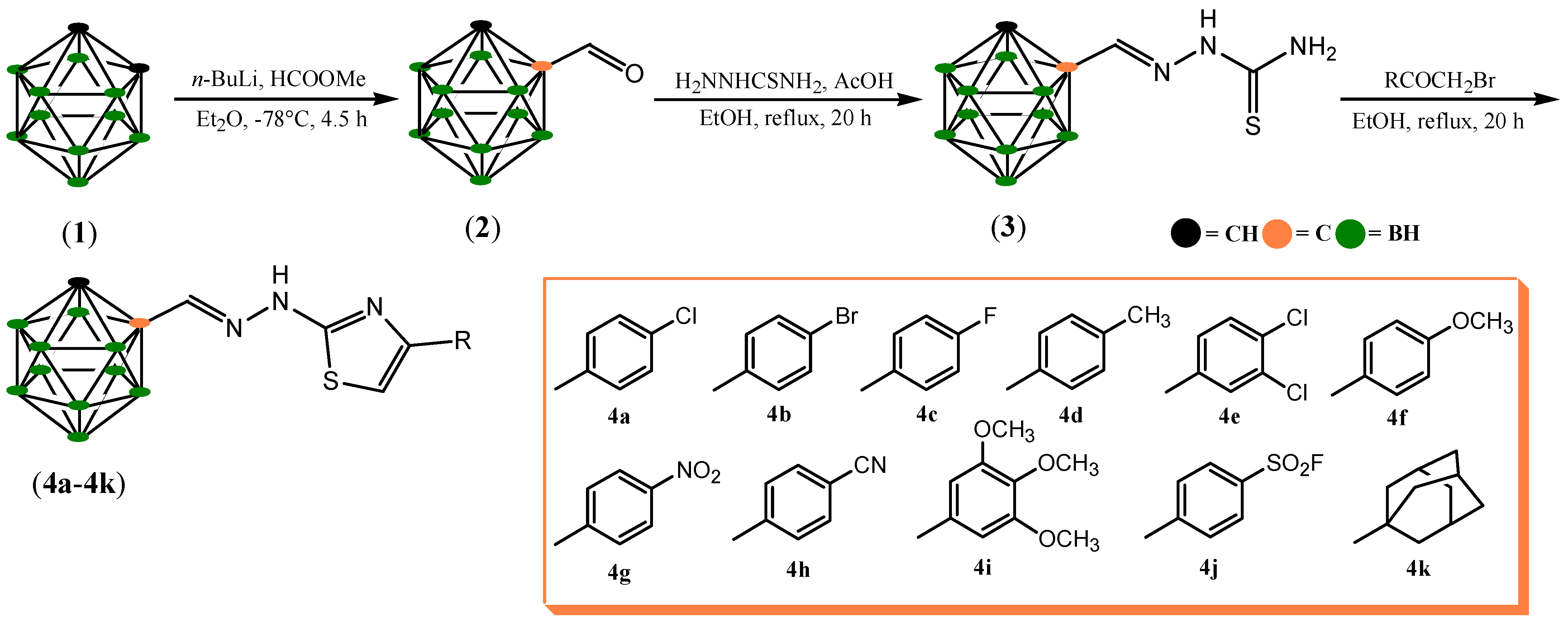
2.1. Chemistry
2.2. Antiproliferative Activity
2.3. Mushroom Tyrosinase Inhibitory Effect and Kinetic Analysis of Compounds
2.4. Inhibition of 11β-HSD1
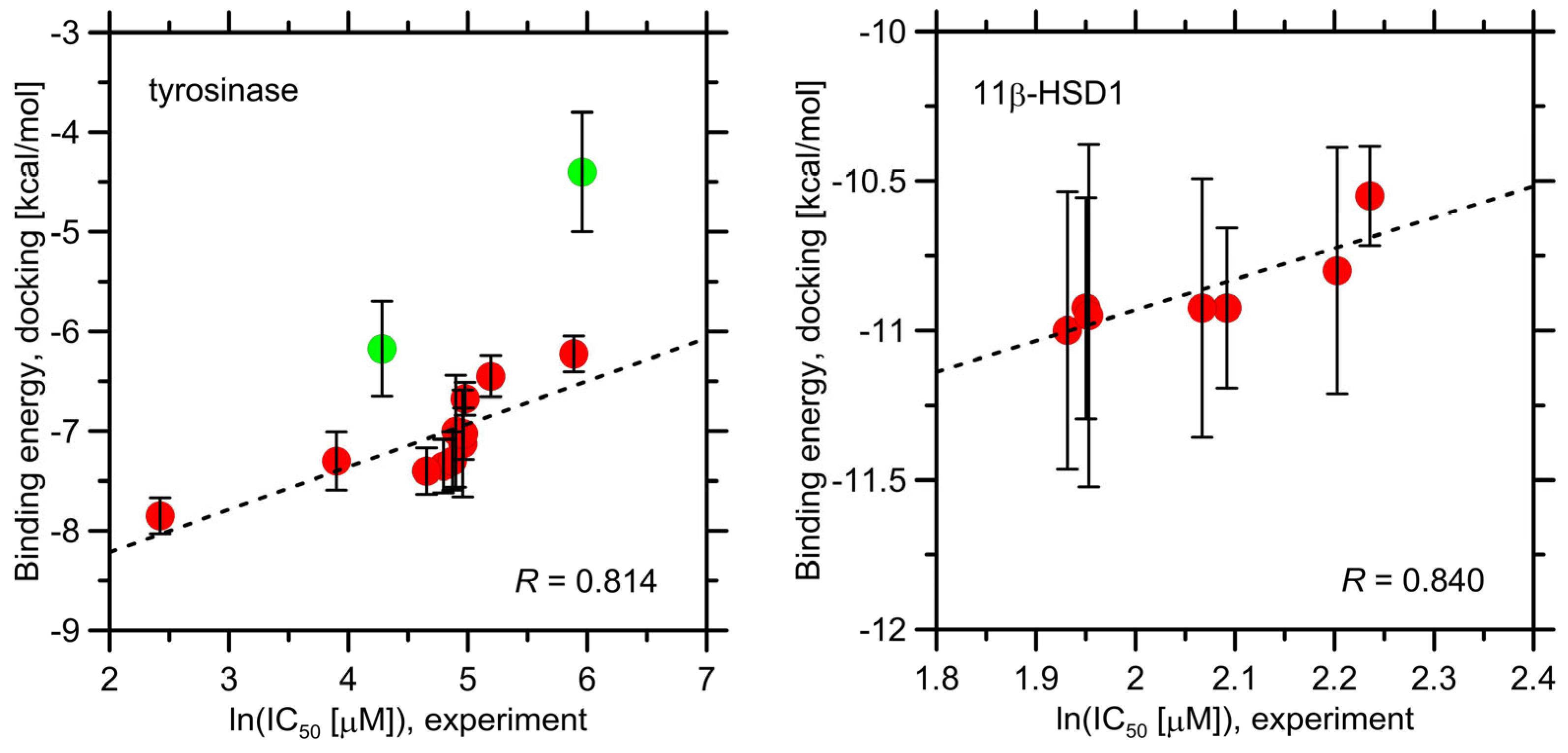
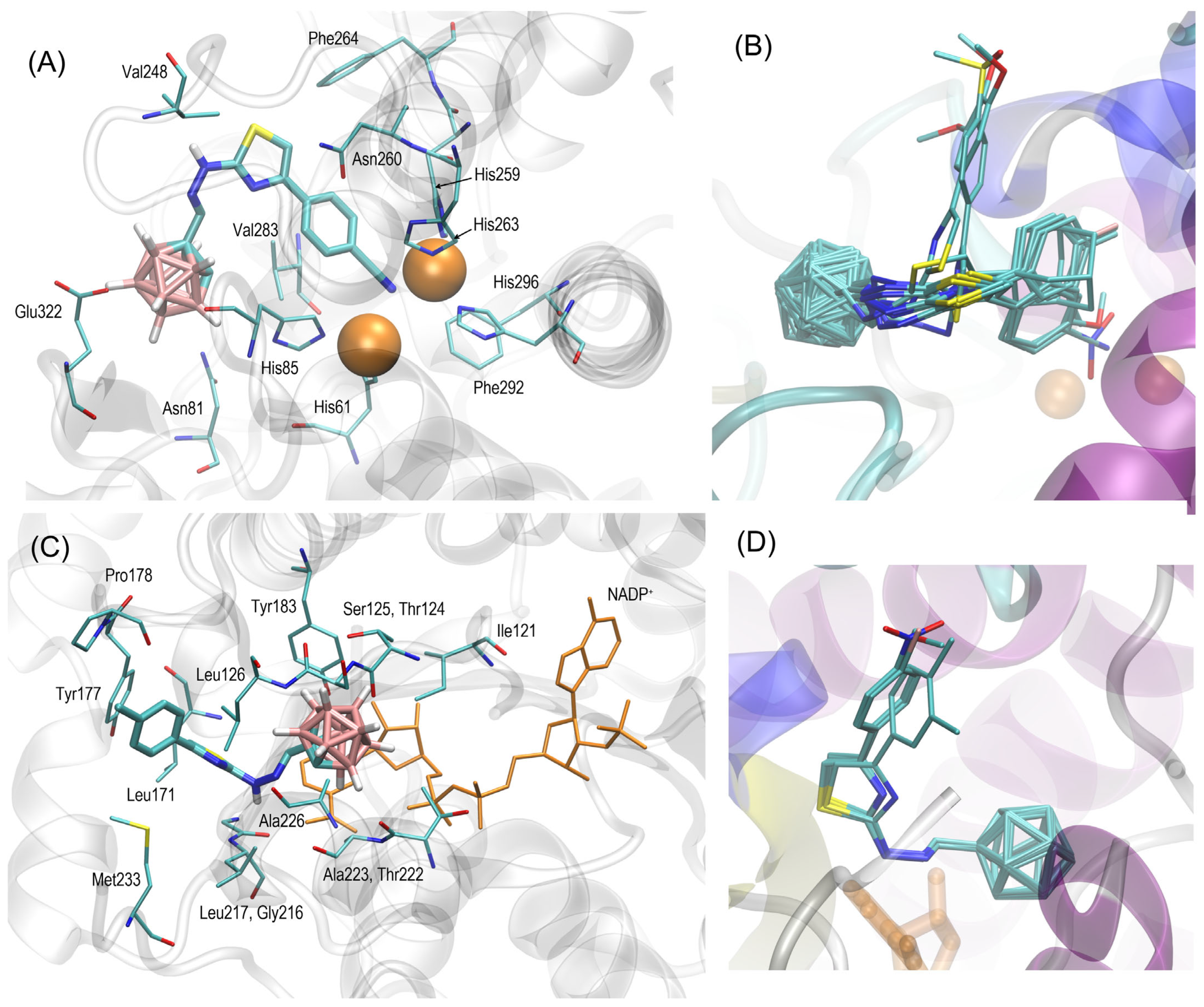
2.5. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. C-Formyl-o-Carborane (2)
3.1.2. 2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinecarbothioamide (3)
3.1.3. 4-(4-Chlorophenyl)-2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)Thiazole (4a)—Typical Procedure
3.1.4. 4-(4-Bromophenyl)-2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)Thiazole (4b)
3.1.5. 2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)-4-(4-Fluorphenyl)Thiazole (4c)
3.1.6. 2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)-4-p-Tolylthiazole (4d)
3.1.7. 2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)-4-(3,4-Dichlorophenyl)Thiazole (4e)
3.1.8. 2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)-4-(4-Methoxyphenyl)Thiazole (4f)
3.1.9. 2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)-4-(4-Nitrophenyl)Thiazole (4g)
3.1.10. 4-(2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)Thiazol-4-yl)Benzonitrile (4h)
3.1.11. 2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)-4-(3,4,5-Trimethoxyphenyl)Thiazole (4i)
3.1.12. 4-(2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)Thiazol-4-yl)Benzene-1-Sulfonyl Fluoride (4j)
3.1.13. 4-(Adamant-1-yl)-2-(2-(1,2-Dicarba-closo-Dodecaboranylmethylene)Hydrazinyl)Thiazole (4k)
3.2. Biological Activity
3.2.1. Antiproliferative Activity
3.2.2. Mushroom Tyrosinase Inhibition Assay
3.2.3. Kinetic Analysis of the Inhibition of Tyrosinase
3.2.4. Reagents and Solvents
3.2.5. Inhibition of 11β-HSD1 Assays
3.2.6. Inhibition of 11β-HSD2 Assays
3.3. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Cancer Burden Growing, Amidst Mounting Need for Services. Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 17 August 2024).
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49, Erratum in CA Cancer J. Clin. 2024, 74, 203. https://doi.org/10.3322/caac.21830. [Google Scholar] [CrossRef] [PubMed]
- Melanoma of the Skin—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 17 August 2024).
- Brożyna, A.A.; Jóźwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A.T. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget 2016, 7, 17844–17853. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Sarna, T.; Płonka, P.M.; Raman, C.; Brożyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045, Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Gautier, J.F.; Chon, S. Assessment of Insulin Secretion and Insulin Resistance in Human. Diabetes Metab. J. 2021, 45, 641–654. [Google Scholar] [CrossRef]
- Zakir, M.; Ahuja, N.; Surksha, M.A.; Sachdev, R.; Kalariya, Y.; Nasir, M.; Kashif, M.; Shahzeen, F.; Tayyab, A.; Khan, M.S.M.; et al. Cardiovascular Complications of Diabetes: From Microvascular to Macrovascular Pathways. Cureus 2023, 15, e45835. [Google Scholar] [CrossRef]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef]
- Geer, E.B.; Islam, J.; Buettner, C. Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrin. Metab. Clin. 2014, 43, 75–102. [Google Scholar] [CrossRef]
- Tomlinson, J.W.; Sherlock, M.; Hughes, B.; Hughes, S.V.; Kilvington, F.; Bartlett, W.; Courtney, R.; Rejto, P.; Carley, W.; Stewart, P.M. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J. Clin. Endocrinol. Metab. 2007, 92, 857–864. [Google Scholar] [CrossRef]
- Qiang, J.K.; Lipscombe, L.L.; Lega, I.C. Association between diabetes, obesity, aging, and cancer: Review of recent literature. Transl. Cancer Res. 2020, 9, 5743–5759. [Google Scholar] [CrossRef]
- Nagore, E.; Martinez-Garcia, M.A.; Gomez-Olivas, J.D.; Manrique-Silva, E.; Martorell, A.; Bañuls, J.; Carrera, C.; Ortiz, P.; Gardeazabal, J.; Boada, A.; et al. Relationship between type 2 diabetes mellitus and markers of cutaneous melanoma aggressiveness: An observational multicentric study in 443 patients with melanoma. Br. J. Dermatol. 2021, 185, 756–763. [Google Scholar] [CrossRef]
- Silva, L.R.; Nunes, J.A.; Zhan, P.; Łączkowski, K.Z.; Cardoso, S.H.; da Silva-Júnior, E.F. Natural coumarin derivatives targeting melanoma. Curr. Med. Chem. 2024, 31, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.A.; Araújo, R.S.A.; Silva, F.N.d.; Cytarska, J.; Łączkowski, K.Z.; Cardoso, S.H.; Mendonça-Júnior, F.J.B.; Silva-Júnior, E.F.d. Coumarin-based compounds as inhibitors of tyrosinase/tyrosine hydroxylase: Synthesis, kinetic studies, and in silico approaches. Int. J. Mol. Sci. 2023, 24, 5216. [Google Scholar] [CrossRef]
- Cytarska, J.; Szulc, J.; Kołodziej-Sobczak, D.A.; Nunes, J.A.; da Silva-Junior, E.F.; Łączkowski, K. Cyrene™ as a tyrosinase inhibitor and anti-browni ng agent. Food Chem. 2024, 442, 138430. [Google Scholar] [CrossRef]
- Baumgart, S.; Kupczyk, D.; Archała, A.; Koszła, O.; Sołek, P.; Płaziński, W.; Płazińska, A.; Studzińska, R. Synthesis of novel 2-(cyclopentylamino)thiazol-4(5H)-one derivatives with potential anticancer, antioxidant, and 11β-HSD inhibitory activities. Int. J. Mol. Sci. 2023, 24, 7252. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, R.; Kołodziejska, R.; Płaziński, W.; Kupczyk, D.; Kosmalski, T.; Jasieniecka, K.; Modzelewska-Banachiewicz, B. Synthesis of the N-methyl derivatives of 2-aminothiazol-4(5H)-one and their interactions with 11βHSD1: Molecular modeling and in vitro studies. Chem. Biodivers. 2019, 16, e1900065. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, R.; Kupczyk, D.; Płazińska, A.; Kołodziejska, R.; Kosmalski, T.; Modzelewska-Banachiewicz, B. Thiazolo [3,2-α]pyrimidin-5-one derivatives as a novel class of 11β-hydroxysteroid dehydrogenase inhibitors. Bioorg Chem. 2018, 81, 21–26. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, G.; He, Y.; Jing Wang, J.J.; Zhao, Y. Tyrosinase inhibitory mechanism and anti-browning properties of novel kojic acid derivatives bearing aromatic aldehyde moiety. Curr. Res. Food Sci. 2023, 6, 100421. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Marfavi, A.; Kavianpour, P.; Rendina, L.M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 2022, 6, 486–504. [Google Scholar] [CrossRef]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sárosi, M.B.; Hey-Hawkins, E. New keys for old locks: Carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef]
- Barth, R.F.; Yang, W.; Al-Madhoun, A.S.; Johnsamuel, J.; Byun, Y.; Chandra, S.; Smith, D.R.; Tjarks, W.; Eriksson, S. Boron-containing nucleosides as potential delivery agents for neutron capture therapy of brain tumors. Cancer Res. 2004, 64, 6287–6295. [Google Scholar] [CrossRef] [PubMed]
- Kabalka, G.W.; Wu, Z.Z.; Yao, M.L.; Natarajan, N. The syntheses and in vivo biodistribution of novel boronated unnatural amino acids. Appl. Radiat. Isot. 2004, 61, 1111–1115. [Google Scholar] [CrossRef]
- Olejniczak, A.B.; Adamska, A.M.; Paradowska, E.; Studzinska, M.; Suski, P.; Leśnikowski, Z.J. Modification of selected anti-HCMV drugs with lipophilic boron cluster modulator. Acta Pol. Pharm. 2013, 70, 489–504. [Google Scholar] [PubMed]
- Fink, K.; Uchman, M. Boron clusters compounds as new chemical leads antimicrobial therapy. Coord. Chem. Rev. 2021, 431, 213684. [Google Scholar] [CrossRef]
- Smith, N.; Quan, D.; Nagalingam, G.; Triccas, J.A.; Rendina, L.M.; Rutledge, P.J. Carborane clusters increase the potency of bis-substituted cyclam derivatives against Mycobacterium tuberculosis. RSC Med. Chem. 2022, 13, 1234–1238. [Google Scholar] [CrossRef]
- Bogucka-Kocka, A.; Kołodziej, P.; Makuch-Kocka, A.; Różycka, D.; Rykowski, S.; Nekvinda, J.; Grüner, B.; Olejniczak, A.B. Nematicidal activity of naphthalimide-boron cluster conjugates. Chem. Commun. 2022, 58, 2528–2531. [Google Scholar] [CrossRef]
- Kugler, M.; Nekvinda, J.; Holub, J.; El Anwar, S.; Das, V.; Šícha, V.; Pospíšilová, K.; Fábry, M.; Král, V.; Brynda, J.; et al. Inhibitors of CA IX Enzyme Based on Polyhedral Boron Compounds. Chembiochem 2021, 22, 2741–2761. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Yasui, Y.; Maruyama, M.; Minegishi, H.; Ban, H.S.; Sato, S. Development of hypoxia-inducible factor (HIF)-1α inhibitors: Effect of ortho-carborane substituents on HIF transcriptional activity under hypoxia. Bioorg. Med. Chem. Lett. 2013, 23, 806–810. [Google Scholar] [CrossRef]
- Li, G.; Azuma, S.; Minegishi, H.; Nakamura, H. Synthesis and biological evaluation of meta-carborane-containing phenoxyacetanilides as inhibitors of hypoxia-inducible factor (HIF)-1 transcriptional activity. J. Organomet. Chem. 2015, 798, 189–195. [Google Scholar] [CrossRef]
- Asawa, Y.; Katsuragi, K.; Sato, A.; Yoshimori, A.; Tanuma, S.I.; Nakamura, H. Structure-based drug design of novel carborane-containing nicotinamide phosphoribosyltransferase inhibitors. Bioorg. Med. Chem. 2019, 27, 2832–2844. [Google Scholar] [CrossRef]
- Neumann, W.; Xu, S.; Sárosi, M.B.; Scholz, M.S.; Crews, B.C.; Ghebreselasie, K.; Banerjee, S.; Marnett, L.J.; Hey-Hawkins, E. nido-Dicarbaborate Induces Potent and Selective Inhibition of Cyclooxygenase-2. ChemMedChem 2016, 11, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Useini, L.; Mojić, M.; Laube, M.; Lönnecke, P.; Dahme, J.; Sárosi, M.B.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; Hey-Hawkins, E. Carboranyl Analogues of Mefenamic Acid and Their Biological Evaluation. ACS Omega 2022, 7, 24282–24291. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, R.; Sárosi, M.B.; George, S.; Lönnecke, P.; Hofmann, B.; Steinhilber, D.; Murganic, B.; Mijatovic, S.; Maksimovic-Ivanic, D.; Hey-Hawkins, E. CarbORev-5901, The First Carborane-Based Inhibitor of the 5-Lipoxygenase Pathway. ChemMedChem 2017, 12, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, R.; Kuhnert, L.; Sárosi, M.B.; George, S.; Draca, D.; Paskas, S.; Hofmann, B.; Steinhilber, D.; Honscha, W.; Mijatović, S.; et al. Borcalein: A Carborane-Based Analogue of Baicalein with 12-Lipoxygenase-Independent Toxicity. ChemMedChem 2022, 17, e202100588. [Google Scholar] [CrossRef]
- Austin, C.J.; Kahlert, J.; Issa, F.; Reed, J.H.; Smith, J.R.; Ioppolo, J.A.; Ong, J.A.; Jamie, J.F.; Hibbs, D.; Rendina, L.M. The first indoleamine-2,3-dioxygenase-1 (IDO1) inhibitors containing carborane. Dalton Trans. 2014, 43, 10719–10724. [Google Scholar] [CrossRef]
- Rosada, B.; Bekier, A.; Cytarska, J.; Płaziński, W.; Zavyalova, O.; Sikora, A.; Dzitko, K.; Łączkowski, K.Z. Benzo[b]thiophene-thiazoles as potent anti-Toxoplasma gondii agents: Design, synthesis, tyrosinase/tyrosine hydroxylase inhibitors, molecular docking study, and antioxidant activity. Eur. J. Med. Chem. 2019, 184, 111765. [Google Scholar] [CrossRef]
- Piechowska, K.; Świtalska, M.; Cytarska, J.; Jaroch, K.; Łuczykowski, K.; Chałupka, J.; Wietrzyk, J.; Misiura, K.; Bojko, B.; Kruszewski, S.; et al. Discovery of tropinone-thiazole derivatives as potent caspase 3/7 activators, and noncompetitive tyrosinase inhibitors with high antiproliferative activity: Rational design, one-pot tricomponent synthesis, and lipophilicity determination. Eur. J. Med. Chem. 2019, 175, 162–171. [Google Scholar] [CrossRef]
- Piechowska, K.; Mizerska-Kowalska, M.; Zdzisińska, B.; Cytarska, J.; Baranowska-Łączkowska, A.; Jaroch, K.; Łuczykowski, K.; Płaziński, W.; Bojko, B.; Kruszewski, S.; et al. Tropinone-derived alkaloids as potent anticancer agents: Synthesis, tyrosinase inhibition, mechanism of action, DFT calculation, and molecular docking studies. Int. J. Mol. Sci. 2020, 21, 9050. [Google Scholar] [CrossRef]
- Dozzo, P.; Kasar, R.A.; Kahl, S.B. Simple, High-yield methods for the synthesis of aldehydes directly from o-, m-, and p-carborane and their further conversions. Inorg. Chem. 2005, 44, 8053–8057. [Google Scholar] [CrossRef]
- Saeed, A.; Mahesar, P.A.; Channar, P.A.; Abbas, Q.; Larik, F.A.; Hassan, M.; Raza, H.; Seo, S.Y. Synthesis, molecular docking studies of coumarinyl-pyrazolinyl substituted thiazoles as non-competitive inhibitors of mushroom tyrosinase. Bioorg Chem. 2017, 74, 187–196. [Google Scholar] [CrossRef]
- Shin, N.H.; Ryu, S.Y.; Choi, E.J.; Kang, S.H.; Chang, I.M.; Min, K.R.; Kim, Y. Oxyresveratrol as the potent inhibitor on dopa oxidase activity of mushroom tyrosinase. Biochem. Biophys. Res. Commun. 1998, 243, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian09 Package, version 09; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Tiwari, R.; Mahasenan, K.; Pavlovicz, R.; Li, C.; Tjarks, W. Carborane clusters in computational drug design: A comparative docking evaluation using AutoDock, FlexX, Glide, and Surflex. J. Chem. Inf. Model. 2009, 49, 1581–1589. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Sraker, R.J.; Tortorello, G.N.; Sharon, C.E.; Keele, L.J.; Chu, E.Y.; Miura, J.T.; Karakousis, G.C.; Ming, M.E. Association of type II diabetes mellitus with characteristics and outcomes for patients undergoing sentinel lymph node biopsy for cutaneous melanoma. J. Surg. Oncol. 2022, 126, 1263–1271. [Google Scholar] [CrossRef]
- Kaneko, A.; Kanemaru, H.; Mizuhashi, S.; Kimura, T.; Kuriyama, H.; Sawamura, S.; Kajihara, I.; Makino, K.; Miyashita, A.; Aoi, J.; et al. Relationship between Type 2 diabetes mellitus and aggressiveness of melanoma. J. Dermatol. Sci. 2022, 106, 65–67. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiazole Derivatives | IC50 ± SD [µM] | SI | |||||
|---|---|---|---|---|---|---|---|
| A172 | B16F10 | MDA-MB-231 | HUVEC | HUVEC/A172 | HUVEC/ B16F10 | HUVEC/ MDA-MB-231 | |
| 4a | 5.77 ± 0.96 | 5.03 ± 0.69 | 6.54 ± 0.52 | 6.87 ± 0.33 | 1.19 | 1.37 | 1.05 |
| 4b | 6.15 ± 0.26 | 5.37 ± 0.11 | 6.56 ± 0.12 | 7.46 ± 0.13 | 1.21 | 1.39 | 1.14 |
| 4c | 7.10 ± 1.90 | 5.57 ± 0.39 | 6.02 ± 0.08 | 7.51 ± 0.64 | 1.06 | 1.35 | 1.25 |
| 4d | 7.88 ± 0.98 | >10 | 6.85 ± 0.86 | 8.32 ± 0.09 | 1.06 | – | 1.21 |
| 4e | 6.44 ± 0.60 | 5.58 ± 0.35 | 6.99 ± 2.27 | 7.86 ± 0.61 | 1.22 | 1.41 | 1.12 |
| 4f | >10 | 5.81 ± 0.82 | 7.32 ± 1.26 | >10 | – | >1.72 | >1.37 |
| 4g | 6.07 ± 1.06 | 5.11 | 6.96 ± 0.74 | >10 | >1.65 | >1.96 | >1.44 |
| 4h | 7.72 ± 1.25 | 5.56 ± 0.30 | 6.64 ± 0.81 | 6.17 ± 0.35 | 0.80 | 1.11 | 0.93 |
| 4i | >10 | 5.88 | >10 | >10 | – | >1.70 | – |
| 4j | 6.40 ± 0.71 | 5.75 ± 0.46 | 6.43 ± 0.23 | >10 | >1.56 | >1.74 | >1.56 |
| 4k | >10 | >10 | >10 | 5.86 ± 0.34 | – | – | – |
| chlorambucil | >10 | 5.44 ± 0.19 | >10 | 7.43 ± 0.54 | – | 1.37 | – |
| Compound at 0.05 mM | IC50 ± SD [µM] | Mechanism of Inhibition | Vmax [mM/min] | KM [mM] |
|---|---|---|---|---|
| 4a | 130.49 ± 20.94 | Mixed | 0.1656 | 0.4368 |
| 4b | 144.88 ± 14.36 | Activator | 0.0912 | 0.1868 |
| 4c | 143.09 ± 9.38 | Activator | 0.0810 | 0.9103 |
| 4d | 120.91 ± 21.88 | Mixed | 0.3820 | 0.5246 |
| 4e | 142.21 ± 12.98 | Activator | 0.1575 | 0.4651 |
| 4f | 49.36 ± 8.01 | Activator | 0.1472 | 0.5206 |
| 4g | 104.96 ± 9.08 | Uncompetitive | 0.0521 | 0.1890 |
| 4h | 11.27 ± 5.90 | Mixed | 0.1258 | 0.5711 |
| 4i | 179.78 ± 7.83 | Mixed | 0.5474 | 0.5868 |
| 4j | 360.51 ± 118.59 | Mixed | 0.7876 | 0.5072 |
| 4k | 134.11 ± 5.84 | Mixed | 0.7863 | 0.6933 |
| Ascorbic acid | 386.50 ± 11.96 | ― | ― | ― |
| Kojic acid | 72.27 ± 3.15 | ― | ― | ― |
| Thiazole Derivatives | % of 11β-HSD1 Inhibition 10 μM | IC50 ± SD (µM) 1 11β-HSD1 | % of 11β-HSD2 Inhibition 10 μM |
|---|---|---|---|
| 4a | 60.07 | 7.03 ± 0.53 | 25.47 |
| 4b | 64.44 | 6.90 ± 1.20 | 27.36 |
| 4c | 55.74 | 8.10 ± 2.00 | 21.70 |
| 4d | 55.78 | 9.05 ± 0.25 | 27.83 |
| 4e | 62.17 | 7.90 ± 1.40 | 19.34 |
| 4f | 55.54 | 9.35 ± 1.85 | 29.25 |
| 4g | 73.51 | 7.05 ± 0.25 | 32.55 |
| Carbenoxolone | 88.93 | <0.5 | |
| 18β-Glycyrrhetinic acid | 43.40 |
| Compound | Binding Energy [kcal/mol] | |
|---|---|---|
| Tyrosinase | 11β-HSD1 | |
| 4a | −7.3 ± 0.3 | −10.9 ± 0.4 |
| 4b | −6.7 ± 0.2 | −11.0 ± 0.5 |
| 4c | −7.0 ± 0.3 | −10.9 ± 0.3 |
| 4d | −7.4 ± 0.3 | −10.8 ± 0.4 |
| 4e | −7.1 ± 0.5 | −10.9 ± 0.4 |
| 4f | −7.3 ± 0.3 | −10.6 ± 0.2 |
| 4g | −7.4 ± 0.2 | −11.0 ± 0.6 |
| 4h | −7.9 ± 0.2 | |
| 4i | −6.2 ± 0.2 | |
| 4j | −6.2 ± 0.2 | |
| 4k | −7.0 ± 0.6 | |
| Ascorbic acid 1 | −4.4 ± 0.6 1 | |
| Kojic acid | −6.2 ± 0.5 | |
| Carbenoxolone | −9.2 ± 0.4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donarska, B.; Cytarska, J.; Kołodziej-Sobczak, D.; Studzińska, R.; Kupczyk, D.; Baranowska-Łączkowska, A.; Jaroch, K.; Szeliska, P.; Bojko, B.; Różycka, D.; et al. Synthesis of Carborane–Thiazole Conjugates as Tyrosinase and 11β-Hydroxysteroid Dehydrogenase Inhibitors: Antiproliferative Activity and Molecular Docking Studies. Molecules 2024, 29, 4716. https://doi.org/10.3390/molecules29194716
Donarska B, Cytarska J, Kołodziej-Sobczak D, Studzińska R, Kupczyk D, Baranowska-Łączkowska A, Jaroch K, Szeliska P, Bojko B, Różycka D, et al. Synthesis of Carborane–Thiazole Conjugates as Tyrosinase and 11β-Hydroxysteroid Dehydrogenase Inhibitors: Antiproliferative Activity and Molecular Docking Studies. Molecules. 2024; 29(19):4716. https://doi.org/10.3390/molecules29194716
Chicago/Turabian StyleDonarska, Beata, Joanna Cytarska, Dominika Kołodziej-Sobczak, Renata Studzińska, Daria Kupczyk, Angelika Baranowska-Łączkowska, Karol Jaroch, Paulina Szeliska, Barbara Bojko, Daria Różycka, and et al. 2024. "Synthesis of Carborane–Thiazole Conjugates as Tyrosinase and 11β-Hydroxysteroid Dehydrogenase Inhibitors: Antiproliferative Activity and Molecular Docking Studies" Molecules 29, no. 19: 4716. https://doi.org/10.3390/molecules29194716
APA StyleDonarska, B., Cytarska, J., Kołodziej-Sobczak, D., Studzińska, R., Kupczyk, D., Baranowska-Łączkowska, A., Jaroch, K., Szeliska, P., Bojko, B., Różycka, D., Olejniczak, A. B., Płaziński, W., & Łączkowski, K. Z. (2024). Synthesis of Carborane–Thiazole Conjugates as Tyrosinase and 11β-Hydroxysteroid Dehydrogenase Inhibitors: Antiproliferative Activity and Molecular Docking Studies. Molecules, 29(19), 4716. https://doi.org/10.3390/molecules29194716