

Ringing the Changes: Effects of Heterocyclic Ring Size on Stereoselectivity in [(η5-C5Me5)RhCl], [(η5-C5Me5)IrCl] and [Ru(η6-cymene)Cl] Complexes of Chiral 3-Amino-1-Azacycles

 , , and
, , and 
Abstract

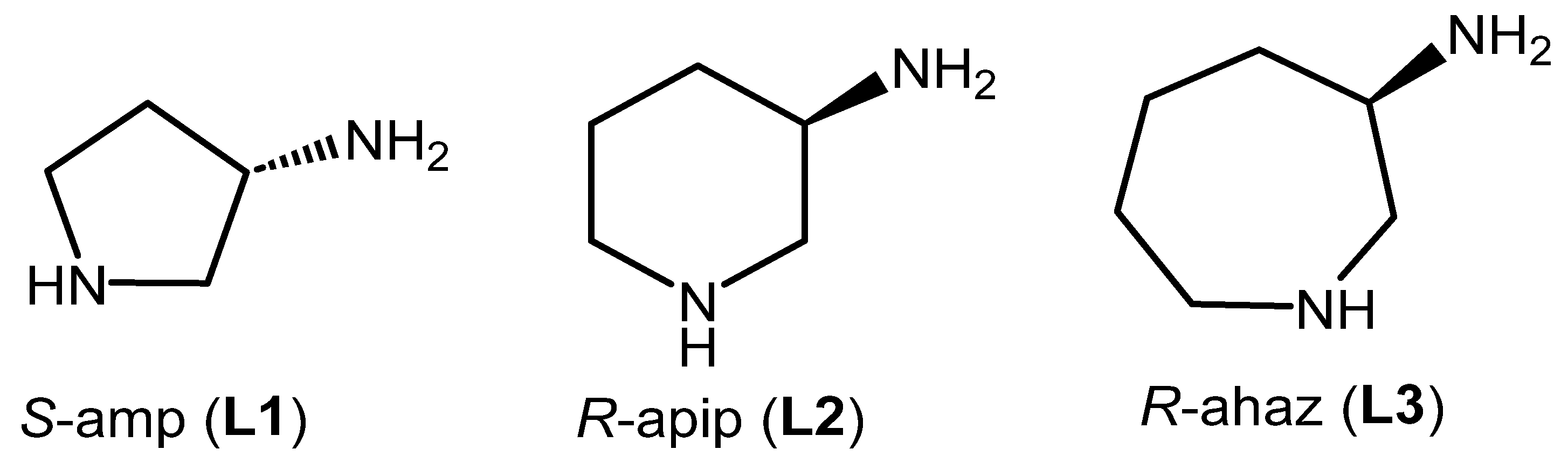
1. Introduction
2. Results and Discussion
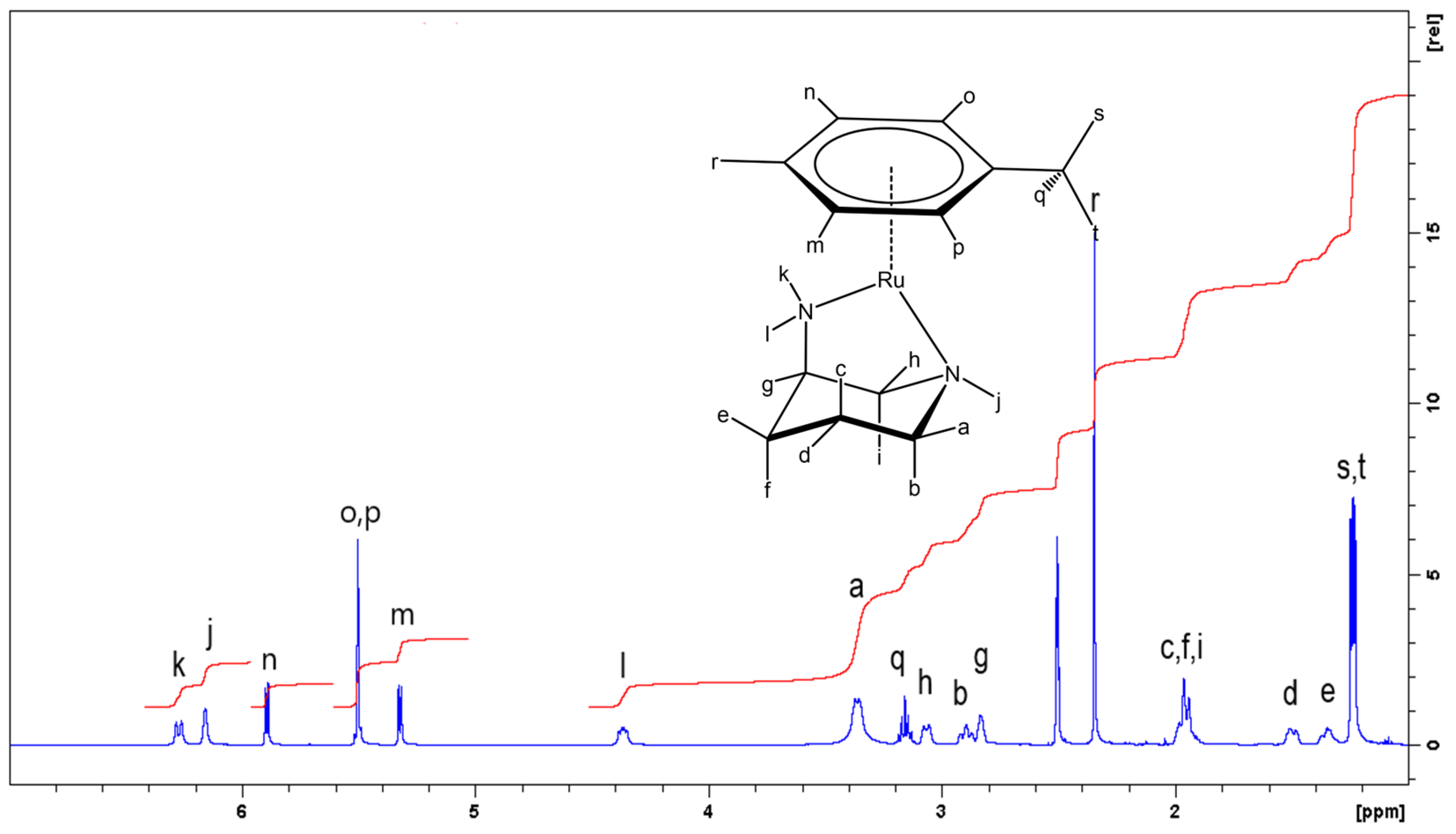
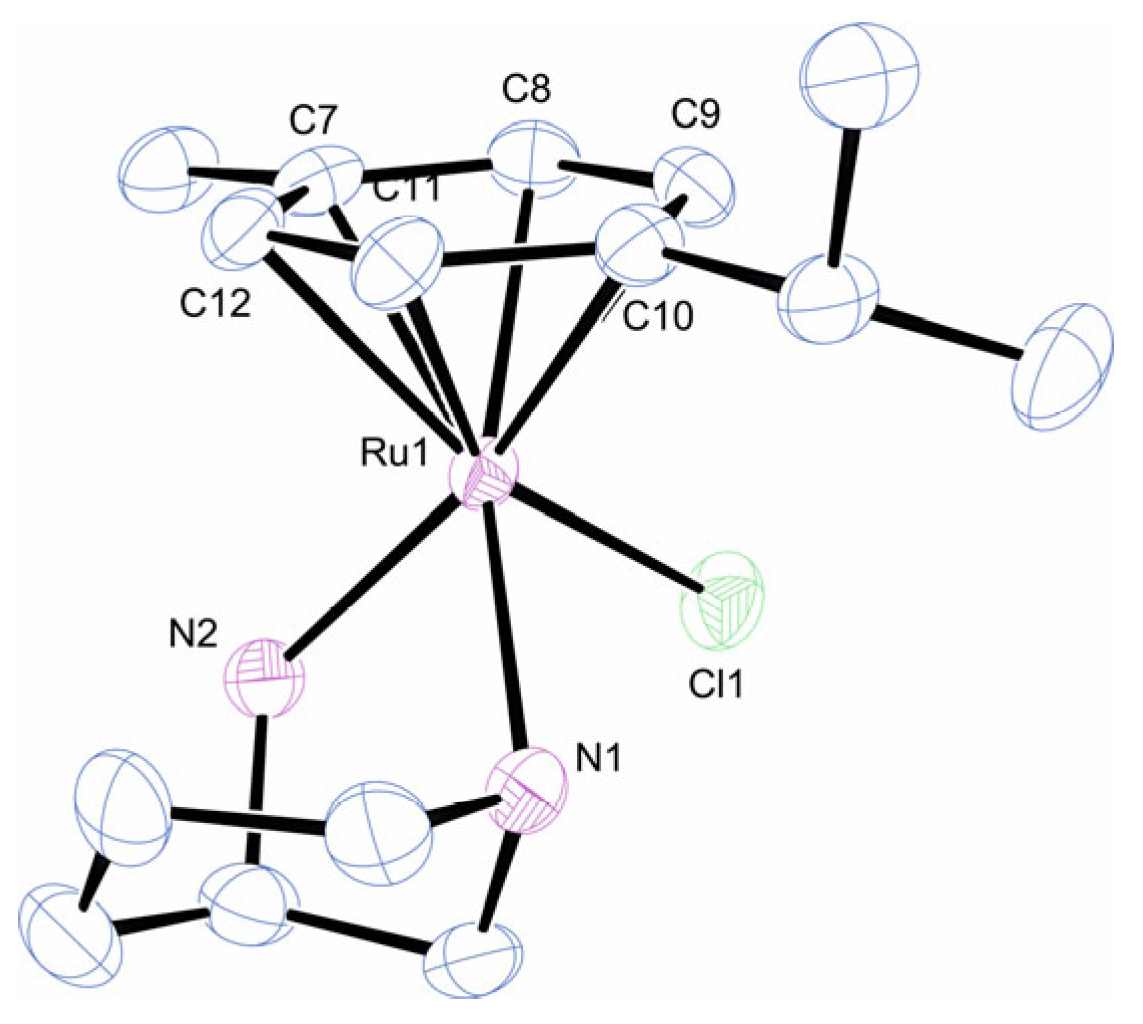
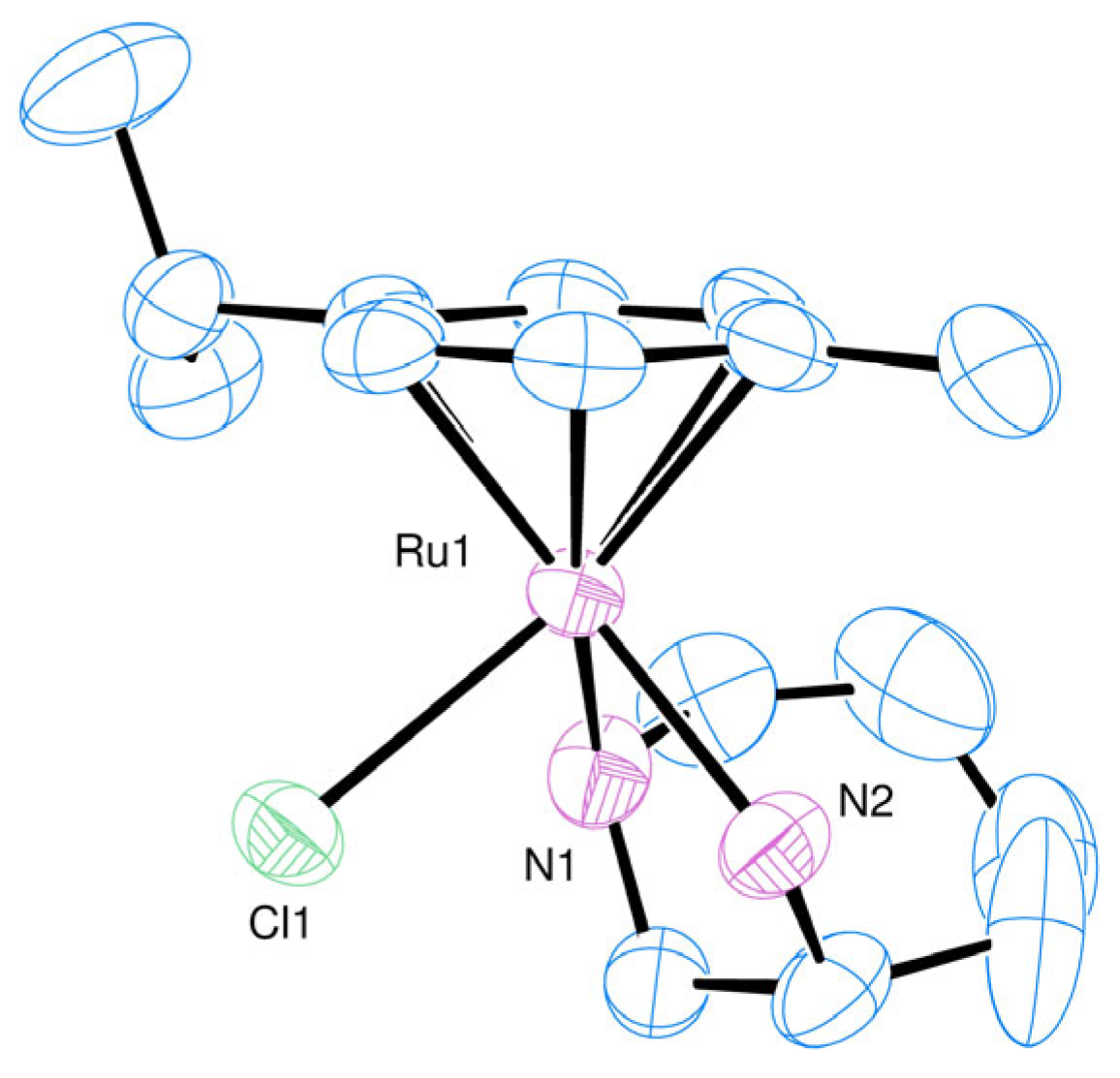
2.1. Synthesis and Characterisation of the Complexes
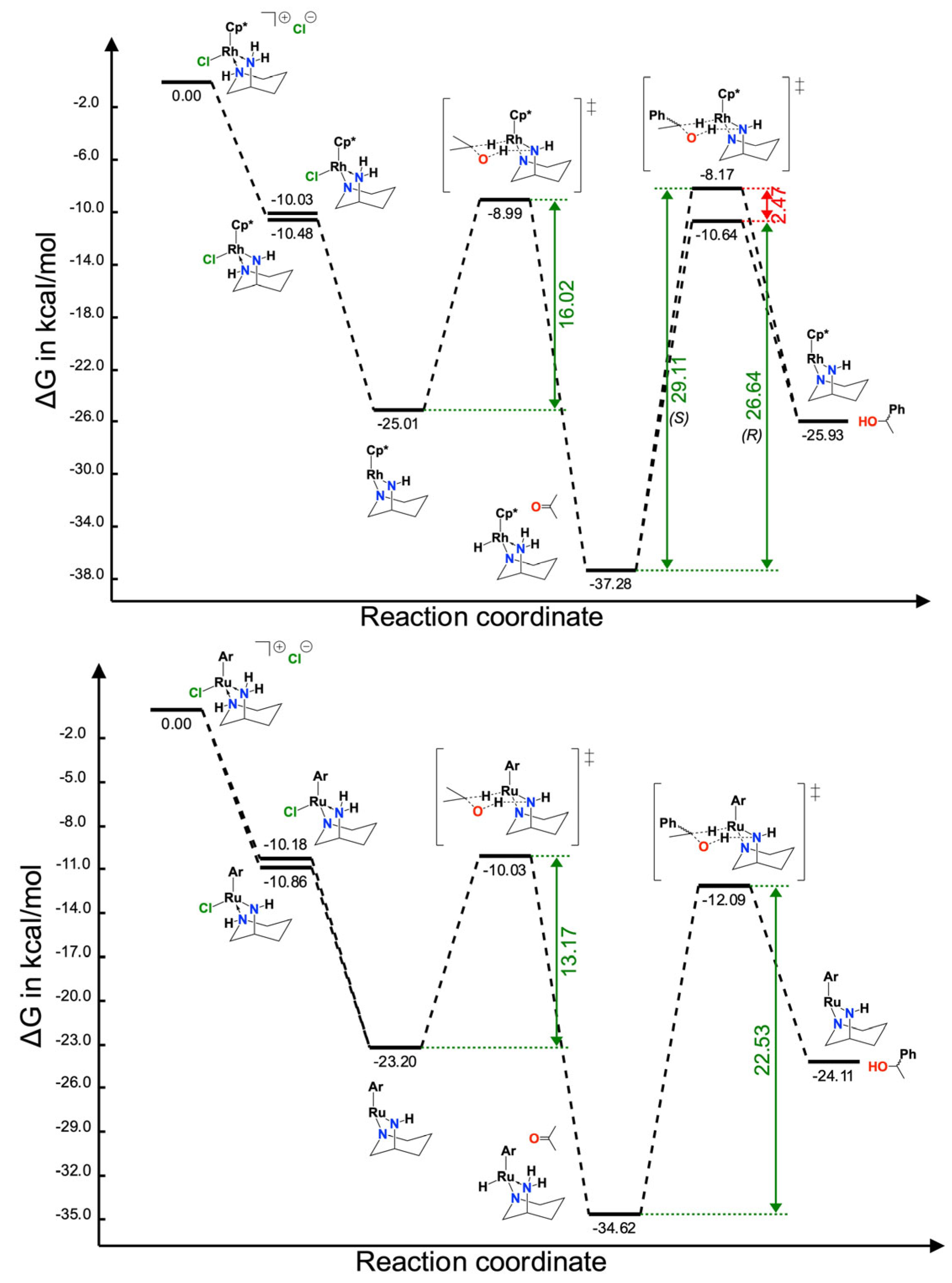
2.2. Computational Studies of the Coordination Stereoselectivity
2.3. Asymmetric Transfer Hydrogenation Studies
3. Materials and Methods
General Synthesis of the [(η5-C5Me5) M (L) Cl] X Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, E.B. Chiral-at-metal complexes and their catalytic applications in organic synthesis. Chem. Soc. Rev. 2012, 41, 3153–3167. [Google Scholar] [CrossRef] [PubMed]
- Cruchter, T.; Larionov, V.A. Asymmetric catalysis with octahedral stereogenic-at-metal complexes featuring chiral ligands. Coord. Chem. Rev. 2018, 376, 95–113. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Cai, J.; Gong, L. Visible-Light-Promoted Asymmetric Catalysis by Chiral Complexes of First-Row Transition Metals. Synthesis 2021, 53, 1570–1583. [Google Scholar]
- Ye, C.-X.; Chen, S.; Han, F.; Xie, X.; Ivlev, S.; Houk, K.N.; Meggers, E. Atroposelective Synthesis of Axially Chiral N-Arylpyrroles by Chiral-at-Rhodium Catalysis. Angew. Chem. Int. Ed. 2020, 59, 13552–13556. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, Z.; Shen, X.; Ivlev, S.; Meggers, E. Asymmetric Catalysis with a Chiral-at-Osmium Complex. Chem. Commun. 2020, 56, 7714–7717. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, X.; Huang, X.; Luo, S.; Meggers, E. Preparation of chiral-at-metal catalysts and their use in asymmetric photoredox chemistry. Nat. Protoc. 2018, 13, 605–632. [Google Scholar] [CrossRef]
- Winterling, E.; Ivlev, S.; Meggers, E. Chiral-at-Ruthenium Catalysts with Mixed Normal and Abnormal N-Heterocyclic Carbene Ligands. Organometallics 2021, 40, 1148–1155. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Ehnbom, A.; Lewis, K.G.; Gladysz, J.A. Hydrogen Bonding Motifs in Structurally Characterized Salts of the Tris(ethylenediamine) Cobalt Trication, [Co(en)3]3+; an Interpretive Review, including Implications for Catalysis. Coord. Chem. Rev. 2017, 350, 30–48. [Google Scholar] [CrossRef]
- Kabes, C.Q.; Maximuck, W.J.; Ghosh, S.K.; Kumar, A.; Bhuvanesh, N.S.; Gladysz, J.A. Tricationic tris(1,2-diphenylethylenediamine) Cobalt(III) Hydrogen Bond Donor Catalysts with Defined Configurations at the Carbon and Metal Stereocenters; The Effect of Chiral Counter Anions upon Enantioselectivities. ACS Catal. 2020, 10, 3249–3263. [Google Scholar] [CrossRef]
- Savel’yeva, T.F.; Khromova, O.V.; Larionov, V.A.; Smol’yakov, A.F.; Fedyanin, I.V.; Belokon, Y.N.; Maleev, V.I. Expanding the Family of Octahedral Chiral-at-Metal Cobalt(III) Catalysts by Introducing Tertiary Amine Moiety into the Ligand. Catalysts 2021, 11, 152. [Google Scholar] [CrossRef]
- Chen, L.-A.; Xu, W.; Huang, B.; Ma, J.; Wang, L.; Xi, J.; Harms, K.; Gong, L.; Meggers, E. Asymmetric Catalysis with Inert Chiral-at-Metal Iridium Complex. J. Am. Chem. Soc. 2013, 135, 10598–10601. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Fu, C.; Harms, K.; Meggers, E. Asymmetric Catalysis with Substitutionally Labile yet Stereochemically Stable Chiral-at-Metal Iridium(III) Complex. J. Am. Chem. Soc. 2014, 136, 2990–2993. [Google Scholar] [CrossRef] [PubMed]
- Wegener, A.R.; Kabes, C.Q.; Gladysz, J.A. Launching Werner Complexes into the Modern Era of Catalytic Enantioselective Organic Synthesis. Acc. Chem. Res. 2020, 53, 2299–2313. [Google Scholar] [CrossRef] [PubMed]
- Larionov, V.A.; Markelova, E.P.; Smol’Yakov, A.F.; Savel’yeva, T.F.; Maleev, V.I.; Belokon, Y.N. Chiral octahedral complexes of Co(iii) as catalysts for asymmetric epoxidation of chalcones under phase transfer conditions. RSC Adv. 2015, 5, 72764–72771. [Google Scholar] [CrossRef]
- Lewis, K.G.; Ghosh, S.K.; Bhuvanesh, N.; Gladysz, J.A. Cobalt(III) Werner Complexes with 1,2-Diphenylethylenediamine Ligands: Readily Available, Inexpensive, and Modular Chiral Hydrogen Bond Donor Catalysts for Enantioselective Organic Synthesis. ACS Cent. Sci. 2015, 1, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Arieno, M.; Löw, H.; Huang, K.; Xie, X.; Cruchter, T.; Ma, Q.; Xi, J.; Huang, B.; Wiest, O.; et al. Metal-Templated Design: Enantioselective Hydrogen-Bond-Driven Catalysis Requiring Only Parts-per-Million Catalyst Loading. J. Am. Chem. Soc. 2016, 138, 8774–8780. [Google Scholar] [CrossRef]
- Kurono, N.; Arai, K.; Uemura, M.; Ohkuma, T. [Ru(phgly)2(binap)]/Li2CO3: A Highly Active, Robust, and Enantioselective Catalyst for the Cyanosilylation of Aldehydes. Angew. Chem. Int. Ed. 2008, 47, 6643–6646. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, T.; Kurono, N. Asymmetric Cyanation with the Chiral Ru–Li Combined Catalysts. Synlett 2012, 23, 1865–1881. [Google Scholar] [CrossRef]
- Suzuki, K.; Oldenburg, P.D.; Que, L., Jr. Iron-Catalyzed Asymmetric Olefin cis-Dihydroxylation with 97 % Enantiomeric Excess. Angew. Chem. Int. Ed. 2008, 47, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Khamis, N.; Clarkson, G.J.; Wills, M. Heterocycle-containing Noyori–Ikariya catalysts for asymmetric transfer hydrogenation of ketones. Dalton Trans. 2022, 51, 13462–13469. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid−Triethylamine Mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Cotman, A.E. Escaping from Flatland: Stereoconvergent Synthesis of Three-Dimensional Scaffolds via Ruthenium(II)-Catalyzed Noyori–Ikariya Transfer Hydrogenation. Chem. Eur. J. 2021, 27, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, H.; Kani, I.; Cetinkaya, B. Transfer Hydrogenation of Aryl Ketones with Half-Sandwich RuII Complexes That Contain Chelating Diamines. Eur. J. Inorg. Chem. 2012, 2012, 4494–4499. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs 2022, 12, e1606. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 64103. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, H.; Noyori, R. The Metal−Ligand Bifunctional Catalysis: A Theoretical Study on the Ruthenium(II)-Catalyzed Hydrogen Transfer between Alcohols and Carbonyl Compounds. J. Am. Chem. Soc. 2000, 122, 1466–1478. [Google Scholar] [CrossRef]
- Furness, J.W.; Kaplan, A.D.; Ning, J.; Perdew, J.P.; Sun, J. Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation. J. Phys. Chem. Lett. 2020, 11, 8208–8215. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Bitzek, E.; Koskinen, P.; Gähler, F.; Moseler, M.; Gumbsch, P. Structural Relaxation Made Simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef]
- Høyvik, I.-M.; Jansik, B.; Jørgensen, P. Trust Region Minimization of Orbital Localization Functions. J. Chem. Theory Comput. 2012, 8, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Helmich-Paris, B. A trust-region augmented Hessian implementation for restricted and unrestricted Hartree–Fock and Kohn–Sham methods. J. Chem. Phys. 2021, 154, 164104. [Google Scholar] [CrossRef] [PubMed]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Conversion (%) b | e.e. (%) c |
|---|---|---|
| Rh-L1 | 98 | 1.8 |
| Rh-L2 | 89 | 0.5 |
| Rh-L3 | 95 | 1.6 |
| Ir-L1 | 98 | 0.4 |
| Ir-L2 | 97 | 0.2 |
| Ir-L3 | 97 | 0.3 |
| Ru-L1 | 90 | 0.6 |
| Ru-L2 | 90 | 1.8 |
| Ru-L3 | 94 | 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vladimirov, V.Y.; Charrier-Chardin, M.; Kariuki, B.M.; Ward, B.D.; Newman, P.D. Ringing the Changes: Effects of Heterocyclic Ring Size on Stereoselectivity in [(η5-C5Me5)RhCl], [(η5-C5Me5)IrCl] and [Ru(η6-cymene)Cl] Complexes of Chiral 3-Amino-1-Azacycles. Molecules 2024, 29, 4659. https://doi.org/10.3390/molecules29194659
Vladimirov VY, Charrier-Chardin M, Kariuki BM, Ward BD, Newman PD. Ringing the Changes: Effects of Heterocyclic Ring Size on Stereoselectivity in [(η5-C5Me5)RhCl], [(η5-C5Me5)IrCl] and [Ru(η6-cymene)Cl] Complexes of Chiral 3-Amino-1-Azacycles. Molecules. 2024; 29(19):4659. https://doi.org/10.3390/molecules29194659
Chicago/Turabian StyleVladimirov, Vladimir Y., Matheo Charrier-Chardin, Benson M. Kariuki, Benjamin D. Ward, and Paul D. Newman. 2024. "Ringing the Changes: Effects of Heterocyclic Ring Size on Stereoselectivity in [(η5-C5Me5)RhCl], [(η5-C5Me5)IrCl] and [Ru(η6-cymene)Cl] Complexes of Chiral 3-Amino-1-Azacycles" Molecules 29, no. 19: 4659. https://doi.org/10.3390/molecules29194659
APA StyleVladimirov, V. Y., Charrier-Chardin, M., Kariuki, B. M., Ward, B. D., & Newman, P. D. (2024). Ringing the Changes: Effects of Heterocyclic Ring Size on Stereoselectivity in [(η5-C5Me5)RhCl], [(η5-C5Me5)IrCl] and [Ru(η6-cymene)Cl] Complexes of Chiral 3-Amino-1-Azacycles. Molecules, 29(19), 4659. https://doi.org/10.3390/molecules29194659