
An Overview of Naphthylimide as Specific Scaffold for New Drug Discovery
Abstract
1. Introduction
2. Progress in Biological Activity of Naphthylimide
2.1. Antimicrobial Activity
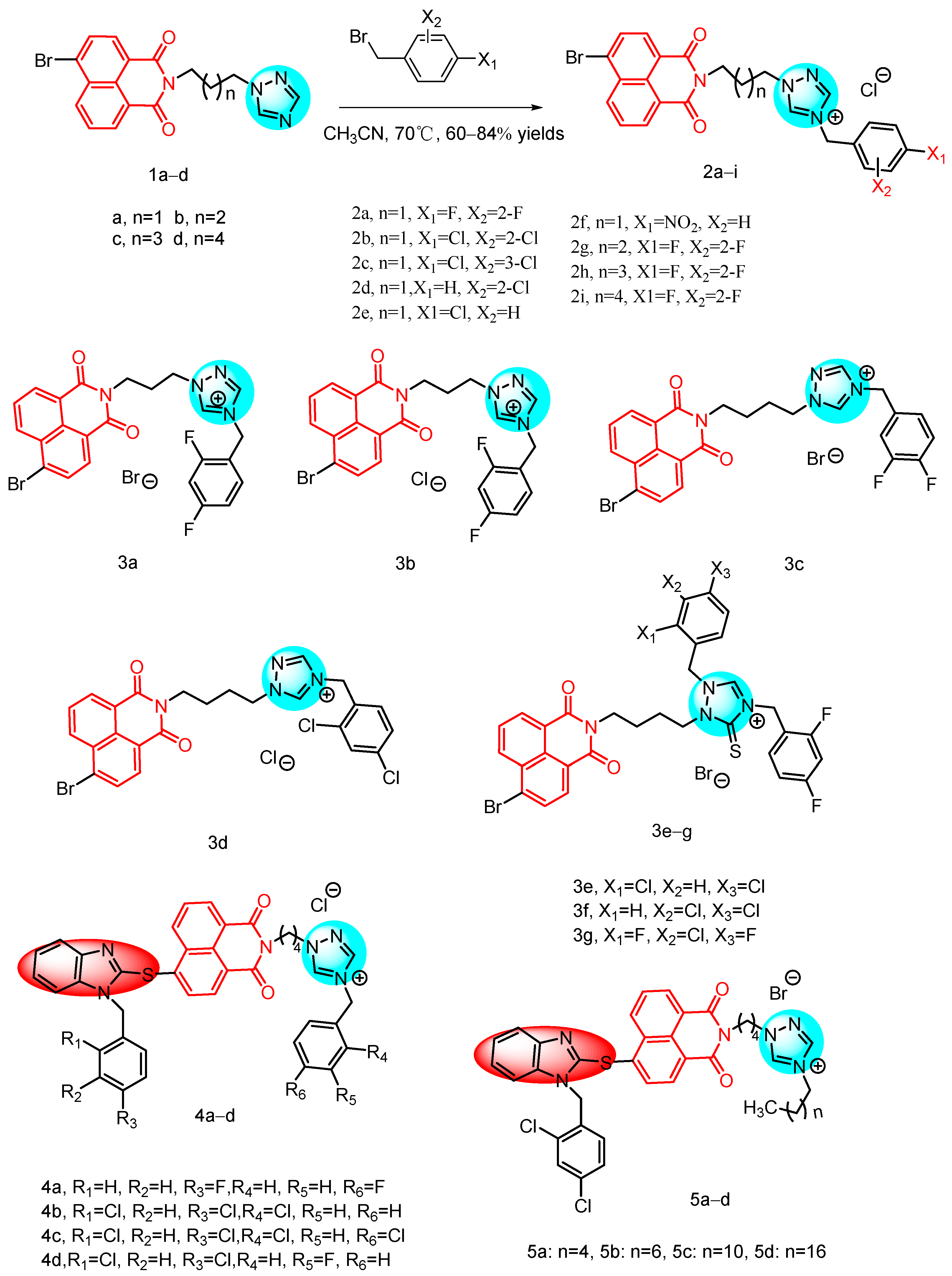
2.1.1. Triazole-Containing Naphthylimide Hybrids
2.1.2. Triazine-Containing Naphthylimide Hybrids
2.1.3. Naphthylimide-Derived Metronidazole
2.1.4. Schiff BASE-Linked Imidazolylnaphthimide
2.1.5. Naphthylimides and Allylenes
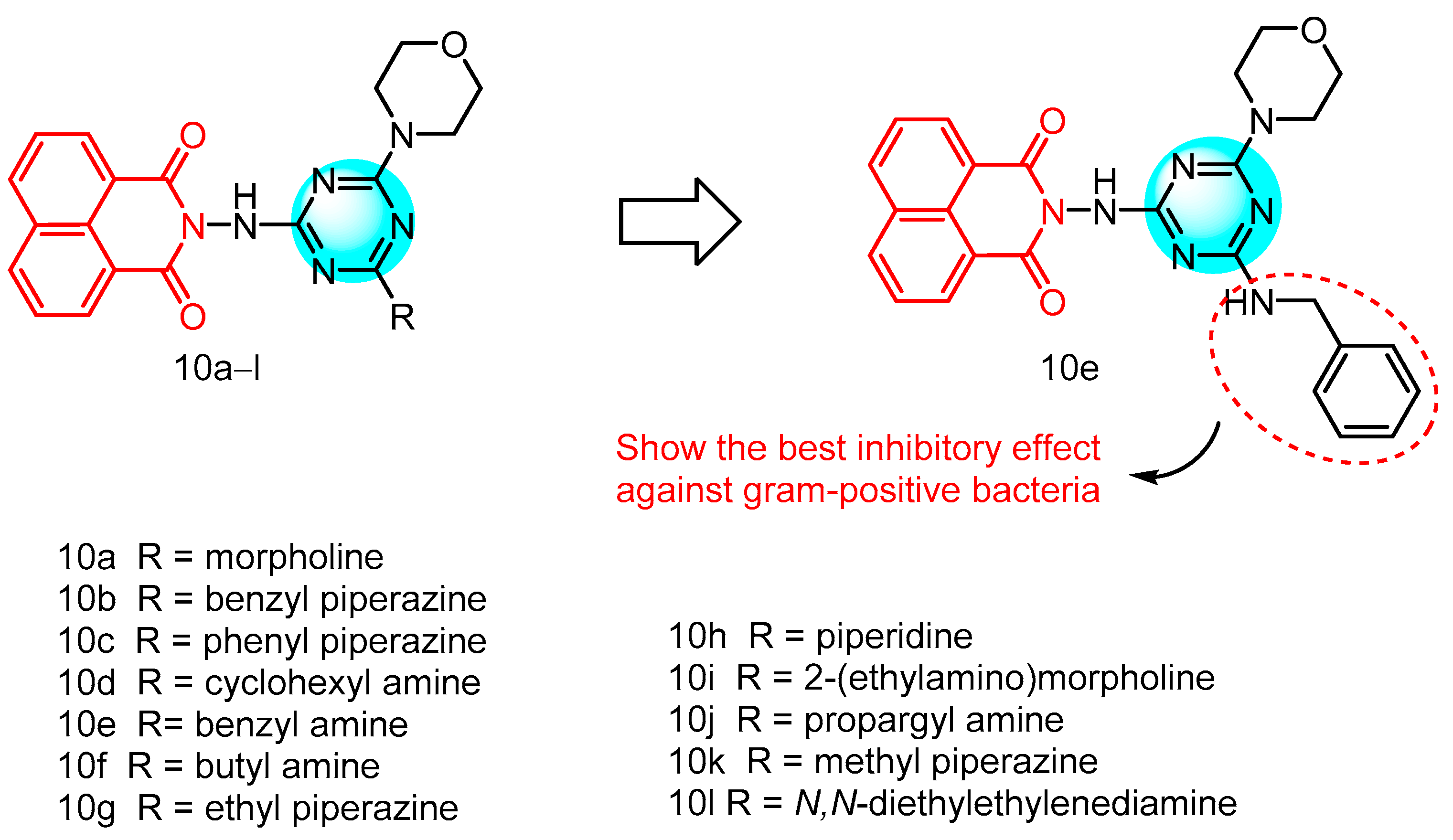
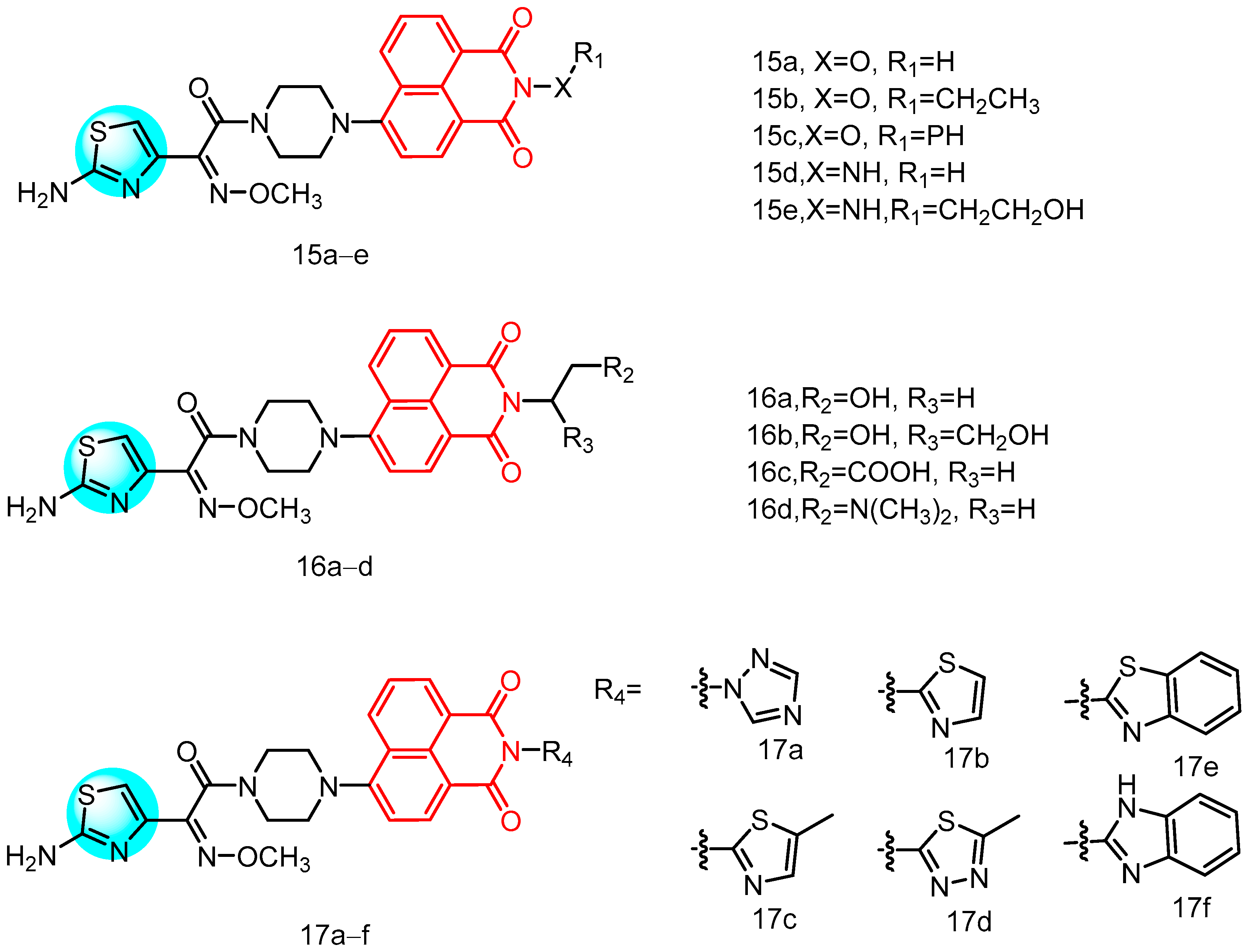
2.1.6. Novel Naphthylimide Aminothiazole
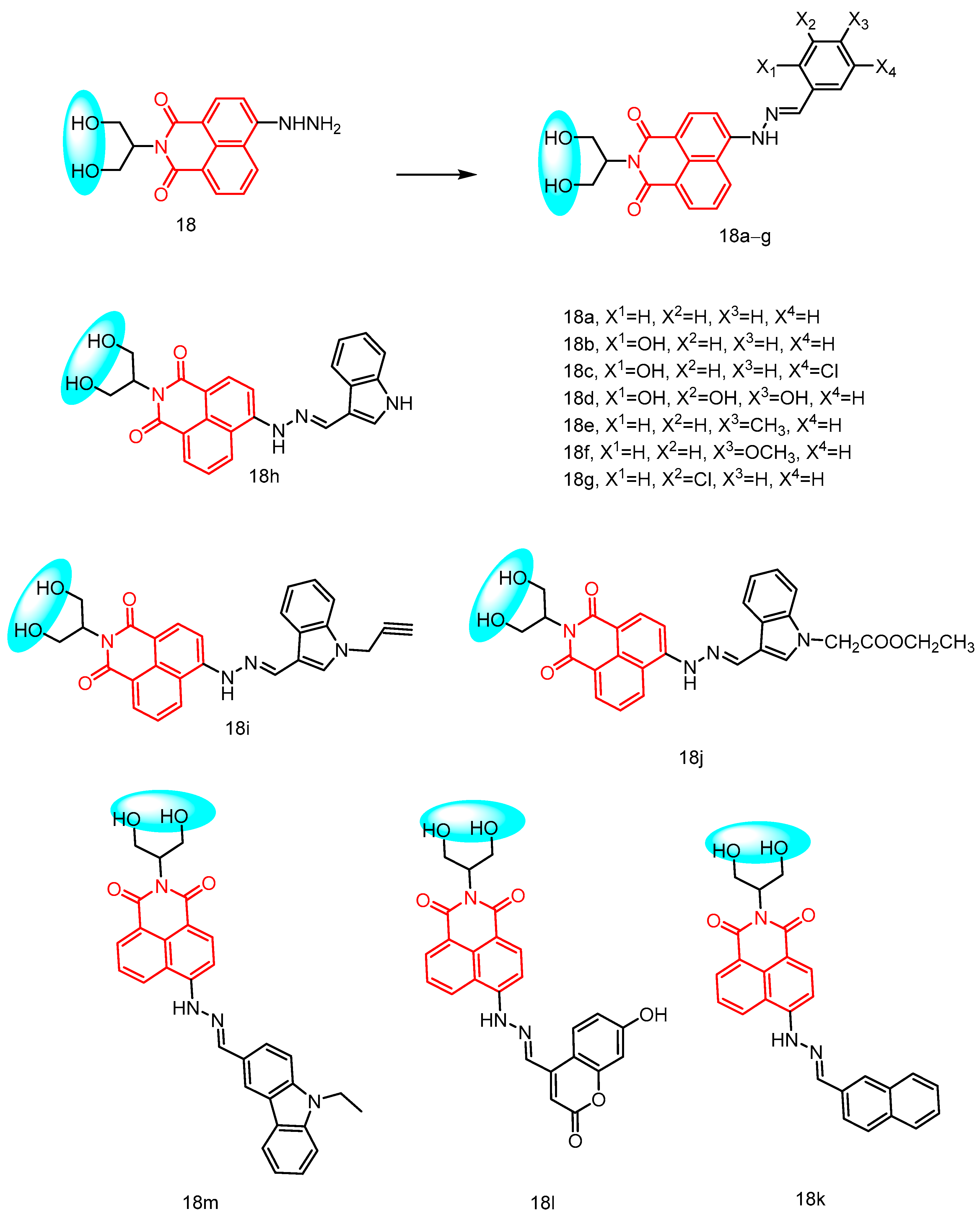
2.1.7. Naphthylimide Derivatives Containing Propylene Glycol
2.1.8. Novel Naphthylimide-Thiourea Derivatives
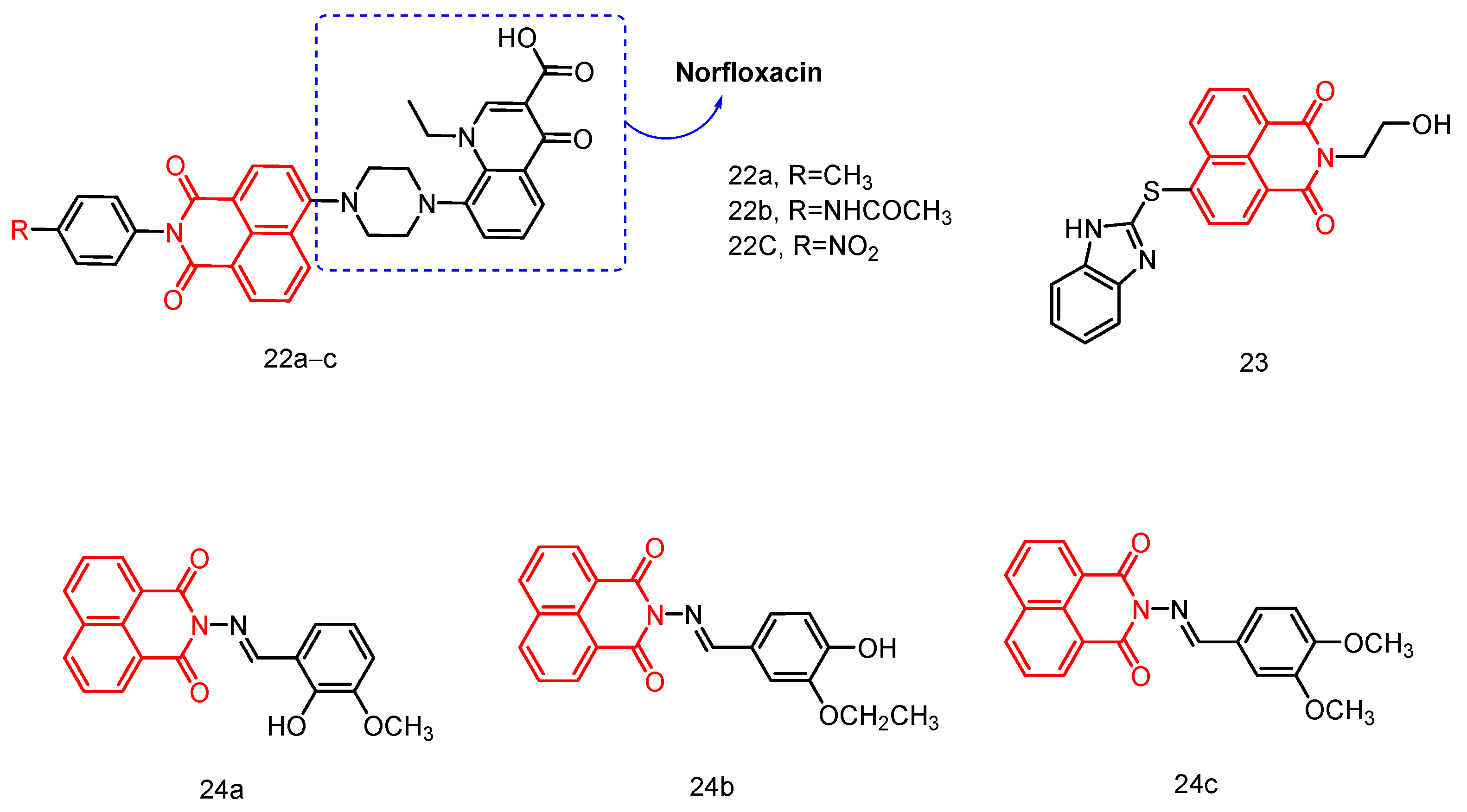
2.1.9. Norfloxacin-Substituted Naphthyl Imide Derivatives
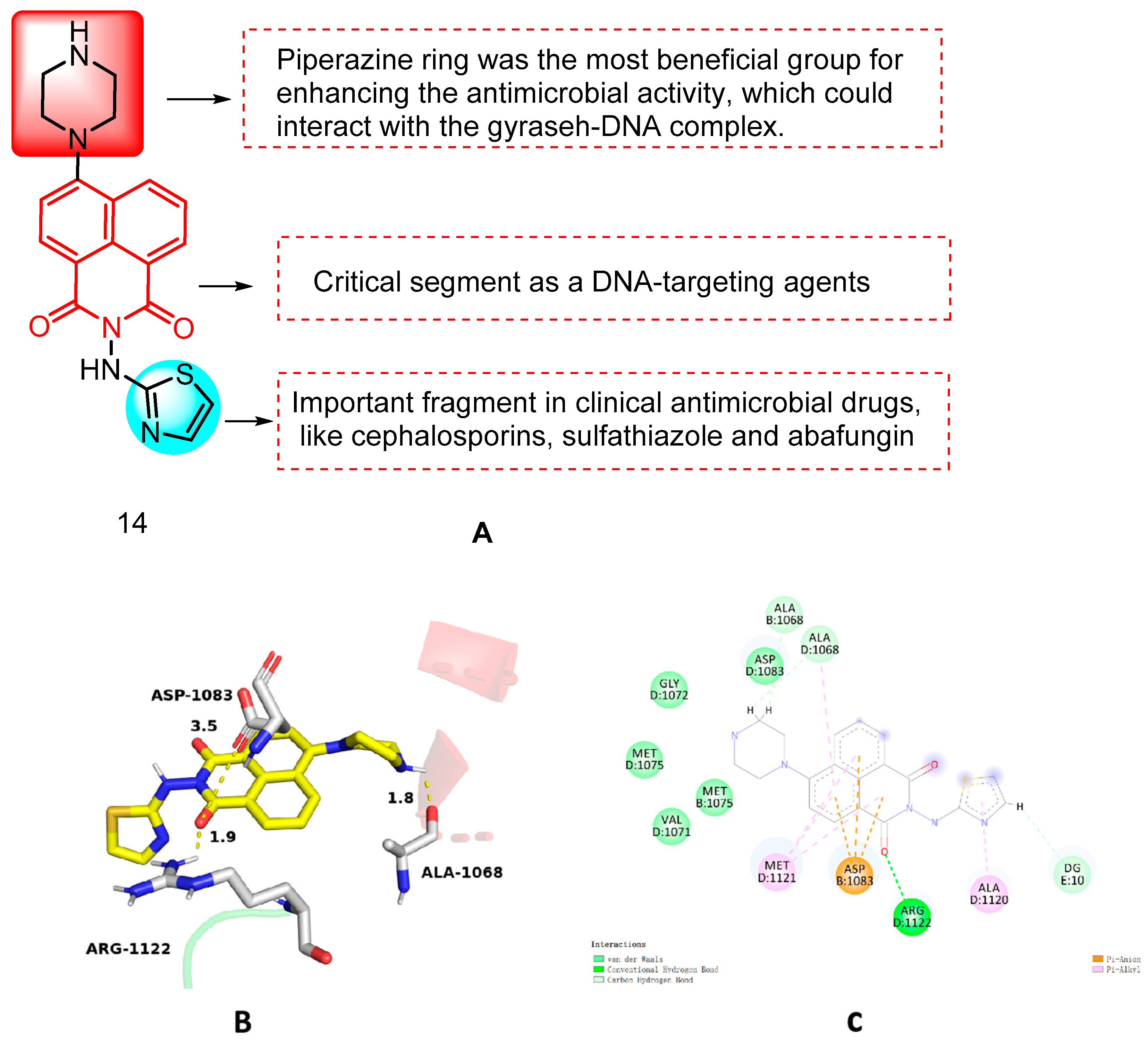
2.1.10. Hydroxyethyl Naphthimide
2.1.11. Naphthalimide-Based Schiff Base Compounds
2.1.12. Naphthalimide–Coumarin Hybrids
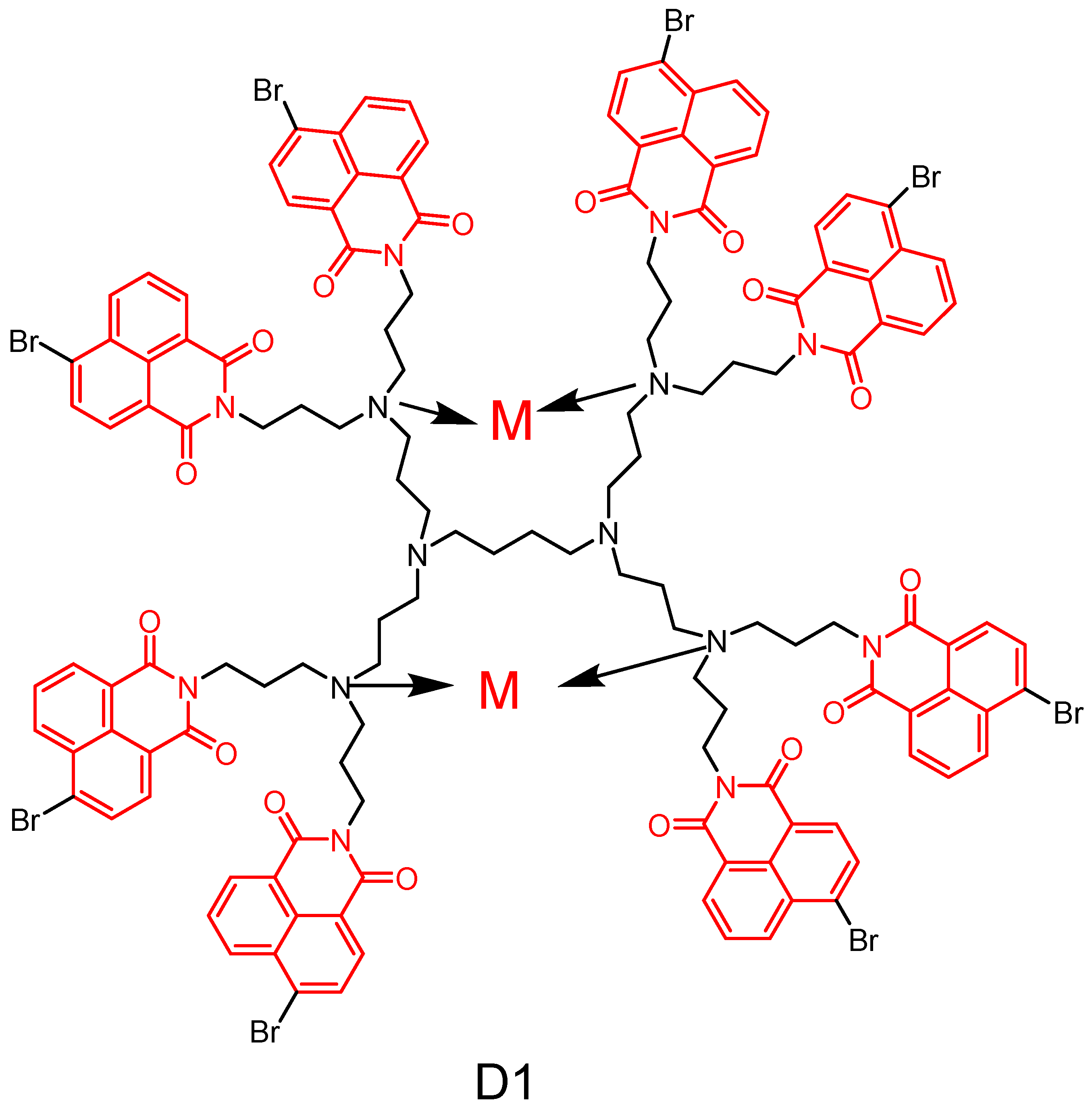
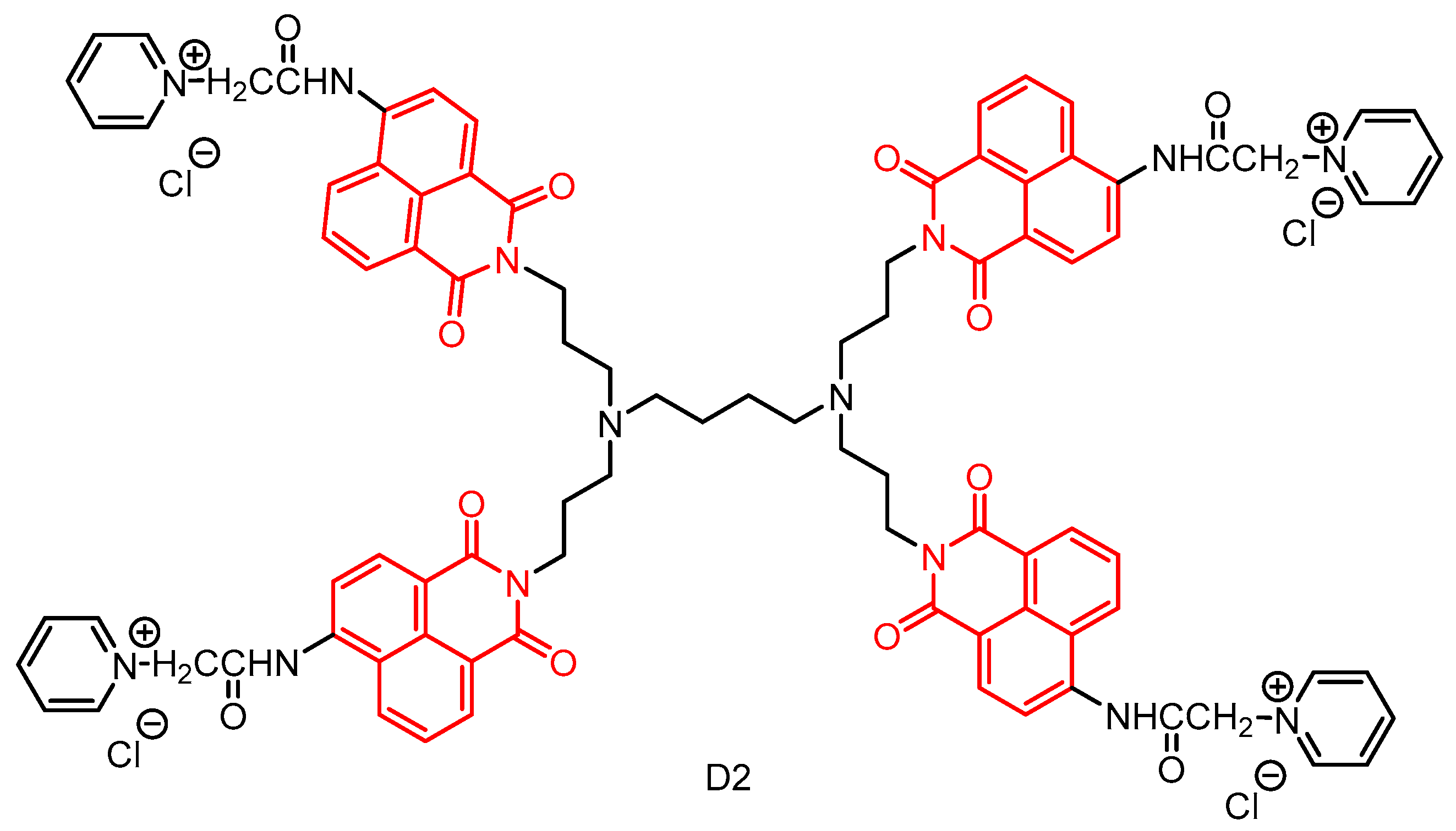
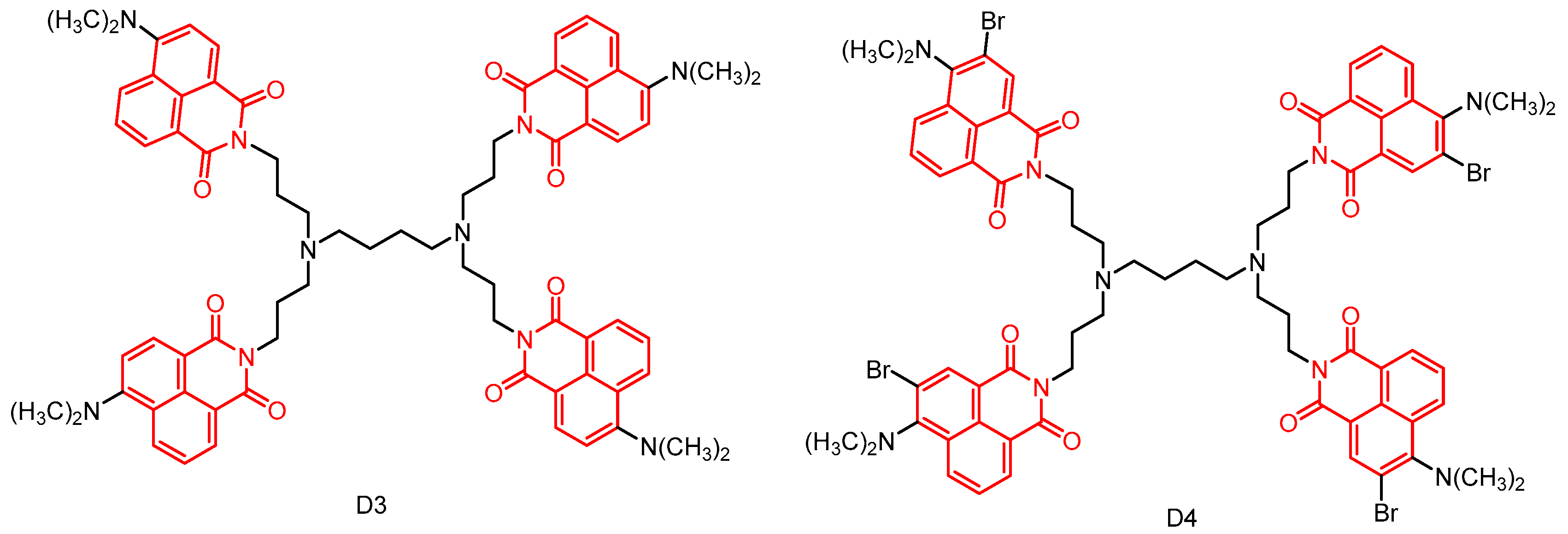
2.2. Naphthylimide Dendrimers with Antimicrobial Activity
2.2.1. Polypropylamine Dendritic Macromolecules
2.2.2. Naphthylimide-Dimer
2.3. Anticancer Activity
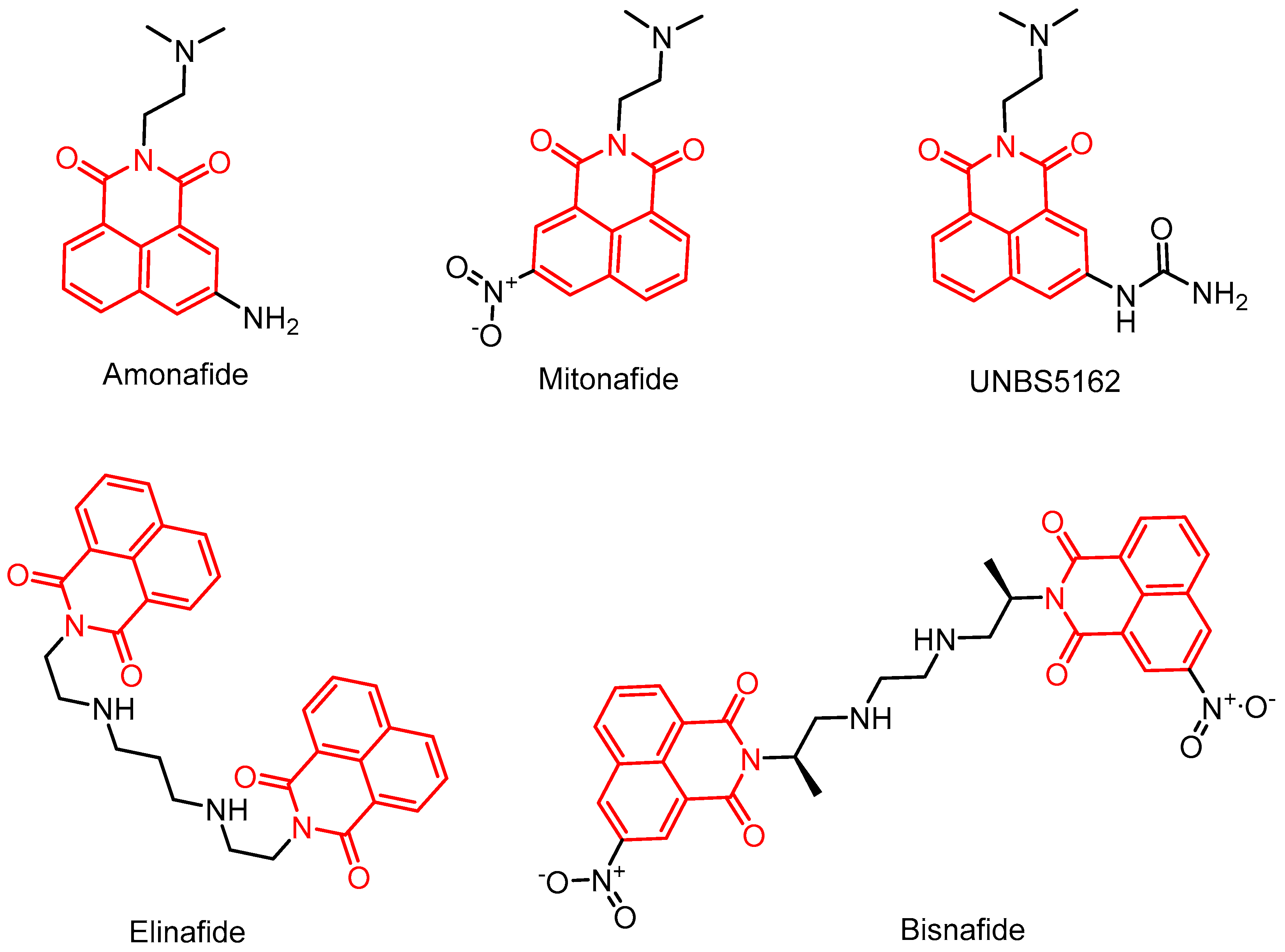
2.3.1. Amonafide Derivatives
2.3.2. Alkylated Naphthylimide Analogues
2.3.3. Study on Bis-Naphthalimide Derivatives
2.3.4. N-Substituted 1,8-Naphthalimide Derivatives
2.3.5. 1,8-Naphthalimide Derivatives
2.3.6. Naphthalimide Metal Complexes
2.4. Other Biological Activities of Naphthalimides (As Antimalarial, Antiviral, Anti-Inflammatory, Antithrombotic, and Antiprotozoal Agents)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Gram-Positive Bacteria | Activities as MIC (μg/mL) | Gram-Negative Bacteria | Activities as MIC (μg/mL) | Fungi | Activities as MIC (μg/mL) | Reference |
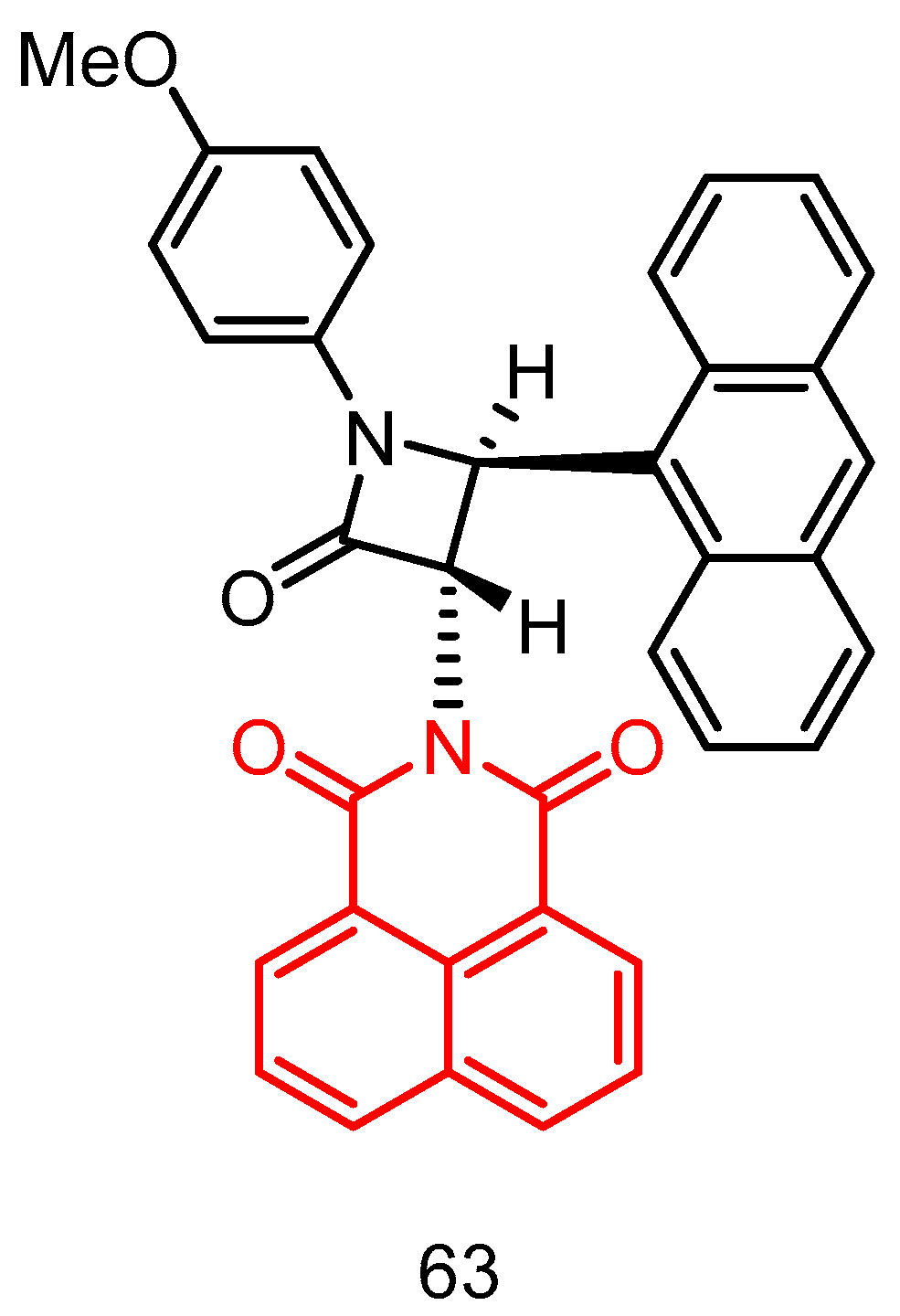
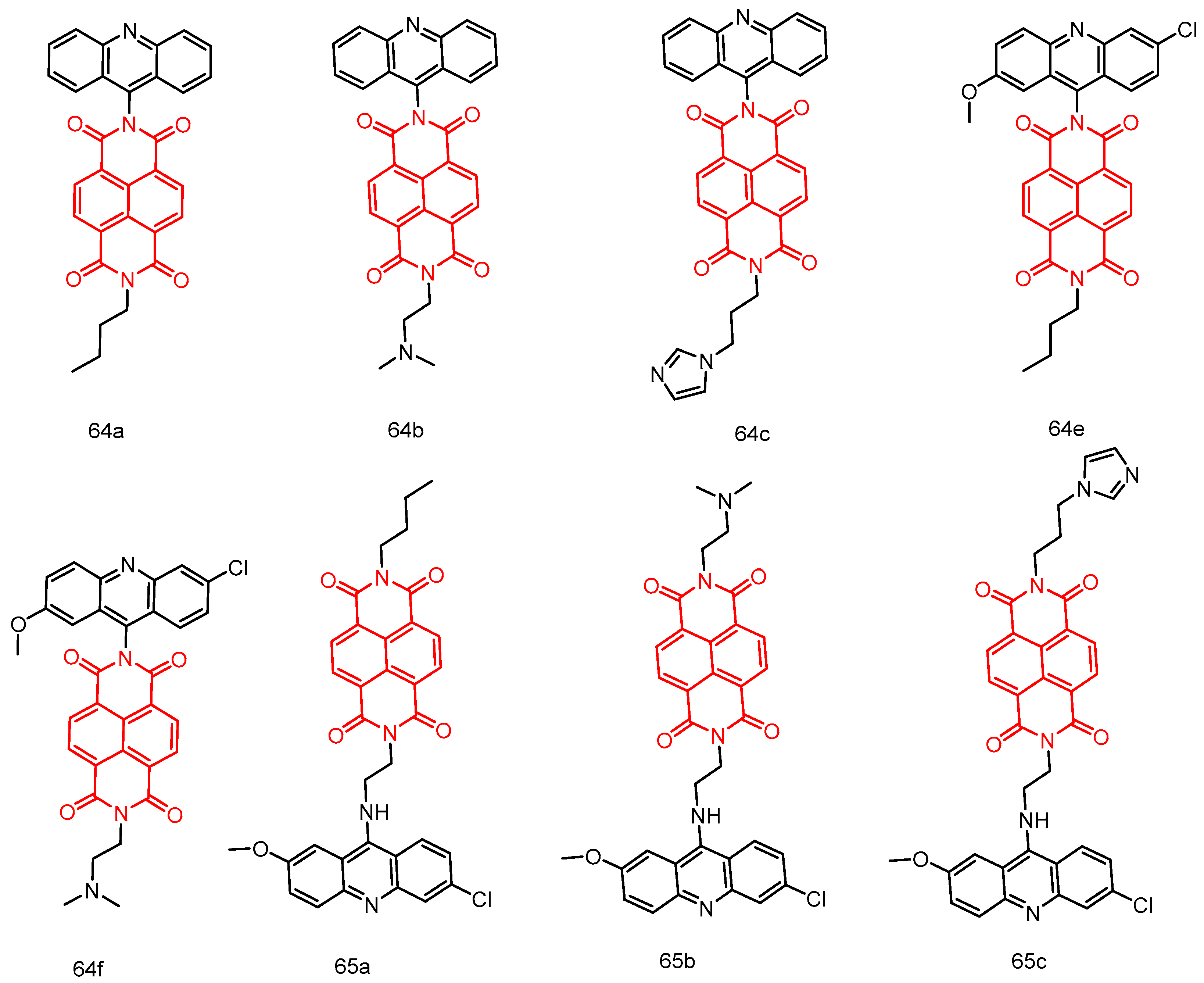
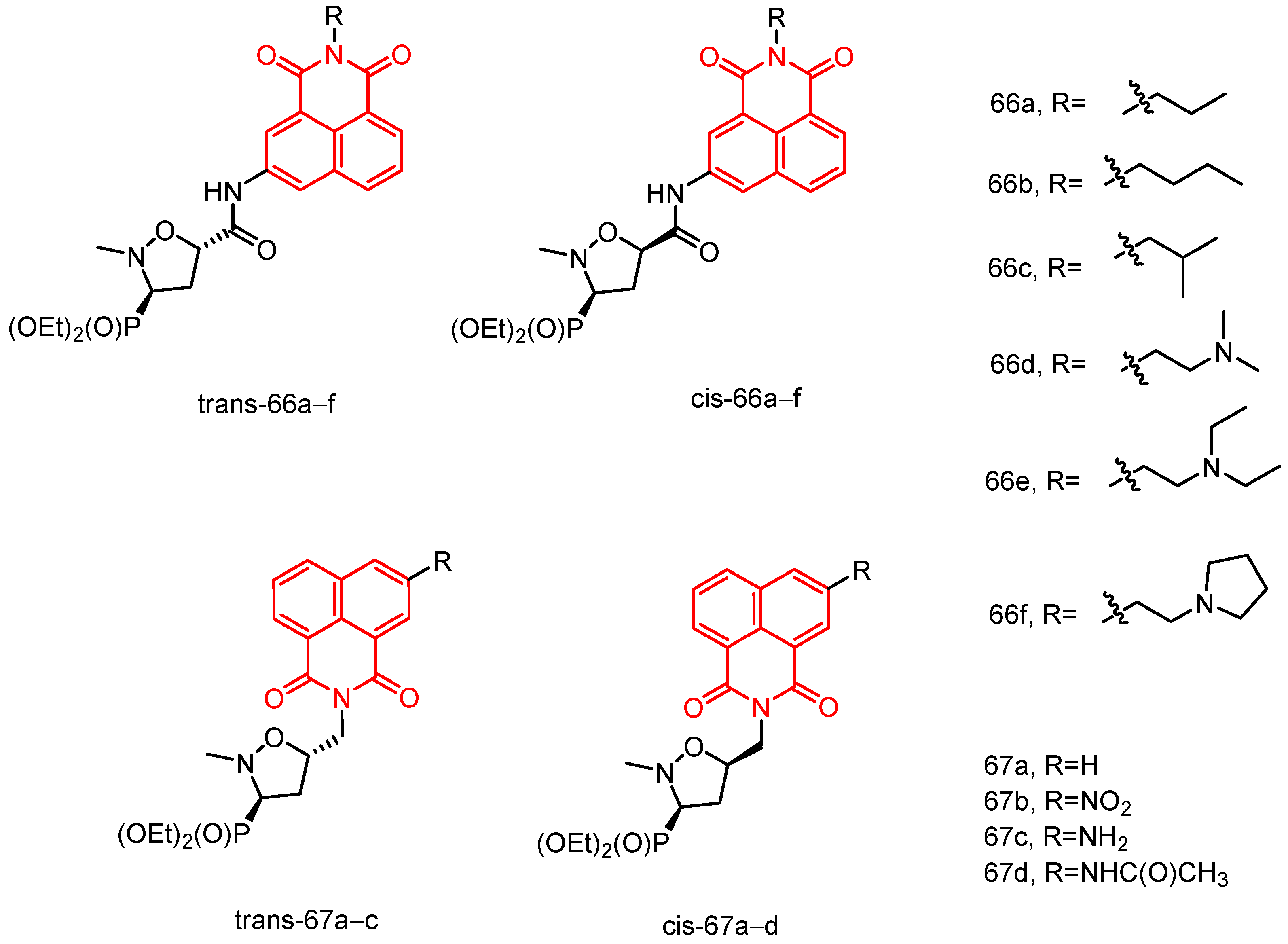
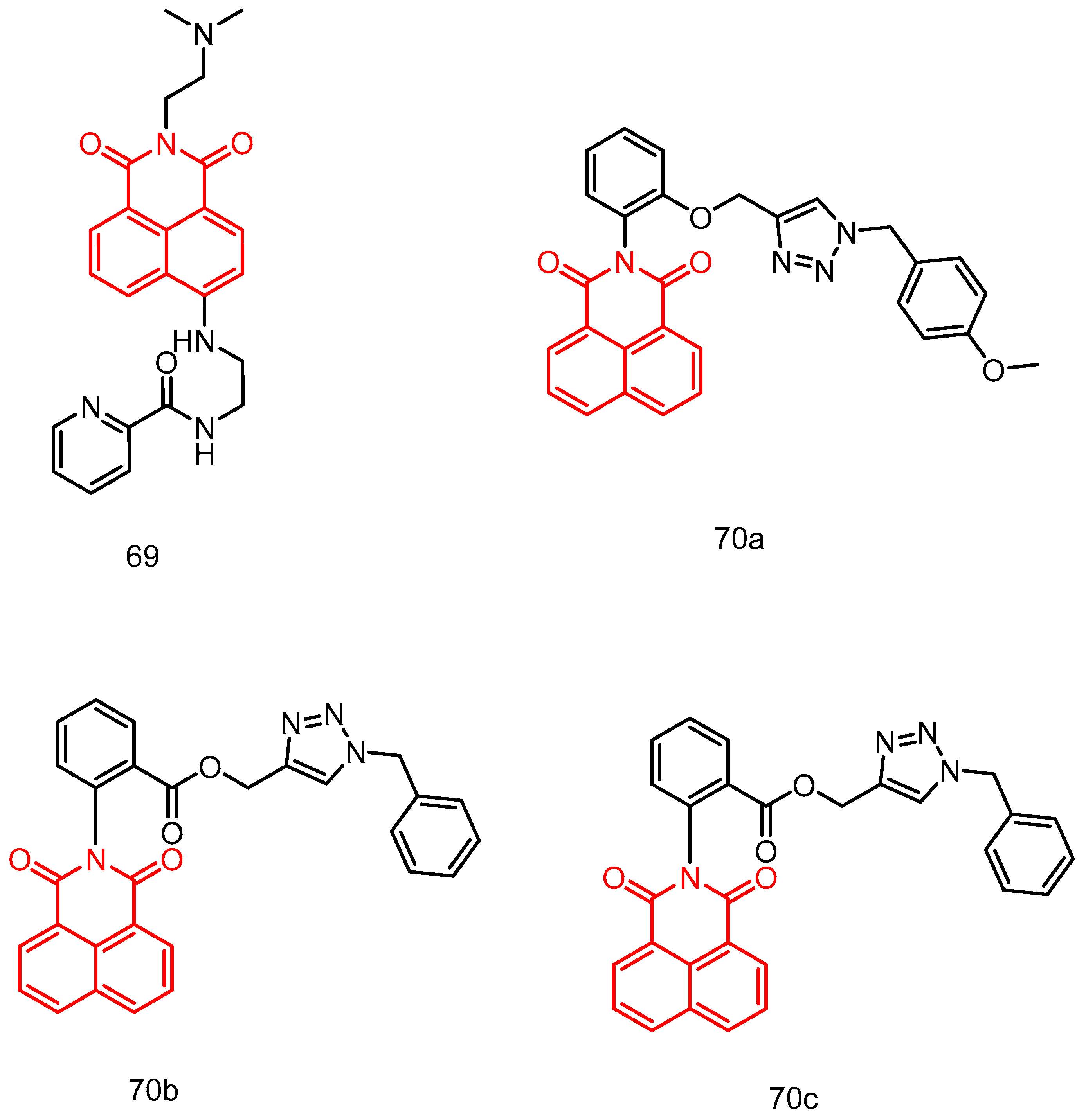
|---|---|---|---|---|---|---|---|
| 1a–d | S. aureus MRSA B. subtilis M. luteus | ≥256 | B. proteus E.coli P. aeruginosa B. typhi | ≥256 | C. albicans C. mycoderma | ≥256 | [24] |
| 2a–i | 4–32 | 1–32 | 4–64 | ||||
| 3a | 4–16 | 2–16 | 4–8 | [21] | |||
| 3b | 8–16 | 4–16 | 8–16 | ||||
| 3c | 4–8 | 2–8 | 8–16 | ||||
| 3d | 8–16 | 2–16 | 16 | ||||
| 3e | 4–512 | 4–8 | 8–16 | ||||
| 3f | 2–512 | 2–4 | 4–8 | ||||
| 3g | 2–512 | 2–4 | 4–8 | ||||
| 4a | 14–29 | 14–512 | C. albicans C. mycoderma C. utilis S. cerevisiae A. flavus | 3.6–512 | [20] | ||
| 4b | 2–14 | 7–14 | 2–14 | ||||
| 4c | 7–230 | 115–230 | 7–29 | ||||
| 4d | 57–205 | 205–512 | 4–14 | ||||
| 5a | 57–115 | 57–205 | 57–512 | ||||
| 5b | 2–29 | 19–29 | 512 | ||||
| 5c | 19–115 | 57–230 | 29–115 | ||||
| 5d | 230–512 | 230–512 | 205–512 | ||||
| 6a | >512 | 1–512 | C. albicans C. mycoderma | 64–512 | [25] | ||
| 6b | 256–512 | 0.5–512 | 32–512 | ||||
| 7 | B. subtilis | 7.6 | - | - | C. albicans | 122 | [26] |
| 8 | - | - | - | - | C. albicans C. albicans 9023 Aspergillus fumigatus Candida tropicalis Candida parapsilosis | 0.772–64 | [27] |
| 9 | B. subtilis S. aureus E. coli | 12–24 | K. pneumoniae | 6.226 | C. albicans A. niger | 6.226–50 | [28] |
| 10a | E. faecalisB. subtilis L. species S. aureus | 1.56–200 | S. enterica A. calcoaceticus S. marcescens | 100–200 | - | - | [29] |
| 10b | 6.25–200 | 50–200 | |||||
| 10c | 0.39–200 | 50–200 | |||||
| 10d | 3.12–100 | 200 | |||||
| 10e | 0.003–6.25 | 100–200 | |||||
| 10f | 0.09–200 | 50–200 | |||||
| 10g | 3.12–200 | 200 | |||||
| 10h | 1.56–50 | 200 | |||||
| 10i | 0.048–200 | 50–200 | |||||
| 10k | 0.048–200 | 12.5–200 | |||||
| 10l | 0.19–200 | 12.5–200 | |||||
| 11a | S. aureus MRSA B. subtilis M. luteus | 0.04–0.08 µmol/mL | B. typhi E. coli (DH52)E. coli (JM109) P. vulgarisP. aeruginosa S. dysenteriae | 0.01–0.16 µmol/mL | C. albicans C. mycoderma B. yeast C. utilis A. flavus | 0.02–0.16 µmol/mL | [31] |
| 11b | 0.04–0.15 µmol/mL | 0.002–0.15 µmol/mL | 0.01–0.04 µmol/mL | ||||
| 11c | 0.02–0.07 µmol/mL | 0.02–1.17 µmol/mL | 0.02–0.29 µmol/mL | ||||
| 11d | 0.04–0.14 µmol/mL | 0.02–1.13 µmol/mL | 0.04–0.57 µmol/mL | ||||
| 11e | 0.91 µmol/mL | 0.45–0.91 µmol/mL | 0.23–0.91 µmol/mL | ||||
| 11f | 0.07–0.29 µmol/mL | 0.04–0.29 µmol/mL | 0.15–0.29 µmol/mL | ||||
| 11g | 0.07–0.29 µmol/mL | 0.07–0.57 µmol/mL | 0.07–0.28 µmol/mL | ||||
| 12a | S. aureus MRSA B. subtilis M. luteus | 0.2–0.8 µmol/mL | B. typhi E. coli (DH52) E. coli (JM109) B. proteusP. aeruginosa S. dysenteriae | 0.01–0.8 µmol/mL | C. albicans C. mycoderma B. yeast C. utilis A. flavus | 0.01–0.81 µmol/mL | [32] |
| 12b | 0.007–0.43 µmol/mL | 0.11–0.43 µmol/mL | 0.22–0.43 µmol/mL | ||||
| 12c | 0.003–0.85 µmol/mL | 0.05–0.42 µmol/mL | 0.003–0.42 µmol/mL | ||||
| 13 | S. aureus B. subtilis B. cereus S. epidermis | <0.65–2.5 | E. coli P. mirabilis | <0.65 | - | - | [33] |
| 14 | S. aureus MRSA S. aureus 25,923 S. aureus 29,213 E. faecalis | 2–16 | K. pneumonia E. coli E. coli 25,922 A. baumanii P. aeruginosa P. aeruginosa ATCC 27,853 | 4–128 | C. albicans C. albicans ATCC 90,023 C. tropicals A. fumigatus C. parapsilosis ATCC 22,019 | 4–64 | [38] |
| 15a | MRSA E. faecalis S. aureus S. aureus ATCC 25,923 S. aureus ATCC 29,213 | 8–64 | K. pneumoniae E. coli P. aeruginosa A. baumanii P. aeruginosa ATCC 27,853 E. coli ATCC 25,922 | 2–64 | - | - | [39] |
| 15b | 8–128 | 2–64 | |||||
| 15c | 1–64 | 2–64 | |||||
| 15d | 4–64 | 2–64 | |||||
| 15e | 4–64 | 2–128 | |||||
| 16a | 2–64 | 16–64 | |||||
| 16b | 4–64 | 4–128 | |||||
| 16c | 8–64 | 8–128 | |||||
| 16d | 0,5–16 | 2–16 | |||||
| 17a | 1–128 | 16–64 | |||||
| 17b | 2–64 | 16–64 | |||||
| 17c | 1–128 | 1–128 | |||||
| 17d | 1–64 | 4–32 | |||||
| 17e | 1–128 | 16–128 | |||||
| 17f | 8–16 | 8–256 | |||||
| 18a | 2–64 | 1–128 | - | - | [40] | ||
| 18b | 2–32 | 4–64 | |||||
| 18c | 1–64 | 2–64 | |||||
| 18d | 4–64 | 2–64 | |||||
| 18e | 0.5–64 | 1–64 | |||||
| 18f | 32–128 | 0.5–64 | |||||
| 18g | 0.25–64 | 2–64 | |||||
| 18h | 2–64 | 4–64 | |||||
| 18i | 0.5–16 | 4–64 | |||||
| 18j | 0.25–64 | 4–64 | |||||
| 18k | 4–128 | 0.5–64 | |||||
| 18m | 0.25–64 | 2–128 | |||||
| 18l | 4–128 | 4–64 | |||||
| 19a | S. aureus ATCC 29,213 | 2 | Mtb H37Rv ATCC 27,294 | >64 | MRSA ATCC 29,213 MRSA NRS 100 MRSA NRS 119 MRSA NRS 129 MRSA NRS 186 MRSA NRS 191 MRSA NRS 192 MRSA NRS 193 MRSA NRS 194 MRSA NRS 198 VRSA VRS 1 VRSA VRS 4 VRSA VRS 12 | 2–4 | [41] |
| 19b–g | >64 | >64 | 0.25–0.5 | ||||
| 19h | 0.125 | 4 | - | ||||
| 19i | 8 | 32 | - | ||||
| 20a–k | 1–>64 | 8–>64 | - | ||||
| 20f | 0.25 | >64 | 0.125–0.5 | ||||
| 20l | 1 | >64 | 1–4 | ||||
| 21a | 0.25 | >64 | 0.125–0.5 | ||||
| 21b | 0.03125 | >64 | 0.25–1 | ||||
| 23 | - | - | - | - | C. albicans C. albicans ATCC 90,023 C. tropicals A. fumigatus C. parapsilosis ATCC 22,019 | 4–128 | [43] |
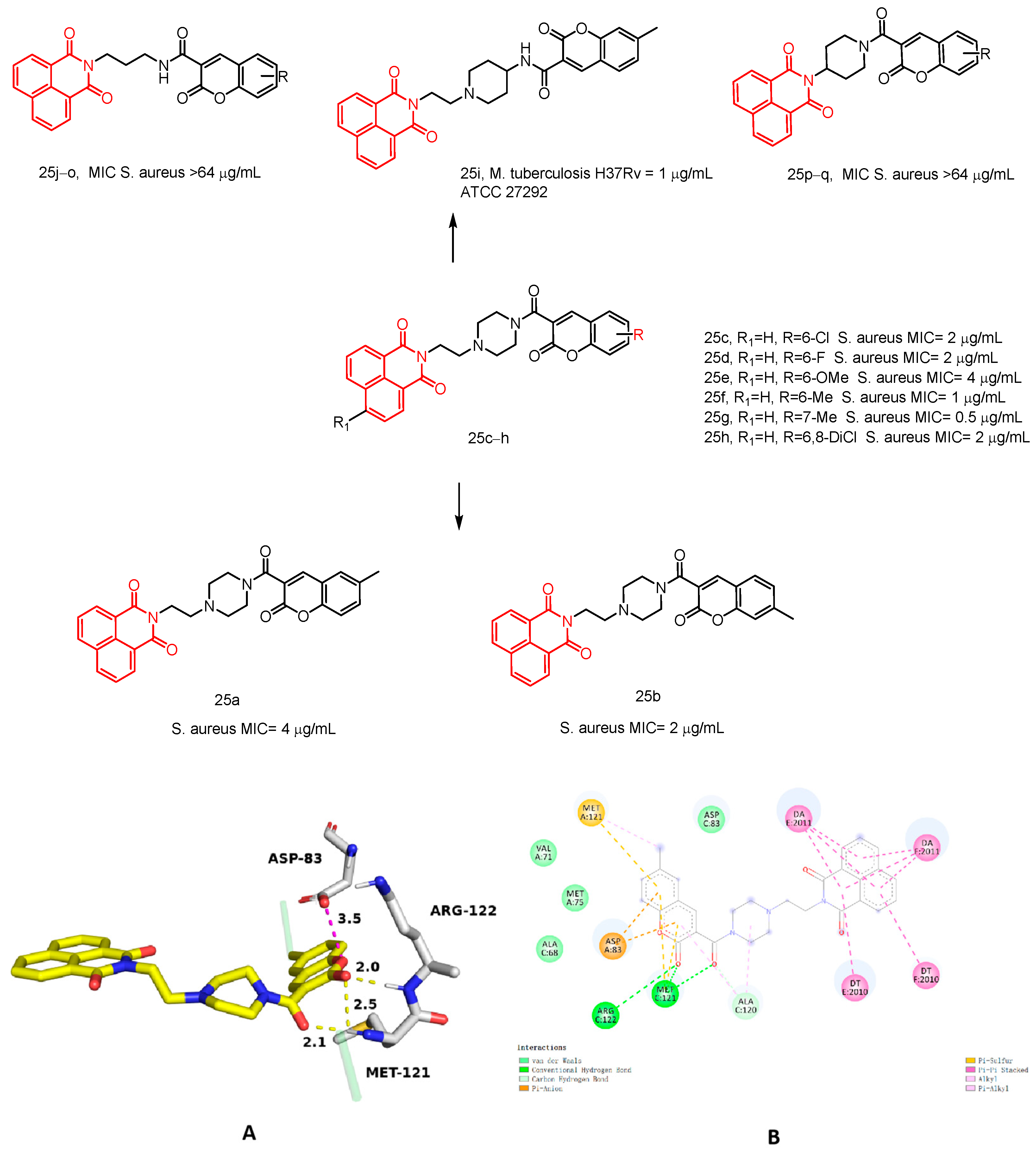
| 25a | S. aureus ATCC 29,213 | 4 | E. coli ATCC 25,922 K. pneumoniae BAA 1705 A. baumannii BAA 1605 P. aeruginosa ATCC 27,853 | >64 | Mtb H37Rv ATCC 27,294 | 16 | [45] |
| 25b | 2 | >64 | 16 | ||||
| 25c | 2 | >64 | >64 | ||||
| 25d | 2 | >64 | 16 | ||||
| 25e | 4 | >64 | 32 | ||||
| 25f | 1 | >64 | 32 | ||||
| 25g | 0.5 | >64 | 64 | ||||
| 25h | 2 | >64 | >64 | ||||
| 25i | >64 | >64 | 1 | ||||
| 25j–o | >64 | >64 | 8–>64 | ||||
| 25p–q | >64 | >64 | >64 |
| Compound | Cancer Cell Lines | IC50/GI50 Values (µM) | Reference |
|---|---|---|---|
| 26 | Hs683, U373MG, HCT-15, LoVo, A549, MCF-7 | 0.8–1.8 | [64] |
| 27 | PC-3, DU-145, U373-MG, Hs683, HCT-15, LoVo, MCF-7, A549, Bx-PC-3 | 4.7–46.5 | [65] |
| 28a | HeLa, P388D1 | 0.62 ± 0.07–0.83 ± 0.08 | [66] |
| 28b | 0.23 ± 0.07–0.71 ± 0.05 | ||
| 28c | 0.43 ± 0.09–1.93 ± 0.06 | ||
| 29 | SK-OV-3, HepG2, A-549, T-24 SMMC-7721, HL-7702 | 4.13 ± 0.9–20.71 ± 2.1 | [67] |
| 30a | A549, A549R, NB-4, A261, HLF | 1.5 ± 0.1–9.9 ± 0.1 | [68] |
| 30b | 2.9 ± 0.04–12.9 ± 0.02 | ||
| 30c | 4.1 ± 0.03–8.4 ± 0.2 | ||
| 31a | HeLa, HepG2, A549 | 5.06 ± 0.25–20.26 ± 0.30 | [69] |
| 31b | 8.48 ± 0.20–38.27 ± 0.26 | ||
| 31c | 17.02 ± 0.16–78.66 ± 0.20 | ||
| 31d | 4.85 ± 0.16–30.95 ± 0.17 | ||
| 31e | 8.95 ± 0.25–74.30 ± 0.12 | ||
| 31f | 24.19 ± 0.11–155.13 ± 0.04 | ||
| 31g | 34.81 ± 0.10–264.17 ± 0.07 | ||
| 31h | 16.74 ± 0.18–39.92 ± 0.13 | ||
| 32a | Jurkat, HeLa, MCF-7, A-549 | 6.53 ± 0.48–>50 | [70] |
| 32b | 5.67 ± 0.12–>50 | ||
| 33 | HCT-116, HepG2, K562, MDA-MB-231, QSG-7701 | 2.86–53.85 | [71] |
| 34 | K562, HepG2, HCT116, SMMC-7721 | 3.30 ± 1.01–18.95 ± 2.17 | [72] |
| 35 | K562, HepG2, HCT116, SMMC-7721 | 4.67 ± 0.87–6.34 ± 1.41 | [73] |
| 36a | Snu-368, Snu-739, MDA-MB-231 MCF-7, A549, A549cisR | 1.19 ± 0.12–3.62 ± 0.15 | [74] |
| 36b | 1.07 ± 0.10–3.89 ± 0.35 | ||
| 36c | 0.83 ± 0.08–1.92 ± 0.23 | ||
| 37 | HepG-2, Huh-7, MDA-MB-231, MCF-7 A549, A549cisR | 0.98 ± 0.19–20.32 ± 0.78 | [75] |
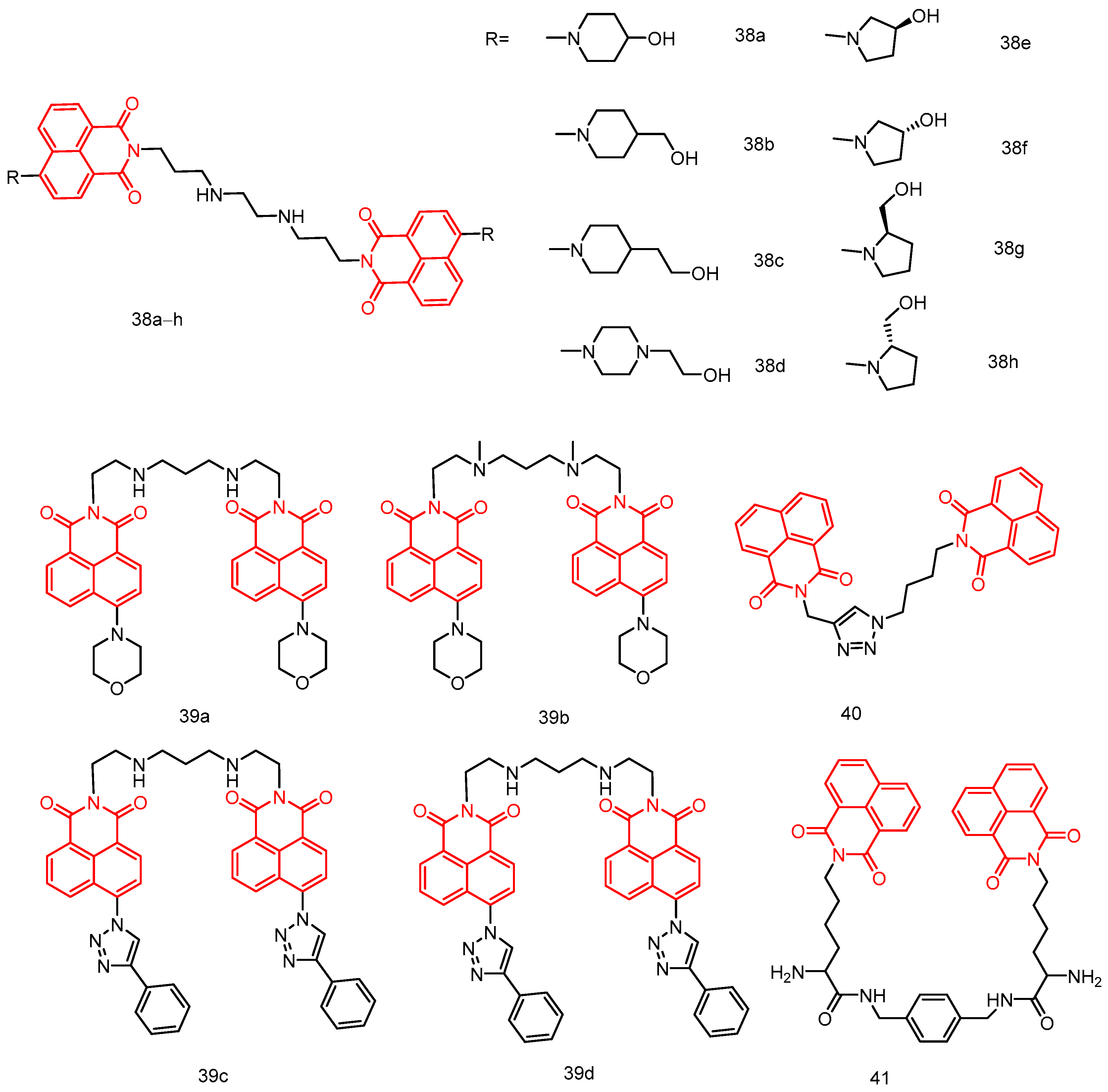
| 38a | Hela, MCF-7, A549, MGC-803 | 0.60 ± 0.10–37.07 ± 0.04 | [76] |
| 38b | 2.03 ± 0.25–22.70 ± 0.02 | ||
| 38c | 6.88 ± 0.05–90.43 ± 0.2 | ||
| 38d | 1.60 ± 0.37–20.01 ± 0.0 | ||
| 38e | 5.96 ± 0.08–141.7 ± 0.02 | ||
| 38f | 9.41 ± 0.27–74.63 ± 0.05 | ||
| 38g | 6.41 ± 0.34–93.53 ± 0.08 | ||
| 38h | 35.05 ± 0.13–146.52 ± 0.0 | ||
| 39a | A549, MRC-5I | 0.51 ± 0.13–1.30 ± 0.23 | [77] |
| 39b | 0.15 ± 0.03–0.95 ± 0.19 | ||
| 39c | 0.89 ± 0.07–1.35 ± 0.43 | ||
| 39d | 1.04 ± 0.07–2.22 ± 0.49 | ||
| 40 | A549, MCF-7, PC-3, Hela, RPE1 | 7.6 ± 0.78–>50 | [78] |
| 41 | EC109, BGC823 | 0.07799–0.14245 | [79] |
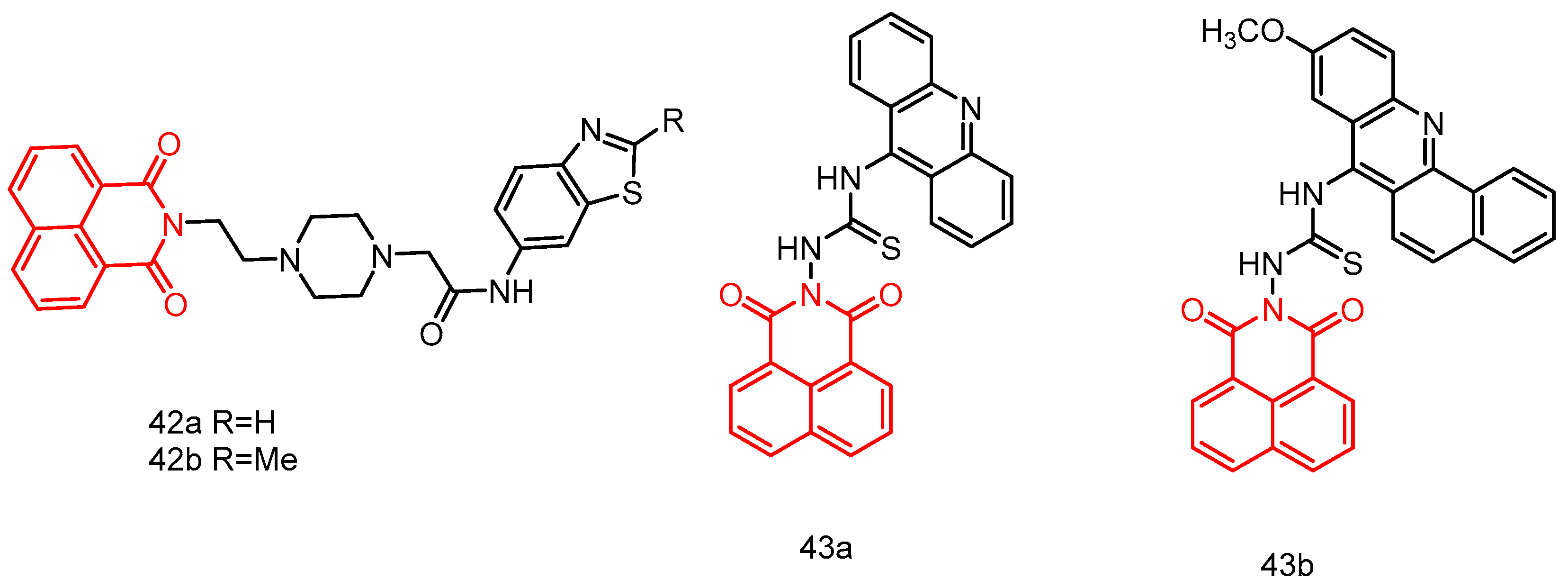
| 42a | HT-29, A549, MCF-7 | 03.72 ± 0.3–07.91 ± 0.4 | [80] |
| 42b | 03.47 ± 0.2–05.08 ± 0.3 | ||
| 43a | HL-60, MCF-7, HepG,2 HeLa, SK-OV-3 MT-4, LO2, BEAS-2B, SH-SY5Y | 46.79 ± 1.96–>100 | [81] |
| 43b | 14.66 ± 0.31–>100 | ||
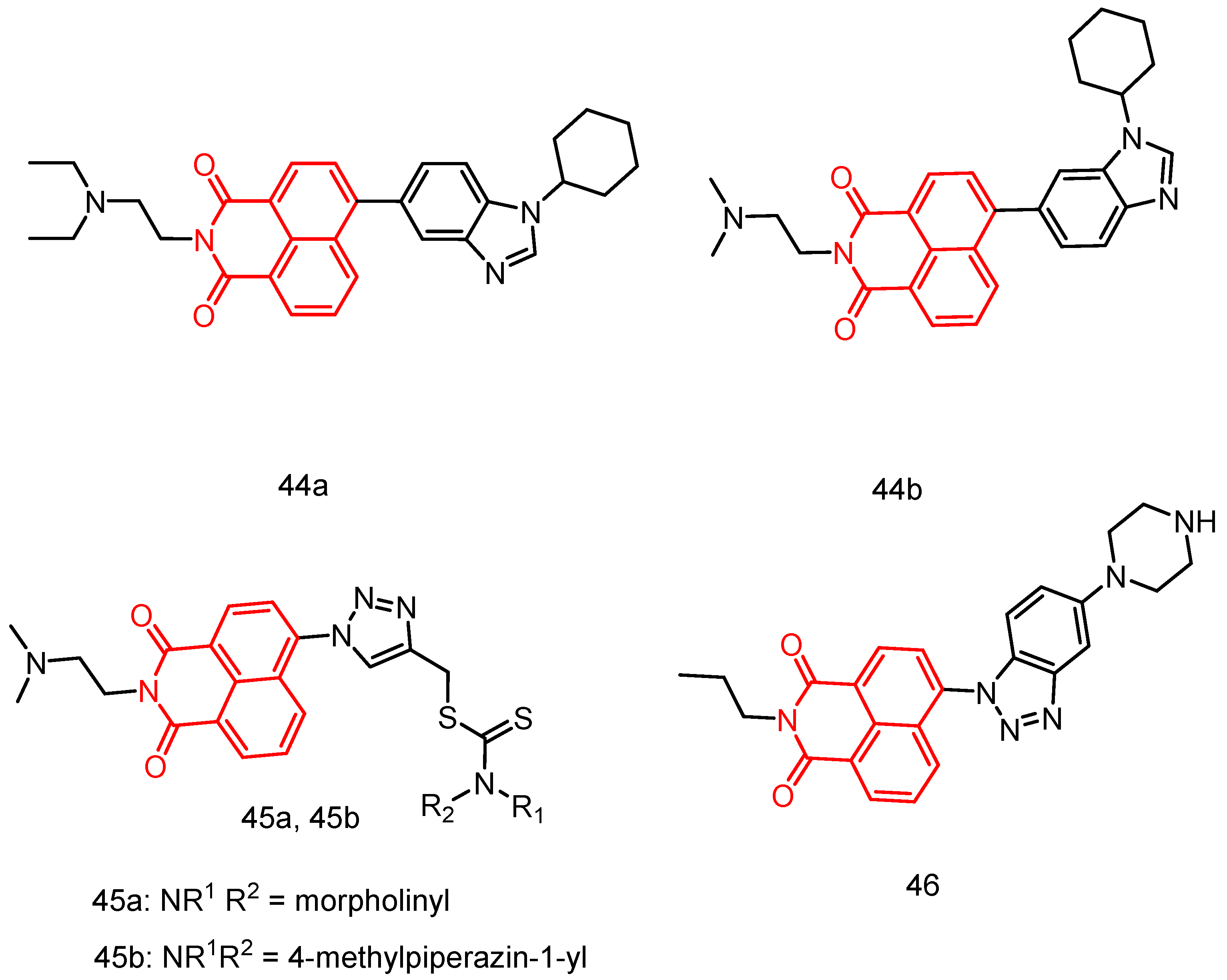
| 44a | Leukemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, breast cancer. | GI50 = 1.09–1.67 | [82] |
| 44b | GI50 = 1.21–2.68 | ||
| 45a | MDA-MB-231, HepG-2, PC12, A549 | 10.86–>100 | [26] |
| 45b | 2.2699–91.453 | ||
| 46 | A549, SK-OV-3, HT-29, HL-60, PC-3, HepG2, MDA-MB-231, MRC-5 | 6.73 ± 0.37–14.00 ± 0.56 | [83] |
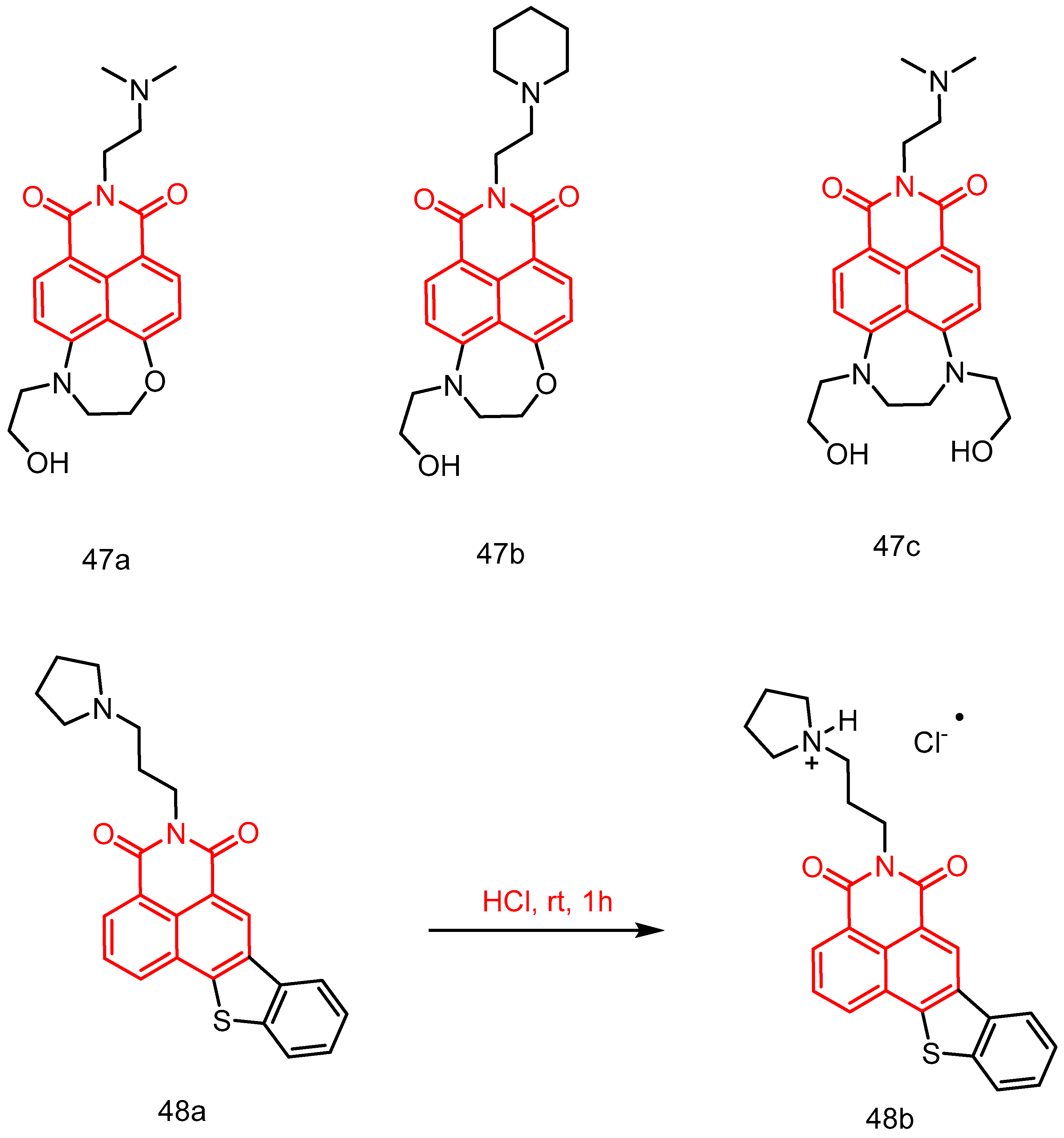
| 47a | A549, HL-60 | 3.1–7.1 | [49] |
| 47b | 1.9–2.5 | ||
| 47c | 1.4–3.6 | ||
| 48a | HepG2, SK-N-SH, MCF-7, PC-3, AGS, A549, MDA-MB-231, K562, A375, 786-O, SH-SY5Y, BE(2)-M17, SK-N-AS, IMR-32 | 0.59 ± 0.08–2.75 ± 0.06 | [84] |
| 48b | 0.80 ± 0.06–4.232 ± 0.07 | ||
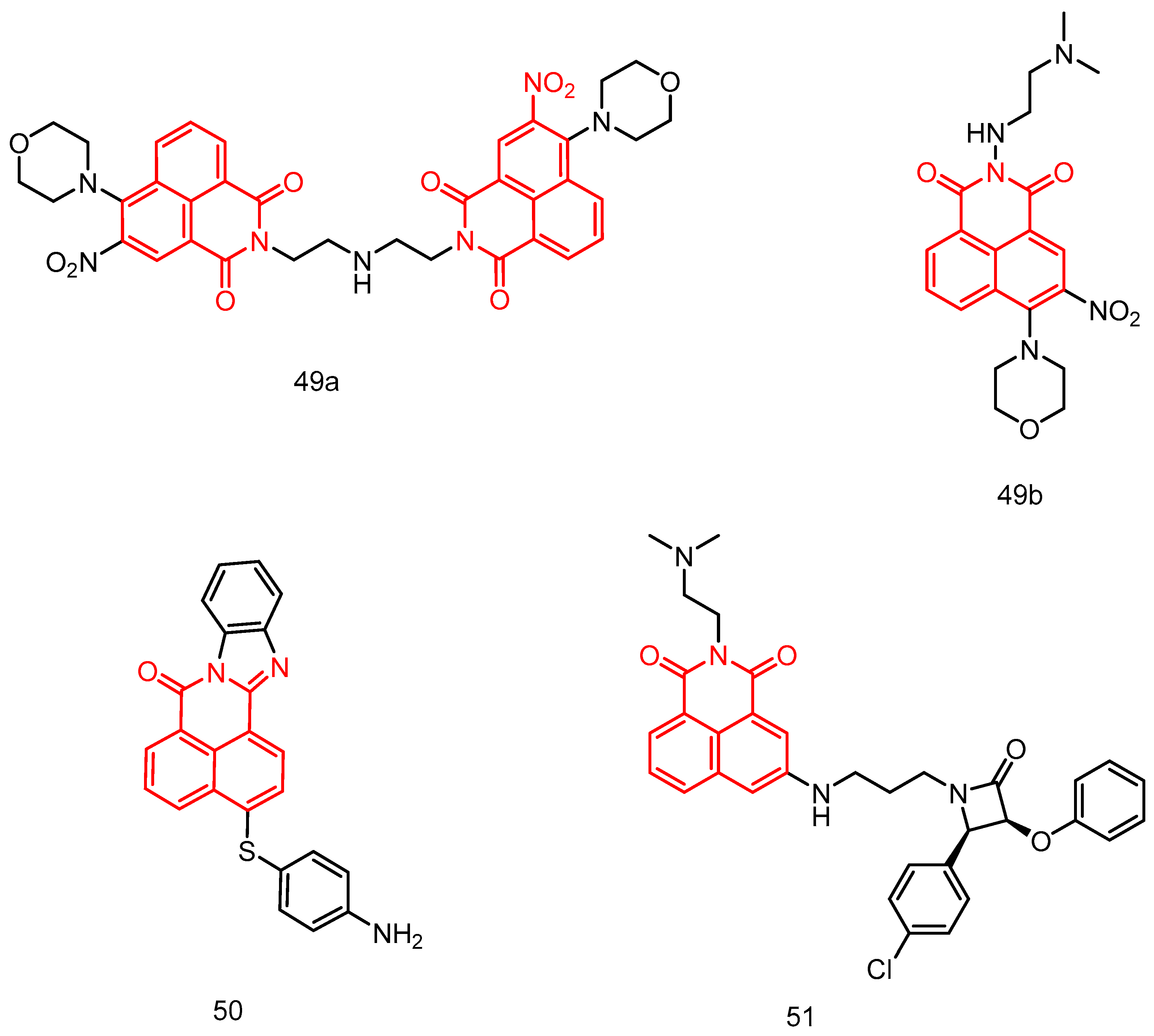
| 49a | T24, MGC-803, CNE-2, A2780 | 0.09 ± 0.06–1.24 ± 0.12 | [85] |
| 49b | 1.48 ± 0.12–4.81 ± 0.22 | ||
| 50 | Leukaemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, breast cancer. a | GI50 = 2.9–6.97 | [86] |
| 51 | HepG2 | 34.2 | [87] |
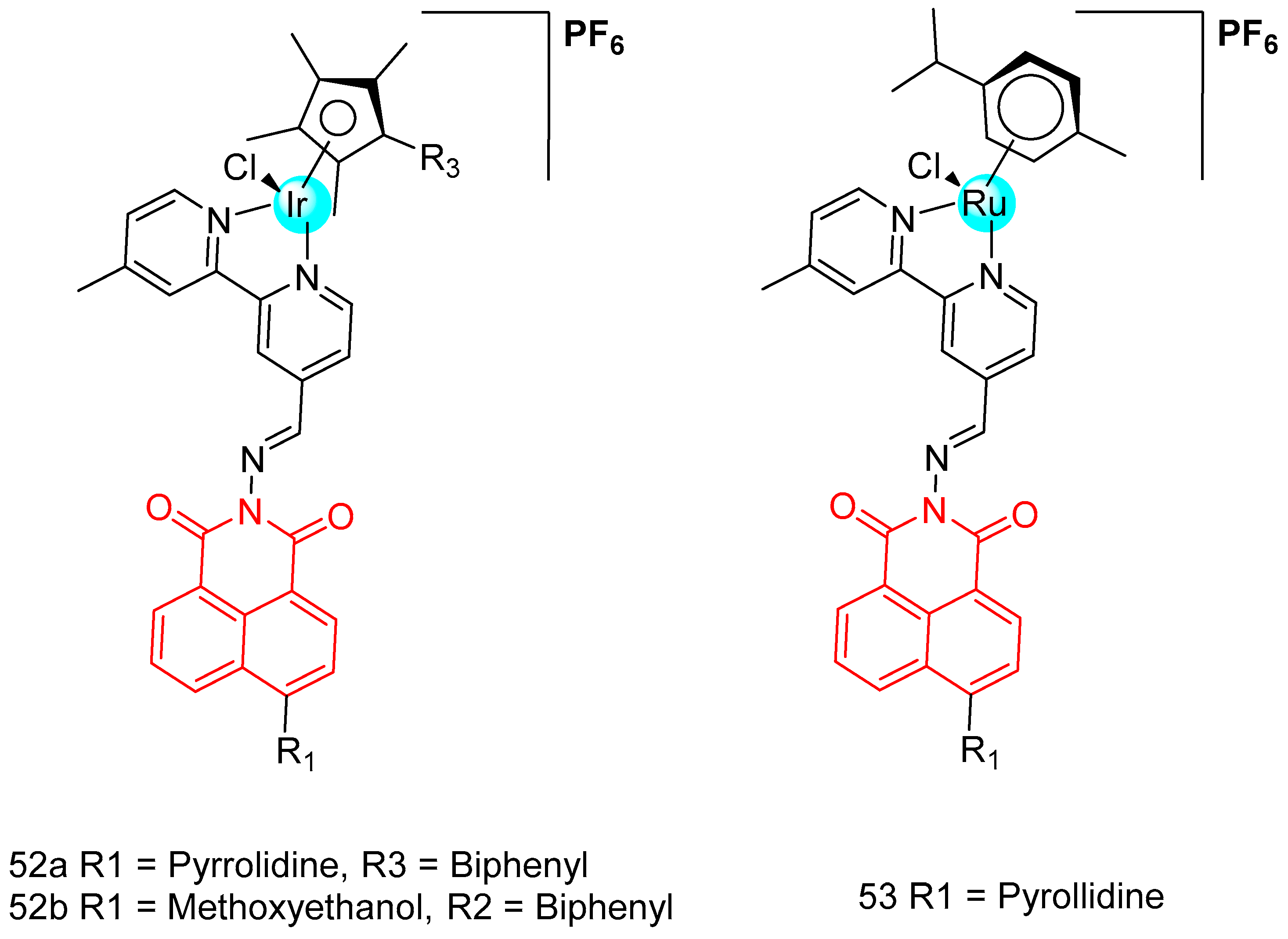
| 52a | A549, HeLa, HepG2, Beas-2b | 11.3 ± 0.1–15.6 ± 1.2 | [88] |
| 52b | 4.9 ± 0.7–17.1 ± 3.6 | ||
| 53 | 10.4 ± 1.3–19.3 ± 6.0 | ||
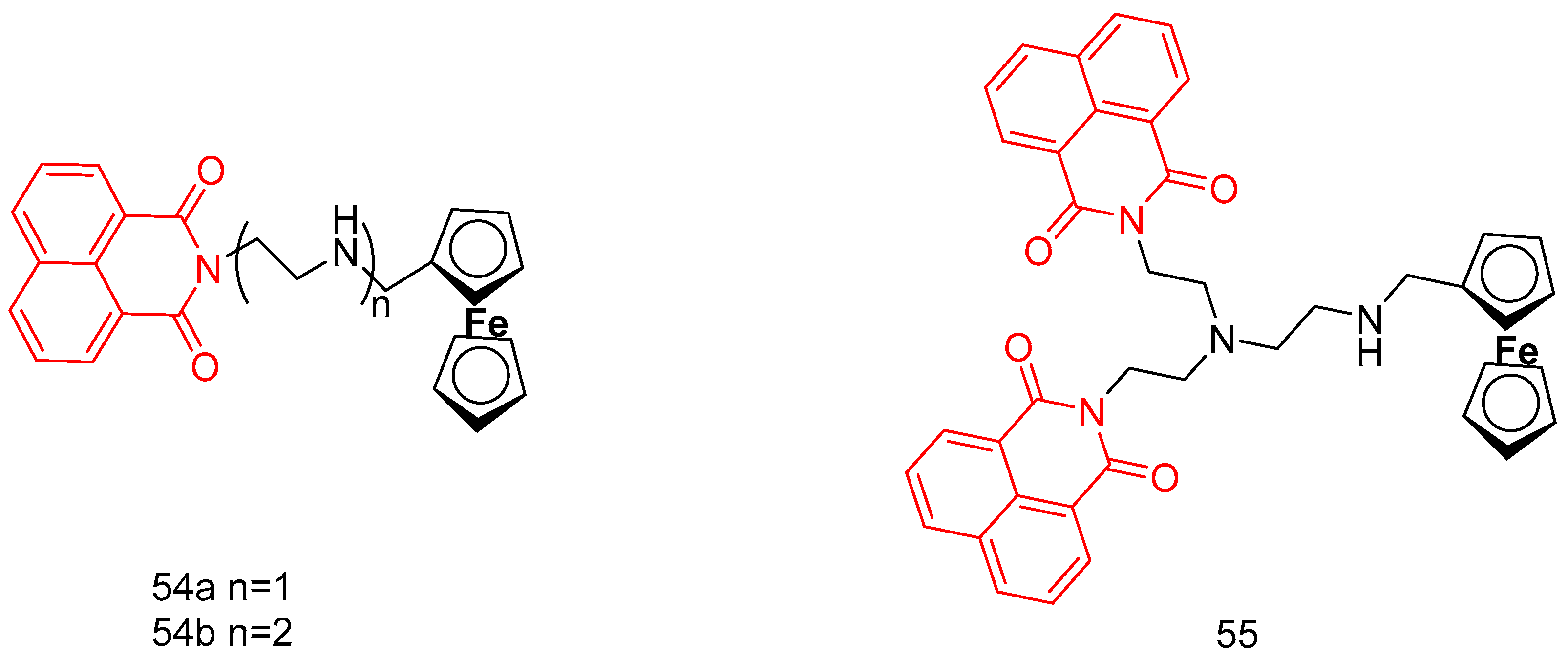
| 54a | EC109, BGC823, SGC 7901, HePG2 | >200 | [89] |
| 54b | 68.54–123.9 | ||
| 55 | 4.33–10.52 | ||
| 56a | MCF-7, HT-29 | 4.6 ± 3.6–27.5 ± 16.2 | [90] |
| 56b | 1.9 ± 0.4–16.0 ± 0.7 | ||
| 56c | 1.5 ± 0.3–9.6 ± 0.2 | ||
| 57a | 5.8 ± 0.5–10.0 ± 0.4 | ||
| 57b | 1.7 ± 0.3–6.5 ± 0.4 | ||
| 57c | 4.0 ± 0.4–6.2 ± 0. 4 | ||
| 58a | 18.6 ± 1.3–36.8 ± 0.48 | ||
| 58b | 11.6 ± 1.0–26.4 ± 1.1 | ||
| 58c | 4.8 ± 0.1–4.9 ± 0.02 | ||
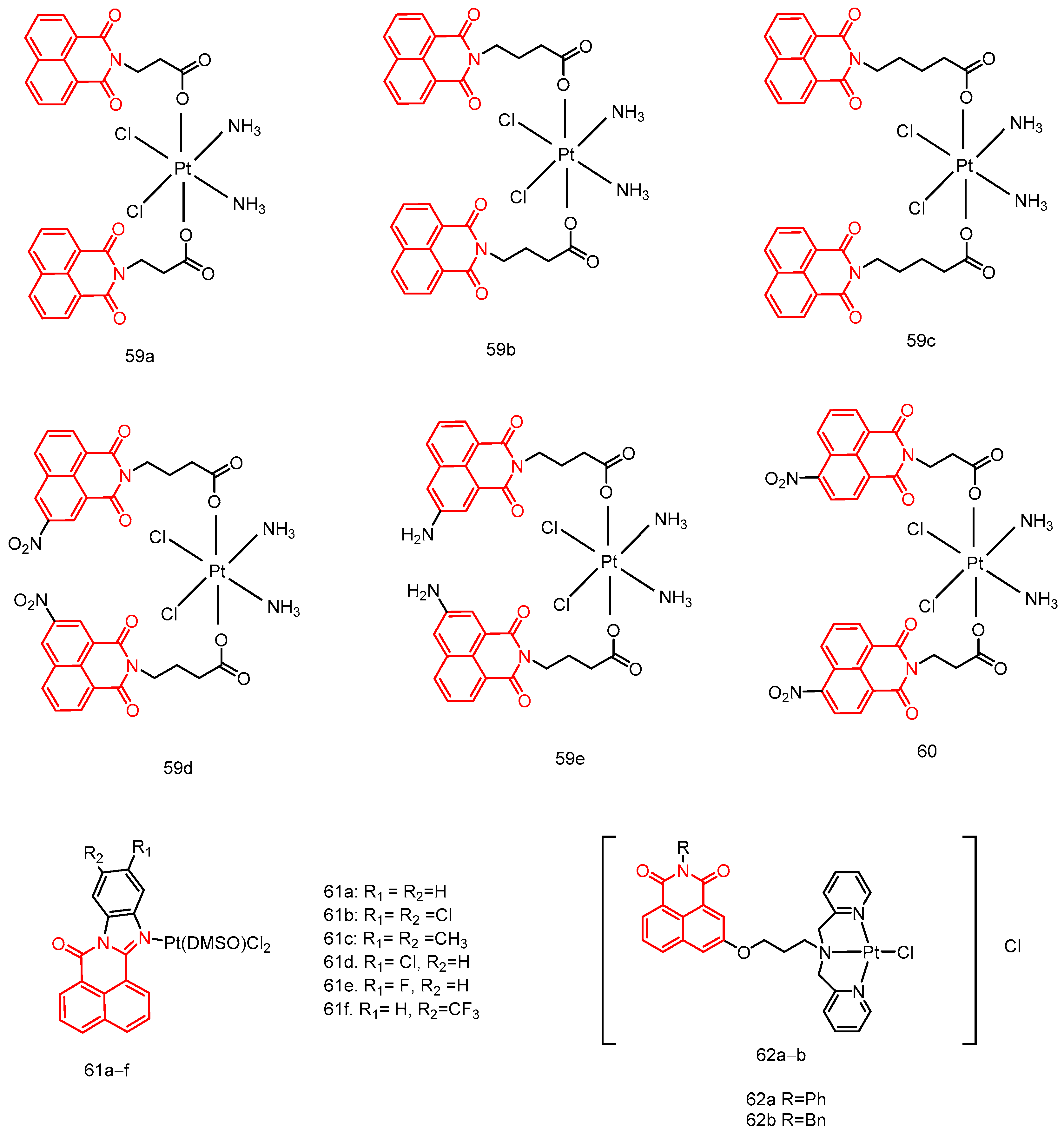
| 59a | SKOV-3, A549, A549R, HeLa Hela/DDP | 8.64 ± 1.41–44.47 ± 12.44 | [91] |
| 59b | 1.47 ± 0.11–14.94 ± 1.20 | ||
| 59c | 3.1 ± 0.43–71.74 ± 10.65 | ||
| 59d | 2.49 ± 0.30–35.35 ± 2.51 | ||
| 59e | 1.28 ± 0.28–6.53 ± 2.90 | ||
| 60 | HT-29, HCT-116, MDA-MB-231, MCF-7, 4T1, A549cisR, A549 | 1.45 ± 0.45–14.17 ± 1.37 | [92] |
| 61a | Hela, HepG-2, NCI-H460, BEL-7404, SMMC-7721, U251 | 6.85 ± 1.41–78.30 ± 1.32 | [93] |
| 61b | 5.46 ± 0.76–>100 | ||
| 61c | 4.92 ± 1.55–>100 | ||
| 61d | 4.99 ± 1.64–>100 | ||
| 61e | 2.36 ± 1.44–>100 | ||
| 61f | 7.33 ± 0.89–>100 | ||
| 62a | SK-OV-3, NCI-H460, HeLa, HL-7702 | 11.32 ± 0.47–45.07 ± 0.37 | [94] |
| 62b | 0.89 ± 0.25–50.22 ± 1.04 |
3. Conclusions and Prospects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Karthikeyan, S.; Grishina, M.; Kandasamy, S.; Mangaiyarkarasi, R.; Ramamoorthi, A.; Chinnathambi, S.; Pandian, G.N.; John Kennedy, L. A review on medicinally important heterocyclic compounds and importance of biophysical approach of underlying the insight mechanism in biological environment. J. Biomol. Struct. Dyn. 2023, 41, 14599–14619. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.P.; Robinson, R.P.; Fobian, Y.M.; Blakemore, D.C.; Jones, L.H.; Fadeyi, O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611–6637. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Park, S.B. Privileged structures: Efficient chemical “navigators” toward unexplored biologically relevant chemical spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef] [PubMed]
- Lachance, H.; Wetzel, S.; Kumar, K.; Waldmann, H. Charting, navigating, and populating natural product chemical space for drug discovery. J. Med. Chem. 2012, 55, 5989–6001. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.B.; Islam, M.I.; Nath, N.; Emran, T.B.; Rahman, M.R.; Sharma, R.; Matin, M.M. Recent advances in pyridine scaffold: Focus on chemistry, synthesis, and antibacterial activities. BioMed Res. Int. 2023, 2023, 9967591. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic scaffolds in medicinal chemistry. J. Med. Chem. 2020, 64, 150–183. [Google Scholar] [CrossRef]
- Passador, K.; Thorimbert, S.; Botuha, C. ‘Heteroaromatic Rings of the Future’: Exploration of Unconquered Chemical Space. Synthesis 2019, 51, 384–398. [Google Scholar]
- Ali, I.; Nadeem Lone, M.; A Al-Othman, Z.; Al-Warthan, A.; Marsin Sanagi, M. Heterocyclic scaffolds: Centrality in anticancer drug development. Curr. Drug Targets 2015, 16, 711–734. [Google Scholar] [CrossRef]
- Mo, F.; Qiu, D.; Zhang, L.; Wang, J. Recent development of aryl diazonium chemistry for the derivatization of aromatic compounds. Chem. Rev. 2021, 121, 5741–5829. [Google Scholar] [CrossRef]
- Dong, H.-Q.; Wei, T.-B.; Ma, X.-Q.; Yang, Q.-Y.; Zhang, Y.-F.; Sun, Y.-J.; Shi, B.-B.; Yao, H.; Zhang, Y.-M.; Lin, Q. 1, 8-Naphthalimide-based fluorescent chemosensors: Recent advances and perspectives. J. Mater. Chem. C 2020, 8, 13501–13529. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, Y.; Qian, X. Naphthalimides and analogues as antitumor agents: A review on molecular design, bioactivity and mechanism of action. Chin. Chem. Lett. 2018, 29, 1741–1756. [Google Scholar] [CrossRef]
- Kamal, A.; Azeeza, S.; Bharathi, E.V.; Malik, M.S.; Shetti, R. Search for new and novel chemotherapeutics for the treatment of human malignancies. Mini Rev. Med. Chem. 2010, 10, 405–435. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.-H.; Addla, D.; Lv, J.-S.; Zhou, C.-H. Heterocyclic naphthalimides as new skeleton structure of compounds with increasingly expanding relational medicinal applications. Curr. Top. Med. Chem. 2016, 16, 3303–3364. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.-A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A review on antibiotic resistance: Alarm bells are ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Dai, M.; Ahmed, S.; Hao, H.; Wang, X.; Yuan, Z. Antimicrobial drugs in fighting against antimicrobial resistance. Front. Microbiol. 2016, 7, 470. [Google Scholar] [CrossRef]
- Xie, M.; Gao, M.; Yun, Y.; Malmsten, M.; Rotello, V.M.; Zboril, R.; Akhavan, O.; Kraskouski, A.; Amalraj, J.; Cai, X. Antibacterial nanomaterials: Mechanisms, impacts on antimicrobial resistance and design principles. Angew. Chem. Int. Ed. 2023, 62, e202217345. [Google Scholar] [CrossRef]
- Wang, Q.; Tan, X.; Liu, Z.; Li, G.; Zhang, R.; Wei, J.; Wang, S.; Li, D.; Wang, B.; Han, J. Design and synthesis of a new series of low toxic naphthalimide platinum (IV) antitumor complexes with dual DNA damage mechanism. Eur. J. Pharm. Sci. 2018, 124, 127–136. [Google Scholar] [CrossRef]
- Shinde, R.G.; Khan, A.A.; Barik, A. Formation of two centre three electron bond by hydroxyl radical induced reaction of thiocoumarin: Evidence from experimental and theoretical studies. Free Radic. Res. 2019, 53, 629–640. [Google Scholar] [CrossRef]
- Luo, Y.-L.; Baathulaa, K.; Kannekanti, V.K.; Zhou, C.-H.; Cai, G.-X. Novel benzimidazole derived naphthalimide triazoles: Synthesis, antimicrobial activity and interactions with calf thymus DNA. Sci. China Chem. 2015, 58, 483–494. [Google Scholar] [CrossRef]
- Damu, G.L.V.; Wang, Q.; Zhang, H.; Zhang, Y.; Lv, J.; Zhou, C. A series of naphthalimide azoles: Design, synthesis and bioactive evaluation as potential antimicrobial agents. Sci. China Chem. 2013, 56, 952–969. [Google Scholar] [CrossRef]
- Zhou, C.H.; Wang, Y. Recent researches in triazole compounds as medicinal drugs. Curr. Med. Chem. 2012, 19, 239–280. [Google Scholar] [CrossRef]
- Guillon, R.; Pagniez, F.; Rambaud, C.; Picot, C.; Duflos, M.; Logé, C.; Le Pape, P. Design, Synthesis, and Biological Evaluation of 1-[(Biarylmethyl) methylamino]-2-(2, 4-difluorophenyl)-3-(1H-1, 2, 4-triazol-1-yl) propan-2-ols as Potent Antifungal Agents: New Insights into Structure–Activity Relationships. ChemMedChem 2011, 6, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Zhou, C.-H. Synthesis and activities of naphthalimide azoles as a new type of antibacterial and antifungal agents. Bioorganic Med. Chem. Lett. 2011, 21, 4349–4352. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.-S.; Peng, X.-M.; Kishore, B.; Zhou, C.-H. 1, 2, 3-Triazole-derived naphthalimides as a novel type of potential antimicrobial agents: Synthesis, antimicrobial activity, interaction with calf thymus DNA and human serum albumin. Bioorganic Med. Chem. Lett. 2014, 24, 308–313. [Google Scholar] [CrossRef]
- Chen, Q.M.; Li, Z.; Tian, G.X.; Chen, Y.; Wu, X.H. 1, 2, 3-triazole-dithiocarbamate-naphthalimides: Synthesis, characterization, and biological evaluation. J. Chem. Res. 2021, 45, 258–264. [Google Scholar] [CrossRef]
- Zhang, P.-L.; Lv, J.-S.; Fawad, A.M.; Narsaiah, B.; Cai, G.-X.; Zhou, C.-H. Synthesis of naphthalimide triazoles with a novel structural framework and their anti-Aspergillus fumigatus effects. Sci. Sin. Chim. 2021, 51, 1094–1103. [Google Scholar] [CrossRef]
- Yadav, P.; Kaushik, C.; Kumar, M.; Kumar, A. Phthalimide/Naphthalimide containing 1, 2, 3-triazole hybrids: Synthesis and antimicrobial evaluation. J. Mol. Struct. 2023, 1276, 134688. [Google Scholar] [CrossRef]
- Gupta, S.; Paul, K. Membrane-active substituted triazines as antibacterial agents against Staphylococcus aureus with potential for low drug resistance and broad activity. Eur. J. Med. Chem. 2023, 258, 115551. [Google Scholar] [CrossRef]
- Giacomazzo, G.E.; Conti, L.; Fagorzi, C.; Pagliai, M.; Andreini, C.; Guerri, A.; Perito, B.; Mengoni, A.; Valtancoli, B.; Giorgi, C. Ruthenium (II) Polypyridyl Complexes and Metronidazole Derivatives: A Powerful Combination in the Design of Photoresponsive Antibacterial Agents Effective under Hypoxic Conditions. Inorg. Chem. 2023, 62, 7716–7727. [Google Scholar] [CrossRef]
- Kang, J.; Tangadanchu, V.K.R.; Gopala, L.; Gao, W.-W.; Cheng, Y.; Liu, H.-B.; Geng, R.-X.; Li, S.; Zhou, C.-H. Novel potentially antibacterial naphthalimide-derived metronidazoles: Design, synthesis, biological evaluation and supramolecular interactions with DNA, human serum albumin and topoisomerase II. Chin. Chem. Lett. 2017, 28, 1369–1374. [Google Scholar] [CrossRef]
- Gong, H.-H.; Baathulaa, K.; Lv, J.-S.; Cai, G.-X.; Zhou, C.-H. Synthesis and biological evaluation of Schiff base-linked imidazolyl naphthalimides as novel potential anti-MRSA agents. MedChemComm 2016, 7, 924–931. [Google Scholar] [CrossRef]
- Kumari, G.; Singh, R.K. Green synthesis, antibacterial activity, and SAR of some novel naphthalimides and allylidenes. Med. Chem. Res. 2015, 24, 171–181. [Google Scholar] [CrossRef]
- Guo, J.; Xie, Z.; Ruan, W.; Tang, Q.; Qiao, D.; Zhu, W. Thiazole-based analogs as potential antibacterial agents against methicillin-resistant Staphylococcus aureus (MRSA) and their SAR elucidation. Eur. J. Med. Chem. 2023, 259, 115689. [Google Scholar] [CrossRef] [PubMed]
- Dive, G.; Bouillon, C.; Sliwa, A.; Valet, B.; Verlaine, O.; Sauvage, E.; Marchand-Brynaert, J. Macrocycle-embedded β-lactams as novel inhibitors of the Penicillin Binding Protein PBP2a from MRSA. Eur. J. Med. Chem. 2013, 64, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Speri, E.; Kim, C.; De Benedetti, S.; Qian, Y.; Lastochkin, E.; Fishovitz, J.; Fisher, J.F.; Mobashery, S. Cinnamonitrile adjuvants restore susceptibility to β-lactams against methicillin-resistant Staphylococcus aureus. ACS Med. Chem. Lett. 2019, 10, 1148–1153. [Google Scholar] [CrossRef]
- Shalaby, M.-A.W.; Dokla, E.M.; Serya, R.A.; Abouzid, K.A. Penicillin binding protein 2a: An overview and a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Gopala, L.; Bheemanaboina, R.R.Y.; Liu, H.-B.; Cheng, Y.; Geng, R.-X.; Zhou, C.-H. Novel naphthalimide aminothiazoles as potential multitargeting antimicrobial agents. ACS Med. Chem. Lett. 2017, 8, 1331–1335. [Google Scholar] [CrossRef]
- Zhang, P.-L.; Gopala, L.; Zhang, S.-L.; Cai, G.-X.; Zhou, C.-H. An unanticipated discovery towards novel naphthalimide corbelled aminothiazoximes as potential anti-MRSA agents and allosteric modulators for PBP2a. Eur. J. Med. Chem. 2022, 229, 114050. [Google Scholar] [CrossRef]
- Zhang, P.-L.; Laiche, M.H.; Li, Y.-L.; Gao, W.-W.; Lin, J.-M.; Zhou, C.-H. An unanticipated discovery of novel naphthalimidopropanediols as potential broad-spectrum antibacterial members. Eur. J. Med. Chem. 2022, 241, 114657. [Google Scholar] [CrossRef]
- Rana, P.; Parupalli, R.; Akhir, A.; Saxena, D.; Maitra, R.; Imran, M.; Malik, P.; Ghouse, S.M.; Joshi, S.V.; Srikanth, D. Synthesis and biological evaluation of new naphthalimide–thiourea derivatives as potent antimicrobial agents active against multidrug-resistant Staphylococcus aureus and Mycobacterium tuberculosis. RSC Med. Chem. 2024, 15, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, G.; Tripathi, A.K.; Seena, S.; Koh, J. Enhanced fluorescence norfloxacin substituted naphthalimide derivatives: Molecular docking and antibacterial activity. J. Mol. Struct. 2018, 1157, 292–299. [Google Scholar] [CrossRef]
- Zhang, P.; Tangadanchu, V.K.R.; Zhou, C. Identification of novel antifungal skeleton of hydroxyethyl naphthalimides with synergistic potential for chemical and dynamic treatments. Molecules 2022, 27, 8453. [Google Scholar] [CrossRef] [PubMed]
- Nayab, P.S.; Pulaganti, M.; Chitta, S.K.; Abid, M.; Uddin, R. Evaluation of DNA binding, radicals scavenging and antimicrobial studies of newly synthesized N-substituted naphthalimides: Spectroscopic and molecular docking investigations. J. Fluoresc. 2015, 25, 1905–1920. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Supriya, M.S.; Kalam, A.; Eedulakanti, C.; Kaul, G.; Akhir, A.; Sindhuja, R.H.; Roy, A.; Agnivesh, P.K.; Saxena, D. Synthesis and antibacterial evaluation of new naphthalimide-coumarin hybrids against multidrug-resistant S. aureus and M. tuberculosis. J. Mol. Struct. 2024, 1307, 137957. [Google Scholar] [CrossRef]
- Irfan, M.; Saeed, A.; Akram, S.; bin Yameen, S. Dendrimers chemistry and applications: A short review. Front. Chem. Sci. 2020, 1, 29–40. [Google Scholar] [CrossRef]
- Patel, H.; Patel, P. Dendrimer applications–a review. Int. J. Pharm. Bio Sci. 2013, 4, 454–463. [Google Scholar]
- Panchenko, P.A.; Fedorova, O.A.E.; Fedorov, Y.V. Fluorescent and colorimetric chemosensors for cations based on 1, 8-naphthalimide derivatives: Design principles and optical signalling mechanisms. Russ. Chem. Rev. 2014, 83, 155. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, X.; Chen, Z.; Xu, Y.; Qian, X.; Zhu, W. Novel seven-membered ring-fused naphthalimide derivatives with potentials for cancer theranostics. Chin. Chem. Lett. 2023, 34, 107696. [Google Scholar] [CrossRef]
- Ingrassia, L.; Lefranc, F.; Kiss, R.; Mijatovic, T. Naphthalimides and azonafides as promising anti-cancer agents. Curr. Med. Chem. 2009, 16, 1192–1213. [Google Scholar] [CrossRef]
- Staneva, D.; Vasileva-Tonkova, E.; Makki, M.S.; Sobahi, T.R.; Abdel-Rahman, R.M.; Boyaci, I.H.; Asiri, A.M.; Grabchev, I. Synthesis and spectral characterization of a new PPA dendrimer modified with 4-bromo-1, 8-naphthalimide and in vitro antimicrobial activity of its Cu (II) and Zn (II) metal complexes. Tetrahedron 2015, 71, 1080–1087. [Google Scholar] [CrossRef]
- Staneva, D.; Manov, H.; Yordanova, S.; Vasileva-Tonkova, E.; Stoyanov, S.; Grabchev, I. Synthesis, spectral properties and antimicrobial activity of a new cationic water-soluble pH-dependent poly (propylene imine) dendrimer modified with 1, 8-naphthalimides. Luminescence 2020, 35, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Staneva, D.; Vasileva-Tonkova, E.; Grozdanov, P.; Vilhelmova-Ilieva, N.; Nikolova, I.; Grabchev, I. Synthesis and photophysical characterisation of 3-bromo-4-dimethylamino-1, 8-naphthalimides and their evaluation as agents for antibacterial photodynamic therapy. J. Photochem. Photobiol. Chem. 2020, 401, 112730. [Google Scholar] [CrossRef]
- Şenkuytu, E.; Öztürk, E.; Aydınoğlu, F.; Eçik, E.T.; Okutan, E. Cyclotriphosphazene cored naphthalimide-BODIPY dendrimeric systems: Synthesis, photophysical and antimicrobial properties. Inorganica Chim. Acta 2020, 502, 119386. [Google Scholar] [CrossRef]
- Eserci, H.; Çetin, M.; Aydınoğlu, F.; Eçik, E.T.; Okutan, E. Naphthalimide-BODIPY dyads: Synthesis, characterization, photophysical properties, live cell imaging and antimicrobial effect. J. Mol. Struct. 2022, 1265, 133440. [Google Scholar] [CrossRef]
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef]
- Lath, A.; Santal, A.R.; Kaur, N.; Kumari, P.; Singh, N.P. Anti-cancer peptides: Their current trends in the development of peptide-based therapy and anti-tumor drugs. Biotechnol. Genet. Eng. Rev. 2023, 39, 45–84. [Google Scholar] [CrossRef]
- Sikora, K.; Advani, S.; Koroltchouk, V.; Magrath, I.; Levy, L.; Pinedo, H.; Schwartsmann, G.; Tattersall, M.; Yan, S. Essential drugs for cancer therapy: A World Health Organization consultation. Ann. Oncol. 1999, 10, 385–390. [Google Scholar] [CrossRef]
- Bunn Jr, P.A.; Kelly, K. New chemotherapeutic agents prolong survival and improve quality of life in non-small cell lung cancer: A review of the literature and future directions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 1087–1100. [Google Scholar]
- Chen, C.; Li, X.; Zhao, H.; Liu, M.; Du, J.; Zhang, J.; Yang, X.; Hou, X.; Fang, H. Discovery of DNA-targeting HDAC inhibitors with potent antitumor efficacy in vivo that trigger antitumor immunity. J. Med. Chem. 2022, 65, 3667–3683. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Han, L.-L.; Wu, H.-Y.; Xu, X.-X.; Yu, M.-B.; Chen, G.-Y.; Qi, X.-L. Research Progress on Structure-Activity Relationship of 1, 8-Naphthalimide DNA Chimeras Against Tumor. Technol. Cancer Res. Treat. 2024, 23, 15330338231225861. [Google Scholar] [CrossRef] [PubMed]
- Van Quaquebeke, E.; Mahieu, T.; Dumont, P.; Dewelle, J.; Ribaucour, F.; Simon, G.; Sauvage, S.; Gaussin, J.-F.; Tuti, J.; El Yazidi, M. 2, 2, 2-Trichloro-N-({2-[2-(dimethylamino) ethyl]-1, 3-dioxo-2, 3-dihydro-1 H-benzo [de] isoquinolin-5-yl} carbamoyl) acetamide (UNBS3157), a Novel Nonhematotoxic Naphthalimide Derivative with Potent Antitumor Activity. J. Med. Chem. 2007, 50, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Mahieu, T.; Bruyère, C.; De Nève, N.; Dewelle, J.; Simon, G.; Dehoux, M.J.; van der Aar, E.; Haibe-Kains, B.; Bontempi, G. UNBS5162, a novel naphthalimide that decreases CXCL chemokine expression in experimental prostate cancers. Neoplasia 2008, 10, 573–586. [Google Scholar] [CrossRef]
- Xie, L.; Xu, Y.; Wang, F.; Liu, J.; Qian, X.; Cui, J. Synthesis of new amonafide analogues via coupling reaction and their cytotoxic evaluation and DNA-binding studies. Bioorganic Med. Chem. 2009, 17, 804–810. [Google Scholar] [CrossRef]
- Xin, M.; Wei, J.-H.; Yang, C.-H.; Liang, G.-B.; Su, D.; Ma, X.-L.; Zhang, Y. Design, synthesis and biological evaluation of 3-nitro-1, 8-naphthalimides as potential antitumor agents. Bioorganic Med. Chem. Lett. 2020, 30, 127051. [Google Scholar] [CrossRef]
- Ge, C.; Liu, L.; Wang, Y.; Di, X.; Luo, X.; Liu, H.; Qian, Y. Novel 1, 8-Naphthalimide Derivatives As Antitumor Agents and Potent Demethylase Inhibitors. ACS Med. Chem. Lett. 2023, 14, 1551–1557. [Google Scholar] [CrossRef]
- Wang, S.-S.; Du, S.-Y.; He, X.; Qi, Y.-M.; Li, X.-L.; Rong, R.-X.; Cao, Z.-R.; Wang, K.-R. Nucleus-targeting imaging and enhanced cytotoxicity based on naphthalimide derivatives. Bioorganic Chem. 2021, 115, 105188. [Google Scholar] [CrossRef]
- Seliga, R.; Pilatova, M.; Sarissky, M.; Viglasky, V.; Walko, M.; Mojzis, J. Novel naphthalimide polyamine derivatives as potential antitumor agents. Mol. Biol. Rep. 2013, 40, 4129–4137. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Zhang, J.; Xie, S.; Wang, C.; Wu, Y. Synthesis and biological evaluation of novel aromatic imide-polyamine conjugates. Molecules 2016, 21, 1637. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; He, H.; Xu, X.; Chen, S.; Wang, C.; Feng, C.; Tian, Z.; Dong, H.; Xie, S. Synthesis and biological evaluation of naphthalimide-polyamine conjugates modified by alkylation as anticancer agents through p53 pathway. Bioorganic Chem. 2018, 77, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Ge, C.; Chang, L.; Wang, C.; Tian, Z.; Wang, S.; Dai, F.; Zhao, L.; Xie, S. Synthesis and biological evaluation of novel alkylated polyamine analogues as potential anticancer agents. Eur. J. Med. Chem. 2018, 143, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, Y.; Li, L.; Yue, K.; Liu, H.; Wang, J.; Xi, Z.; Shi, M.; Zhao, S.; Ma, Q. A polyamine-based dinitro-naphthalimide conjugate as substrates for polyamine transporters preferentially accumulates in cancer cells and minimizes side effects in vitro and in vivo. Front. Chem. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, L.; Yue, K.; Zhang, Z.; Su, S.; Chen, Y.; Yu, L.; Zhang, P.; Ma, R.; Li, Y. A naphthalimide-polyamine conjugate preferentially accumulates in hepatic carcinoma metastases as a lysosome-targeted antimetastatic agent. Eur. J. Med. Chem. 2021, 221, 113469. [Google Scholar] [CrossRef] [PubMed]
- Rong, R.-X.; Wang, S.-S.; Liu, X.; Li, R.-F.; Wang, K.-R.; Cao, Z.-R.; Li, X.-L. Lysosomes-targeting imaging and anticancer properties of novel bis-naphthalimide derivatives. Bioorganic Med. Chem. Lett. 2018, 28, 742–747. [Google Scholar] [CrossRef]
- Ou, Z.; Li, Z.; Gao, Y.; Xing, W.; Jia, H.; Zhang, H.; Yi, N. Novel triazole and morpholine substituted bisnaphthalimide: Synthesis, photophysical and G-quadruplex binding properties. J. Mol. Struct. 2019, 1185, 27–37. [Google Scholar] [CrossRef]
- Shankaraiah, N.; Kumar, N.P.; Tokala, R.; Gayatri, B.S.; Talla, V.; Santos, L.S. Synthesis of new 1,2,3-triazolo-naphthalimide/phthalimide conjugates via ‘Click’Reaction: DNA intercalation and cytotoxic studies. J. Braz. Chem. Soc. 2019, 30, 454–461. [Google Scholar]
- Huang, Y.; Wu, C.-X.; Song, Y.; Huang, M.; Tian, D.-N.; Yang, X.-B.; Fan, Y.-R. Synthesis, DNA binding, and anticancer properties of bis-naphthalimide derivatives with lysine-modified polyamine linkers. Molecules 2018, 23, 266. [Google Scholar] [CrossRef]
- Rao, N.S.; Nagesh, N.; Nayak, V.L.; Sunkari, S.; Tokala, R.; Kiranmai, G.; Regur, P.; Shankaraiah, N.; Kamal, A. Design and synthesis of DNA-intercalative naphthalimide-benzothiazole/cinnamide derivatives: Cytotoxicity evaluation and topoisomerase-IIα inhibition. MedChemComm 2019, 10, 72–79. [Google Scholar]
- Chen, R.; Yuan, C.; Jaiswal, Y.; Huo, L.; Li, D.; Williams, L.; Zhong, J.; Liang, Y. Synthesis and Biological Evaluation of Some 1, 8-Naphthalimide-Acridinyl Hybrids. J. Chem. 2020, 2020, 7989852. [Google Scholar] [CrossRef]
- Singh, I.; Luxami, V.; Paul, K. Synthesis and in vitro evaluation of naphthalimide–benzimidazole conjugates as potential antitumor agents. Org. Biomol. Chem. 2019, 17, 5349–5366. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, M.; Xiong, X.-Q.; Yang, H.; Wang, P.; Zhang, K.; Awadasseid, A.; Narva, S.; Wu, Y.-L.; Zhang, W. Design, synthesis and bioactivity of novel naphthalimide-benzotriazole conjugates against A549 cells via targeting BCL2 G-quadruplex and inducing autophagy. Life Sci. 2022, 302, 120651. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, X.; Chen, A.; Zhang, H.; Yu, Q.; Shen, C.; Awadasseid, A.; Zhao, X.; Xiong, X.; Wu, Y. Design, synthesis and anti-tumor activity of novel benzothiophenonaphthalimide derivatives targeting mitochondrial DNA (mtDNA) G-quadruplex. Biochem. Pharmacol. 2022, 201, 115062. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-M.; Zhou, J.-Y.; Liu, S.-Q.; Song, L.-H.; Wang, H.-L.; Wang, Q.; Liang, S.-M.; Lu, L.; Wei, J.-H.; Huang, R. Design, synthesis, and antitumor evaluation of morpholine substituted bisnaphthalimides as DNA targeting agents. Bioorganic Med. Chem. Lett. 2023, 85, 129218. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Luxami, V.; Paul, K. Synthesis, in vitro evaluation and molecular modelling of naphthalimide analogue as anticancer agents. Eur. J. Med. Chem. 2013, 68, 352–360. [Google Scholar] [CrossRef]
- Rad, J.A.; Jarrahpour, A.; Aseman, M.D.; Nabavizadeh, M.; Pournejati, R.; Karbalaei-Heidari, H.R.; Turos, E. Design, synthesis, DNA binding, cytotoxicity, and molecular docking studies of amonafide-linked β-lactam. ChemistrySelect 2019, 4, 2741–2746. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, S.; Tian, Z.; Xu, Z.; Zhang, Y.; Xia, X.; Chen, X.; Liu, Z. Potential anticancer agent for selective damage to mitochondria or lysosomes: Naphthalimide-modified fluorescent biomarker half-sandwich iridium (III) and ruthenium (II) complexes. Eur. J. Med. Chem. 2019, 181, 111599. [Google Scholar] [CrossRef]
- Jia, D.-G.; Zheng, J.-A.; Fan, Y.-R.; Yu, J.-Q.; Wu, X.-L.; Wang, B.-J.; Yang, X.-B.; Huang, Y. Ferrocene appended naphthalimide derivatives: Synthesis, DNA binding, and in vitro cytotoxic activity. J. Organomet. Chem. 2019, 888, 16–23. [Google Scholar] [CrossRef]
- Streciwilk, W.; Terenzi, A.; Cheng, X.; Hager, L.; Dabiri, Y.; Prochnow, P.; Bandow, J.E.; Wölfl, S.; Keppler, B.K.; Ott, I. Fluorescent organometallic rhodium (I) and ruthenium (II) metallodrugs with 4-ethylthio-1, 8-naphthalimide ligands: Antiproliferative effects, cellular uptake and DNA-interaction. Eur. J. Med. Chem. 2018, 156, 148–161. [Google Scholar] [CrossRef]
- Wang, Q.; Li, G.; Liu, Z.; Tan, X.; Ding, Z.; Ma, J.; Li, L.; Li, D.; Han, J.; Wang, B. Naphthalimide platinum (IV) compounds as antitumor agents with dual DNA damage mechanism to overcome cisplatin resistance. Eur. J. Inorg. Chem. 2018, 2018, 4442–4451. [Google Scholar] [CrossRef]
- Li, Y.; Yue, K.; Li, L.; Niu, J.; Liu, H.; Ma, J.; Xie, S. A Pt (IV)-based mononitro-naphthalimide conjugate with minimized side-effects targeting DNA damage response via a dual-DNA-damage approach to overcome cisplatin resistance. Bioorganic Chem. 2020, 101, 104011. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.-B.; Yu, Y.-C.; Wei, J.-H.; Kuang, W.-B.; Chen, Z.-F.; Zhang, Y. Design, synthesis and biological evaluation of naphthalenebenzimidizole platinum (II) complexes as potential antitumor agents. Eur. J. Med. Chem. 2020, 188, 112033. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-B.; Chen, S.; Qin, Q.-P.; Luo, J.-R.; Tan, M.-X.; Wang, Z.-F.; Zou, B.-Q.; Liang, H. Preparation of platinum (II) complexes with naphthalene imide derivatives and exploration of their in vitro cytotoxic activities. Inorg. Chem. Commun. 2019, 104, 124–128. [Google Scholar] [CrossRef]
- Rad, J.A.; Jarrahpour, A.; Latour, C.; Sinou, V.; Brunel, J.M.; Zgou, H.; Mabkhot, Y.; Hadda, T.B.; Turos, E. Synthesis and antimicrobial/antimalarial activities of novel naphthalimido trans-β-lactam derivatives. Med. Chem. Res. 2017, 26, 2235–2242. [Google Scholar] [CrossRef]
- Dana, S.; Keshri, S.K.; Shukla, J.; Vikramdeo, K.S.; Mondal, N.; Mukhopadhyay, P.; Dhar, S.K. Design, synthesis and evaluation of bifunctional acridinine− naphthalenediimide redox-active conjugates as antimalarials. ACS Omega 2016, 1, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Kokosza, K.; Andrei, G.; Schols, D.; Snoeck, R.; Piotrowska, D.G. Design, antiviral and cytostatic properties of isoxazolidine-containing amonafide analogues. Bioorganic Med. Chem. 2015, 23, 3135–3146. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Alswaidan, I.; Ghabbour, H.A.; Ezzeldin, E.; Elaasser, M.; Marzouk, M. Docking and antiherpetic activity of 2-aminobenzo [de]-isoquinoline-1, 3-diones. Molecules 2015, 20, 5099–5111. [Google Scholar] [CrossRef]
- Shih, T.L.; Lin, K.H.; Chen, R.J.; Chen, T.Y.; Kao, W.T.; Liu, J.W.; Wang, H.H.; Peng, H.Y.; Sun, Y.Y.; Lu, W.J. A novel naphthalimide derivative reduces platelet activation and thrombus formation via suppressing GPVI. J. Cell. Mol. Med. 2021, 25, 9434–9446. [Google Scholar] [CrossRef]
- Begam, R.; Shajahan, A.; Shefin, B.; Murugan, V. Synthesis of novel naphthalimide tethered 1,2,3-triazoles: In vitro biological evaluation and docking study of anti-inflammatory inhibitors. J. Mol. Struct. 2022, 1254, 132364. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, W.; Xie, Z.; Wang, Y.; Xia, L.; Guo, Y.; Qiao, D. An Overview of Naphthylimide as Specific Scaffold for New Drug Discovery. Molecules 2024, 29, 4529. https://doi.org/10.3390/molecules29194529
Ruan W, Xie Z, Wang Y, Xia L, Guo Y, Qiao D. An Overview of Naphthylimide as Specific Scaffold for New Drug Discovery. Molecules. 2024; 29(19):4529. https://doi.org/10.3390/molecules29194529
Chicago/Turabian StyleRuan, Wei, Zhouling Xie, Ying Wang, Lulu Xia, Yuping Guo, and Dan Qiao. 2024. "An Overview of Naphthylimide as Specific Scaffold for New Drug Discovery" Molecules 29, no. 19: 4529. https://doi.org/10.3390/molecules29194529
APA StyleRuan, W., Xie, Z., Wang, Y., Xia, L., Guo, Y., & Qiao, D. (2024). An Overview of Naphthylimide as Specific Scaffold for New Drug Discovery. Molecules, 29(19), 4529. https://doi.org/10.3390/molecules29194529