
Adsorptive Stripping Voltammetric Quercetin Determination in Pharmaceuticals and Urine Samples Using a Long Service-Life Array of Carbon Composite Microelectrodes


Abstract
1. Introduction
2. Discussion
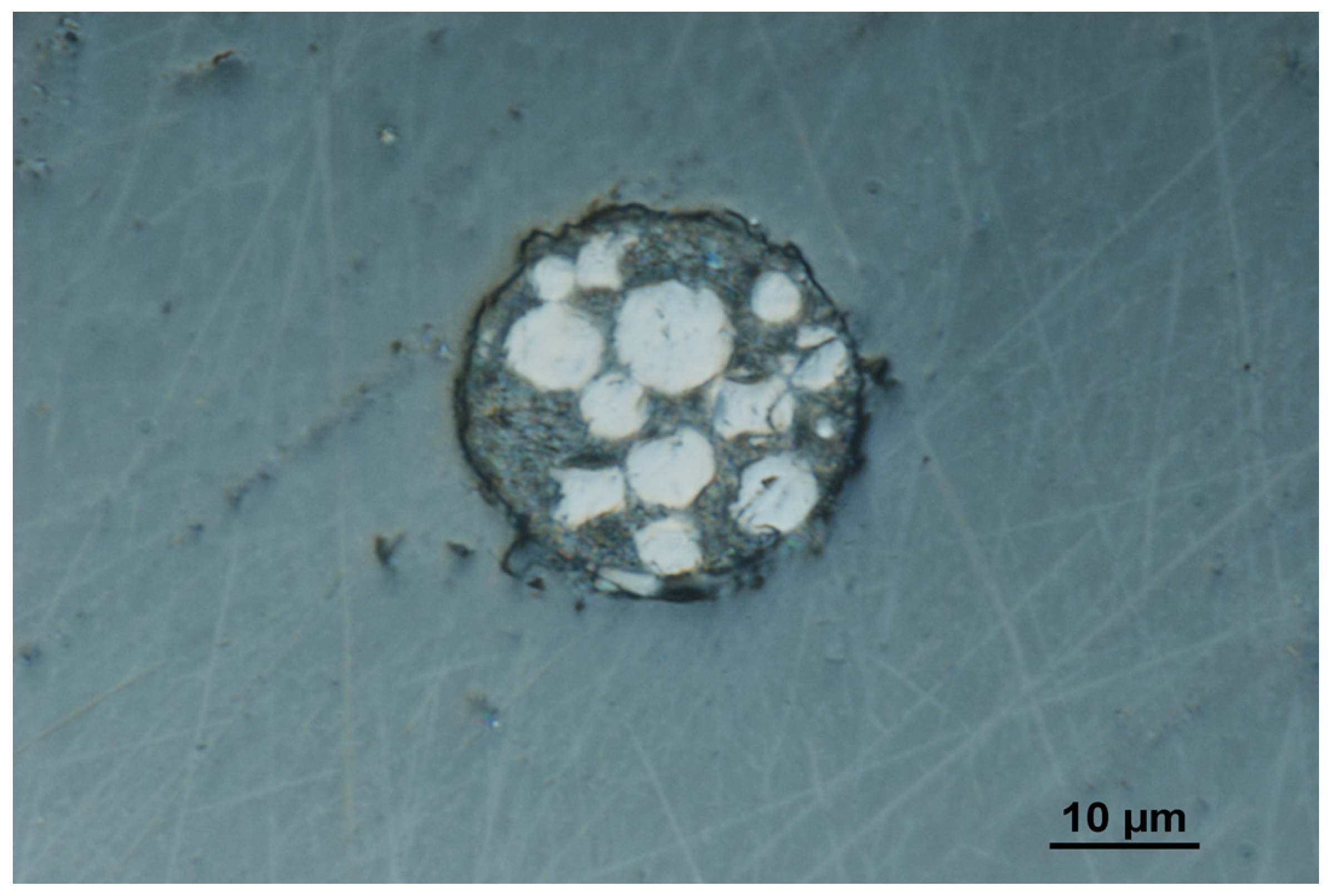
2.1. Microelectrode Properties of the Array of Carbon Composite Microelectrodes
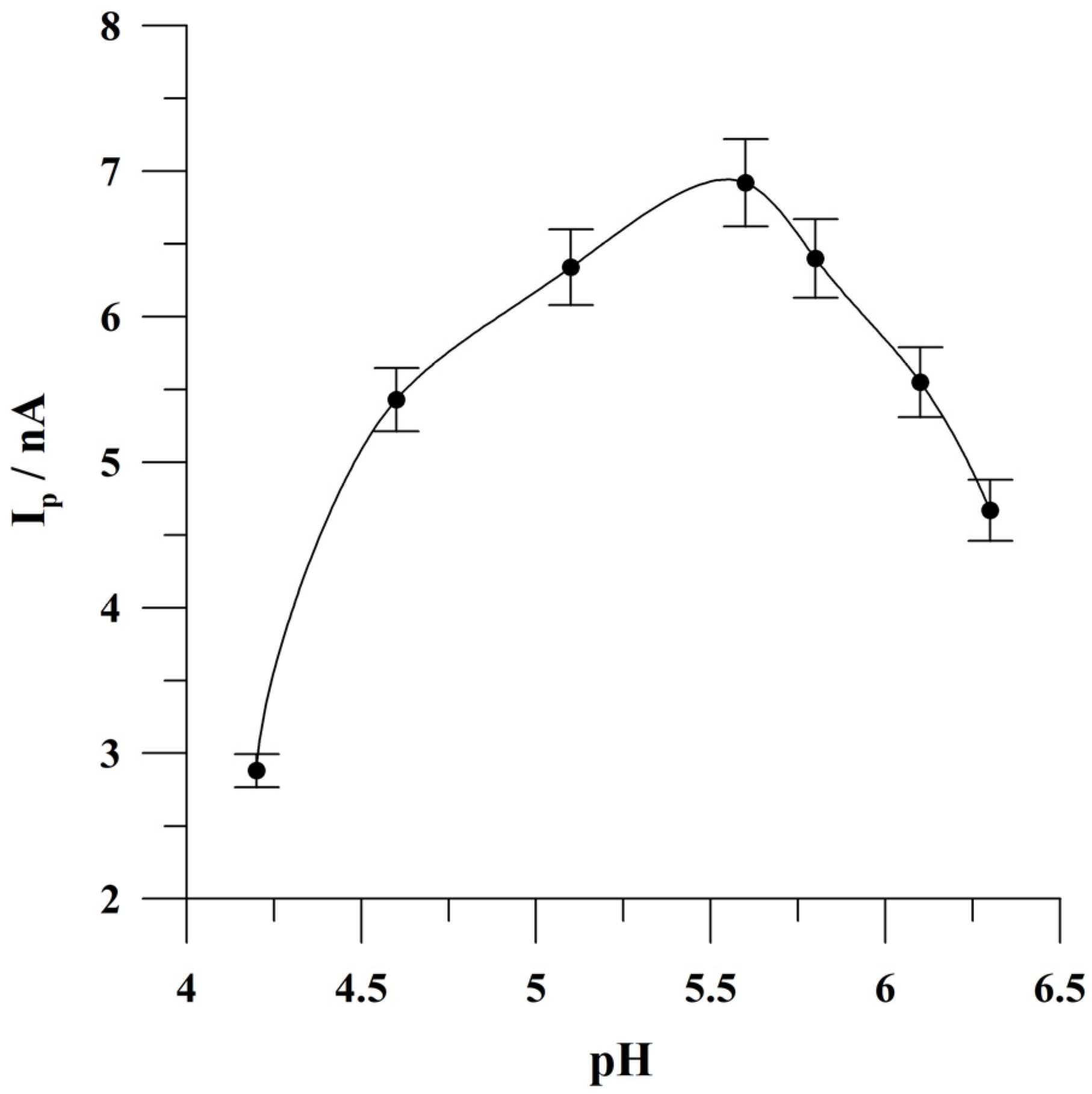
2.2. Optimization of the pH of the Supporting Electrolyte
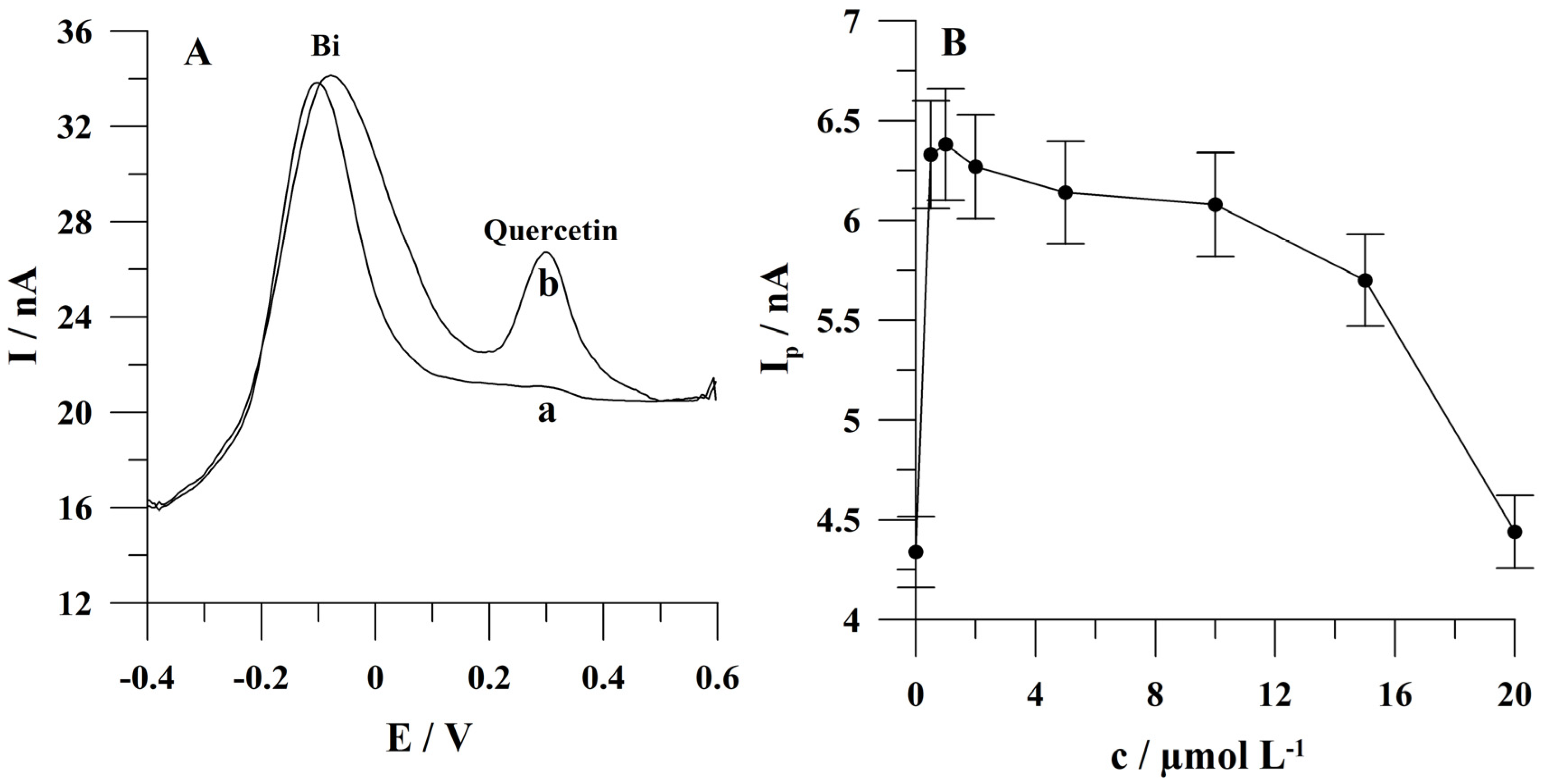
2.3. Optimization of a Concentration of Bi(III)
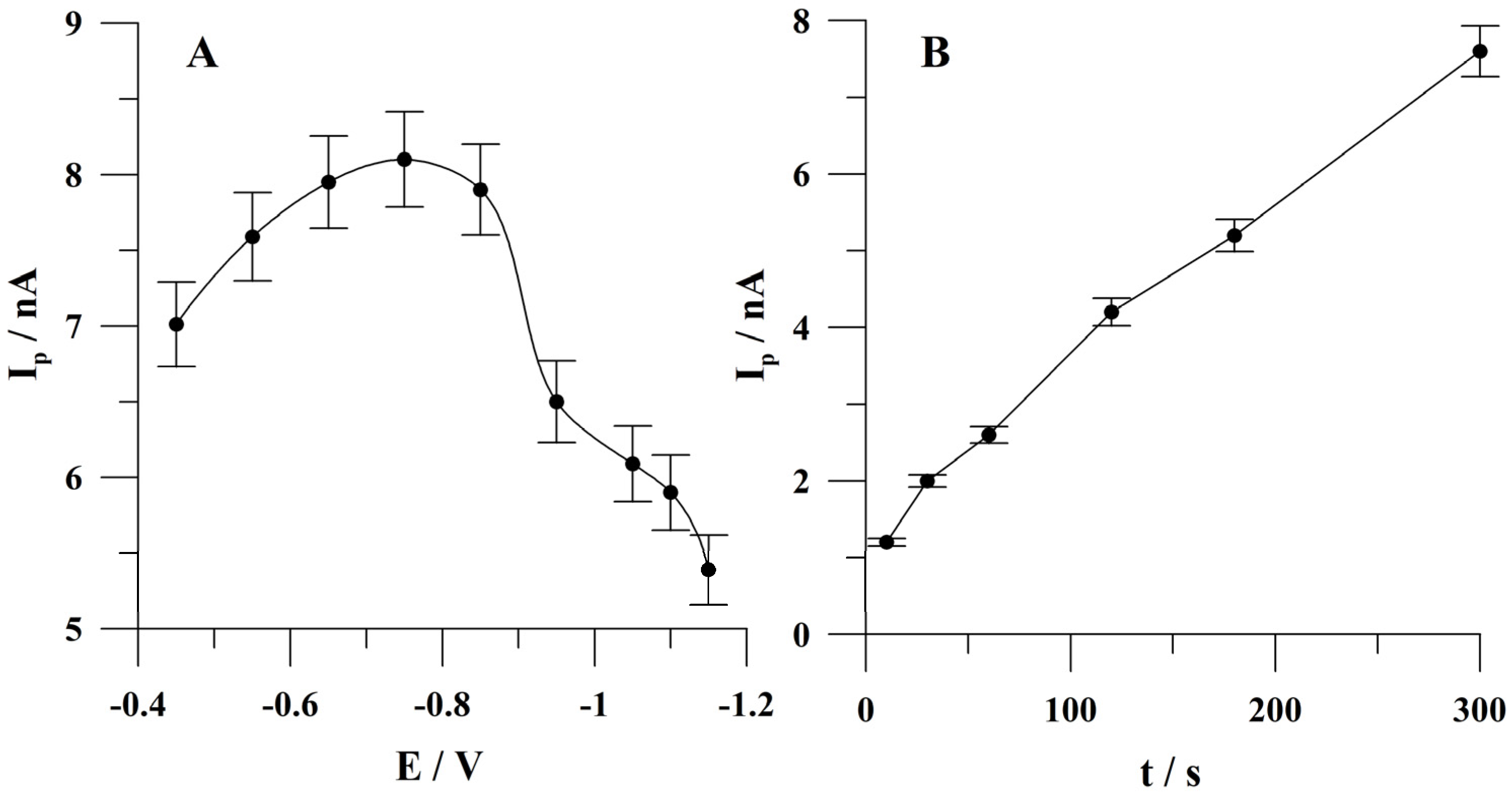
2.4. Optimization of the Potential of Bismuth Plating
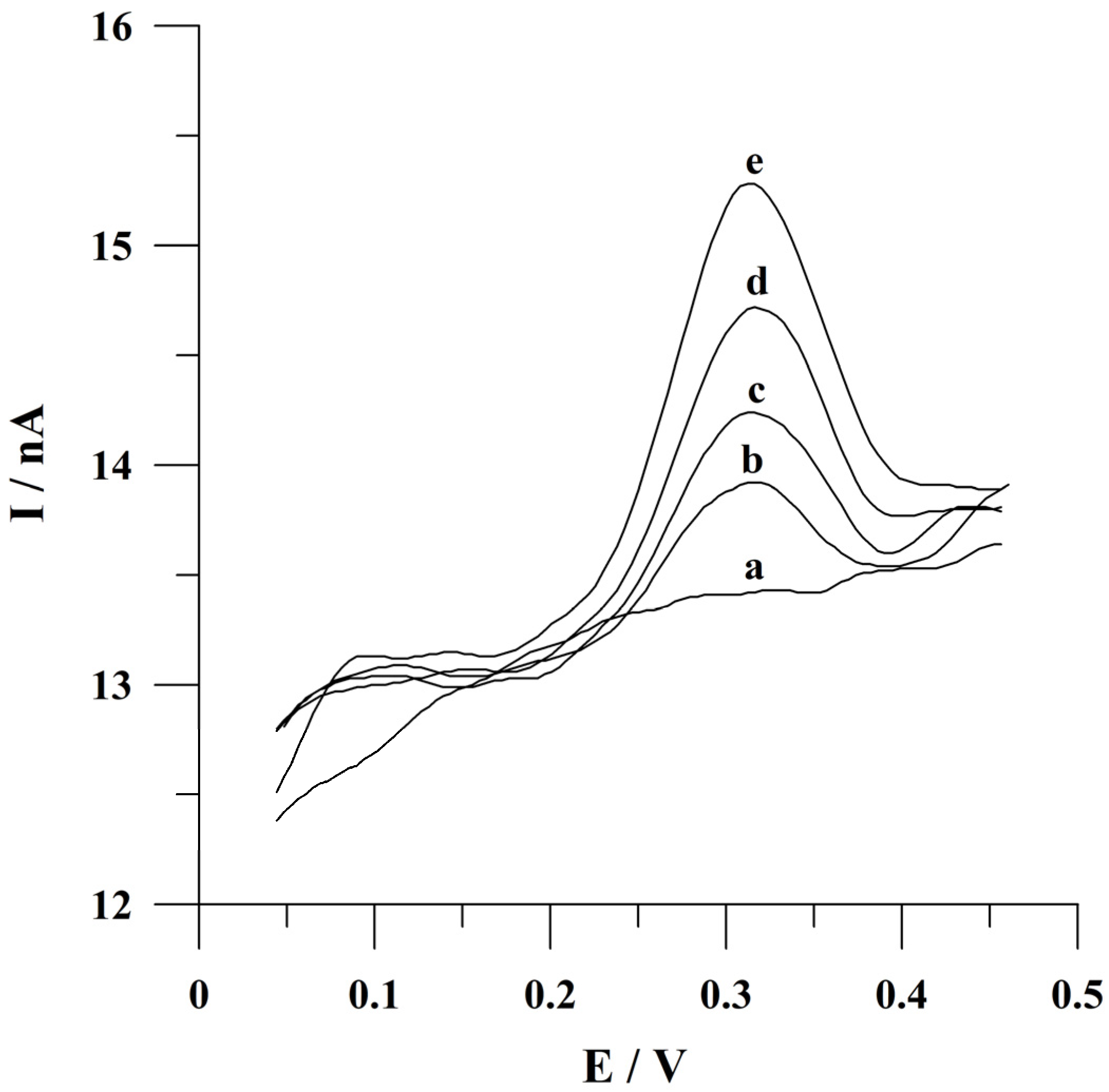
2.5. Optimization of Quercetin Accumulation Conditions
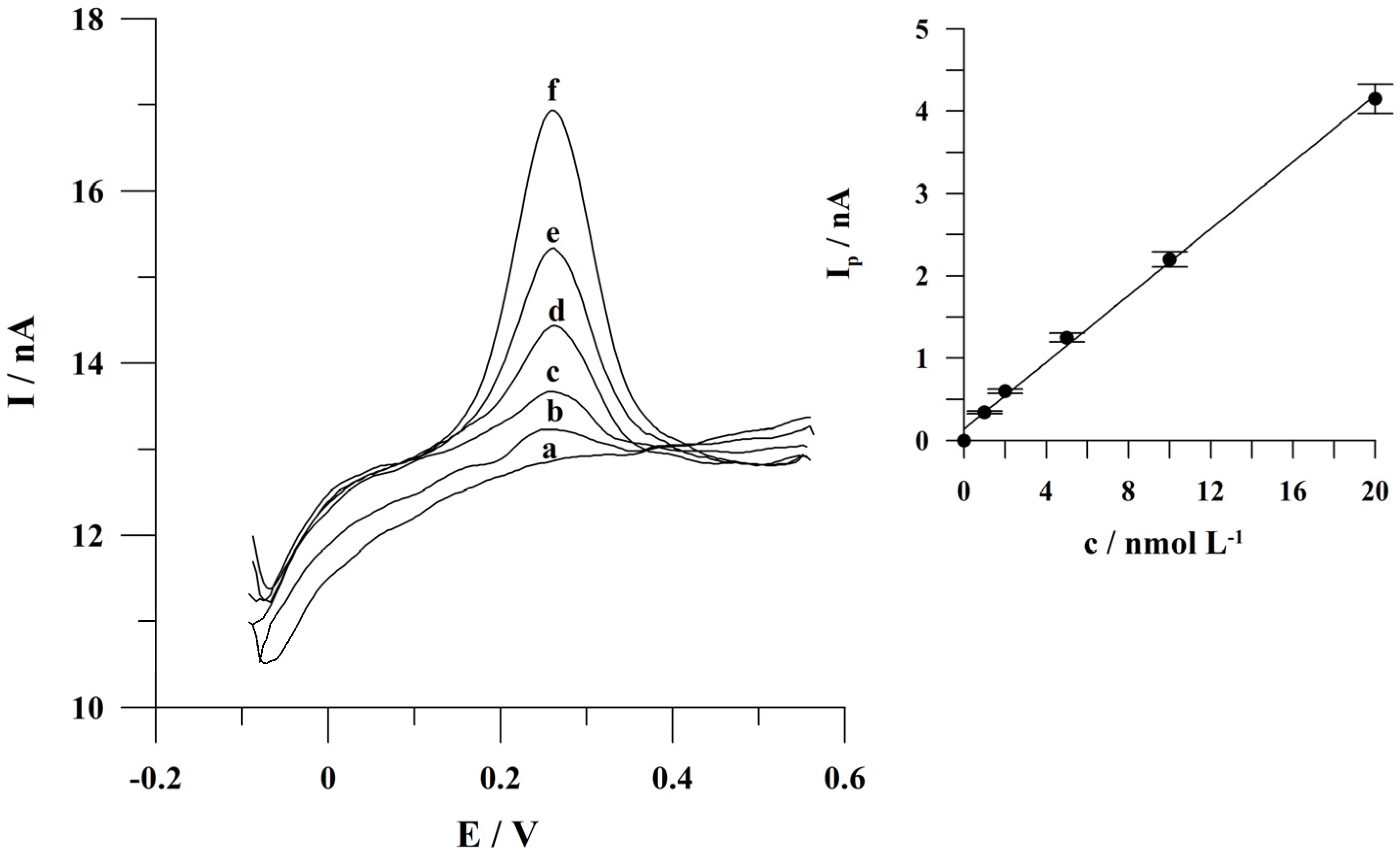
2.6. Calibration Studies
2.7. Reproducibility Studies
2.8. Interferences Studies
2.9. Analytical Application
3. Materials and Methods
3.1. Instrumentation
3.2. Reagents
3.3. Preparation of Pharmaceutical and Urine Samples
3.4. Standard Procedure of the Measurements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zielinska, D.; Pierozynski, B. Electrooxidation of Quercetin at Glassy Carbon Electrode Studied by a.c. Impedance Spectroscopy. J. Electroanal. Chem. 2009, 625, 149–155. [Google Scholar] [CrossRef]
- Oliveira-Brett, A.M.; Diculescu, V.C. Electrochemical Study of Quercetin–DNA Interactions: Part II. In Situ Sensing with DNA Biosensors. Bioelectrochemistry 2004, 64, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Volikakis, G.J.; Efstathiou, C.E. Determination of Rutin and Other Flavonoids by Flow-Injection/Adsorptive Stripping Voltammetry Using Nujol-Graphite and Diphenylether-Graphite Paste Electrodes. Talanta 2000, 51, 775–785. [Google Scholar] [CrossRef]
- Carvalho, D.; Pinho, C.; Oliveira, R.; Moreira, F.; Oliveira, A.I. Chromatographic Methods Developed for the Quantification of Quercetin Extracted from Natural Sources: Systematic Review of Published Studies from 2018 to 2022. Molecules 2023, 28, 7714. [Google Scholar] [CrossRef]
- Liu, B.; Anderson, D.; Ferry, D.R.; Seymour, L.W.; de Takats, P.G.; Kerr, D.J. Determination of Quercetin in Human Plasma Using Reversed-Phase High-Performance Liquid Chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1995, 666, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Abdelkawy, K.S.; Balyshev, M.E.; Elbarbry, F. A New Validated HPLC Method for the Determination of Quercetin: Application to Study Pharmacokinetics in Rats. Biomed. Chromatogr. 2017, 31, 3819. [Google Scholar] [CrossRef]
- Alipour, F.; Raoof, J.B.; Ghani, M. Determination of Quercetin via Thin Film Microextraction Using the in Situ Growth of Co–Al-Layered Double Hydroxide Nanosheets on an Electrochemically Anodized Aluminum Substrate Followed by HPLC. Anal. Methods 2020, 12, 799–806. [Google Scholar] [CrossRef]
- Carvalho, D.; Jesus, Â.; Pinho, C.; Oliveira, R.F.; Moreira, F.; Oliveira, A.I. Validation of an HPLC-DAD Method for Quercetin Quantification in Nanoparticles. Pharmaceuticals 2023, 16, 1736. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.M.; Abouelenein, D.; Angeloni, S.; Maggi, F.; Navarini, L.; Sagratini, G.; Santanatoglia, A.; Torregiani, E.; Vittori, S.; Caprioli, G. A New HPLC-MS/MS Method for the Simultaneous Determination of Quercetin and Its Derivatives in Green Coffee Beans. Foods 2022, 11, 3033. [Google Scholar] [CrossRef]
- Pilařová, V.; Plachká, K.; Chrenková, L.; Najmanová, I.; Mladěnka, P.; Švec, F.; Novák, O.; Nováková, L. Simultaneous Determination of Quercetin and Its Metabolites in Rat Plasma by Using Ultra-High Performance Liquid Chromatography Tandem Mass Spectrometry. Talanta 2018, 185, 71–79. [Google Scholar] [CrossRef]
- Yunita, E.; Yulianto, D.; Fatimah, S.; Firanita, T. Validation of UV-Vis Spectrophotometric Method of Quercetin in Ethanol Extract of Tamarind Leaf. J. Fundam. Appl. Pharm. Sci. 2020, 1, 11–18. [Google Scholar] [CrossRef]
- Soylak, M.; Ozdemir, B.; Yilmaz, E. An Environmentally Friendly and Novel Amine-Based Liquid Phase Microextraction of Quercetin in Food Samples Prior to Its Determination by UV-Vis Spectrophotometry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 243, 118806. [Google Scholar] [CrossRef] [PubMed]
- Alva-Ensastegui, J.C.; Palomar-Pardavé, M.; Romero-Romo, M.; Ramírez-Silva, M.T. Quercetin Spectrofluorometric Quantification in Aqueous Media Using Different Surfactants as Fluorescence Promoters. RSC Adv. 2018, 8, 10980–10986. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, H.; Ye, J. Determination of Rutin and Quercetin in Plants by Capillary Electrophoresis with Electrochemical Detection. Anal. Chim. Acta 2000, 423, 69–76. [Google Scholar] [CrossRef]
- Zoulis, N.E.; Efstathiou, C.E. Preconcentration at a Carbon-Paste Electrode and Determination by Adsorptive-Stripping Voltammetry of Rutin and Other Flavonoids. Anal. Chim. Acta 1996, 320, 255–261. [Google Scholar] [CrossRef]
- Farghaly, O.A. Voltammetric Determination of Quercetin in Urine at Carbon Paste Electrode. Anal. Lett. 2005, 38, 2247–2258. [Google Scholar] [CrossRef]
- He, J.B.; Lin, X.Q.; Pan, J. Multi-Wall Carbon Nanotube Paste Electrode for Adsorptive Stripping Determination of Quercetin: A Comparison with Graphite Paste Electrode via Voltammetry and Chronopotentiometry. Electroanalysis 2005, 17, 1681–1686. [Google Scholar] [CrossRef]
- Lin, X.Q.; He, J.B.; Zha, Z.G. Simultaneous Determination of Quercetin and Rutin at a Multi-Wall Carbon-Nanotube Paste Electrodes by Reversing Differential Pulse Voltammetry. Sens. Actuators B Chem. 2006, 119, 608–614. [Google Scholar] [CrossRef]
- Xiao, P.; Zhao, F.; Zeng, B. Voltammetric Determination of Quercetin at a Multi-Walled Carbon Nanotubes Paste Electrode. Microchem. J. 2007, 85, 244–249. [Google Scholar] [CrossRef]
- Pliuta, K.; Chebotarev, A.; Koicheva, A.; Bevziuk, K.; Snigur, D. Development of a Novel Voltammetric Sensor for the Determination of Quercetin on an Electrochemically Pretreated Carbon-Paste Electrode. Anal. Methods 2018, 10, 1472–1479. [Google Scholar] [CrossRef]
- Kuyumcu Savan, E. Square Wave Voltammetric (SWV) Determination of Quercetin in Tea Samples at a Single-Walled Carbon Nanotube (SWCNT) Modified Glassy Carbon Electrode (GCE). Anal. Lett. 2019, 53, 858–872. [Google Scholar] [CrossRef]
- Xu, G.R.; Kim, S. Selective Determination of Quercetin Using Carbon Nanotube-Modified Electrodes. Electroanalysis 2006, 18, 1786–1792. [Google Scholar] [CrossRef]
- Gutiérrez, F.; Ortega, G.; Cabrera, J.L.; Rubianes, M.D.; Rivas, G.A. Quantification of Quercetin Using Glassy Carbon Electrodes Modified with Multiwalled Carbon Nanotubes Dispersed in Polyethylenimine and Polyacrylic Acid. Electroanalysis 2010, 22, 2650–2657. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, D.; Tong, Z.; Xu, X.; Yang, X. Voltammetric Behavior and the Determination of Quercetin at a Flowerlike Co3O4 Nanoparticles Modified Glassy Carbon Electrode. J. Appl. Electrochem. 2011, 41, 189–196. [Google Scholar] [CrossRef]
- Pereira, E.R.d.C.V.; Bessegato, G.G.; Yamanaka, H.; Zanoni, M.V.B. Determination of Quercetin by a Siloxane-Polyester/Poly-L-Lysine Nanocomposite Modified Glassy Carbon Electrode. Anal. Lett. 2016, 49, 1398–1411. [Google Scholar] [CrossRef]
- Guss, E.V.; Ziyatdinova, G.K.; Zhupanova, A.S.; Budnikov, H.C. Voltammetric Determination of Quercetin and Rutin on Their Simultaneous Presence on an Electrode Modified with Polythymolphthalein. J. Anal. Chem. 2020, 75, 526–535. [Google Scholar] [CrossRef]
- Manokaran, J.; Muruganantham, R.; Muthukrishnaraj, A.; Balasubramanian, N. Platinum-Polydopamine@SiO2 Nanocomposite Modified Electrode for the Electrochemical Determination of Quercetin. Electrochim. Acta 2015, 168, 16–24. [Google Scholar] [CrossRef]
- Yola, M.L.; Gupta, V.K.; Eren, T.; Şen, A.E.; Atar, N. A Novel Electro Analytical Nanosensor Based on Graphene Oxide/Silver Nanoparticles for Simultaneous Determination of Quercetin and Morin. Electrochim. Acta 2014, 120, 204–211. [Google Scholar] [CrossRef]
- Tesfaye, G.; Hailu, T.; Ele, E.; Negash, N.; Tessema, M. Square Wave Voltammetric Determination of Quercetin in Wine and Fruit Juice Samples at Poly (Safranine O) Modified Glassy Carbon Electrode. Sens. Bio-Sens. Res. 2021, 34, 100466. [Google Scholar] [CrossRef]
- Zhang, Z.; Gu, S.; Ding, Y.; Shen, M.; Jiang, L. Mild and Novel Electrochemical Preparation of β-Cyclodextrin/Graphene Nanocomposite Film for Super-Sensitive Sensing of Quercetin. Biosens. Bioelectron. 2014, 57, 239–244. [Google Scholar] [CrossRef]
- Reddaiah, K.; Swamy, K.; Reddy, T.M.; Raghu, P.; Kumra Swamy, B.E. Electrochemical Determination of Quercetin at β-Cyclodextrin Modified Chemical Sensor: A Voltammetric Study. Anal. Bioanal. Electrochem 2012, 4, 122–134. [Google Scholar]
- Yola, M.L.; Atar, N.; Üstündaǧ, Z.; Solak, A.O. A Novel Voltammetric Sensor Based on P-Aminothiophenol Functionalized Graphene Oxide/Gold Nanoparticles for Determining Quercetin in the Presence of Ascorbic Acid. J. Electroanal. Chem. 2013, 698, 9–16. [Google Scholar] [CrossRef]
- Vilian, A.T.E.; Puthiaraj, P.; Kwak, C.H.; Choe, S.R.; Huh, Y.S.; Ahn, W.S.; Han, Y.K. Electrochemical Determination of Quercetin Based on Porous Aromatic Frameworks Supported Au Nanoparticles. Electrochim. Acta 2016, 216, 181–187. [Google Scholar] [CrossRef]
- Yola, M.L.; Atar, N. A Novel Voltammetric Sensor Based on Gold Nanoparticles Involved in P-Aminothiophenol Functionalized Multi-Walled Carbon Nanotubes: Application to the Simultaneous Determination of Quercetin and Rutin. Electrochim. Acta 2014, 119, 24–31. [Google Scholar] [CrossRef]
- Gupta, V.K.; Golestani, F.; Ahmadzadeh, S.; Karimi-Maleh, H.; Fazli, G.; Khosravi, S. NiO/CNTs Nanocomposite Modified Ionic Liquid Carbon Paste Electrode as a Voltammetric Sensor for Determination of Quercetin. Int. J. Electrochem. Sci. 2015, 10, 3657–3667. [Google Scholar] [CrossRef]
- Elçin, S.; Yola, M.L.; Eren, T.; Girgin, B.; Atar, N. Highly Selective and Sensitive Voltammetric Sensor Based on Ruthenium Nanoparticle Anchored Calix[4]Amidocrown-5 Functionalized Reduced Graphene Oxide: Simultaneous Determination of Quercetin, Morin and Rutin in Grape Wine. Electroanalysis 2016, 28, 611–619. [Google Scholar] [CrossRef]
- Leau, S.A.; Marin, M.; Toader, A.M.; Anastasescu, M.; Matei, C.; Lete, C.; Lupu, S. MeNPs-PEDOT Composite-Based Detection Platforms for Epinephrine and Quercetin. Biosensors 2024, 14, 320. [Google Scholar] [CrossRef]
- Bond, A.M. Past, Present and Future Contributions of Microelectrodes to Analytical Studies Employing Voltammetric Detection. A Review. Analyst 1994, 119, 1R–21R. [Google Scholar] [CrossRef]
- Zoski, C.G. Ultramicroelectrodes: Design, Fabrication, and Characterization. Electroanalysis 2002, 14, 1041–1051. [Google Scholar] [CrossRef]
- Xie, X.; Stueben, D.; Berner, Z. The Application of Microelectrodes for the Measurements of Trace Metals in Water. Anal. Lett. 2005, 38, 2281–2300. [Google Scholar] [CrossRef]
- Stulík, K.; Amatore, C.; Holub, K.; Marecek, V.; Kutner, W. Microelectrodes. Definitions, Characterization, and Applications (Technical Report). Pure Appl. Chem. 2000, 72, 1483–1492. [Google Scholar] [CrossRef]
- Magnuszewska, J.; Krogulec, T. Application of Hot Platinum Microelectrodes for Determination of Flavonoids in Flow Injection Analysis and Capillary Electrophoresis. Anal. Chim. Acta 2013, 786, 39–46. [Google Scholar] [CrossRef]
- Sokolová, R.; Degano, I.; Ramešová, Š.; Bulíčková, J.; Hromadová, M.; Gál, M.; Fiedler, J.; Valášek, M. The Oxidation Mechanism of the Antioxidant Quercetin in Nonaqueous Media. Electrochim. Acta 2011, 56, 7421–7427. [Google Scholar] [CrossRef]
- Guo, J.; Lindner, E. Cyclic Voltammograms at Coplanar and Shallow Recessed Microdisk Electrode Arrays: Guidelines for Design and Experiment. Anal. Chem. 2009, 81, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Yazdanshenas, R.; Gharib, F. Protonation Equilibria Studies of Quercetin in Aqueous Solutions of Ethanol and Dimethyl Sulphoxide. J. Mol. Liq. 2016, 224, 1227–1232. [Google Scholar] [CrossRef]
- Yazdanshenas, R.; Gharib, F. Thermodynamic Studies on Protonation Constant of Quercetin at Different Ionic Strengths. J. Solut. Chem. 2016, 45, 1246–1258. [Google Scholar] [CrossRef]
- Rutyna, I.; Korolczuk, M. Catalytic Adsorptive Stripping Voltammetry of Cobalt in the Presence of Nitrite at an In Situ Plated Bismuth Film Electrode. Electroanalysis 2011, 23, 637–641. [Google Scholar] [CrossRef]
- Rutyna, I. Determination of Folic Acid at a Bismuth Film Electrode by Adsorptive Stripping Voltammetry. Anal. Lett. 2015, 48, 1593–1603. [Google Scholar] [CrossRef]
- Brett, A.M.O.; Ghica, M.E. Electrochemical Oxidation of Quercetin. Electroanalysis 2003, 15, 1745–1750. [Google Scholar] [CrossRef]
- Abdel-Hamid, R.; Rabia, M.K.; Newair, E.F. Electrochemical Behaviour of Antioxidants: Part 2. Electrochemical Oxidation Mechanism of Quercetin at Glassy Carbon Electrode Modified with Multi-Wall Carbon Nanotubes. Arab. J. Chem. 2016, 9, 350–356. [Google Scholar] [CrossRef]
- Svancara, I.; Baldrianova, L.; Vlcek, M.; Metelka, R.; Vytras, K. A Role of the Plating Regime in the Deposition of Bismuth Films onto a Carbon Paste Electrode. Microscopic Study. Electroanalysis 2005, 17, 120–126. [Google Scholar] [CrossRef]
- Vladislavić, N.; Buzuk, M.; Brinić, S.; Buljac, M.; Bralić, M. Morphological Characterization of Ex Situ Prepared Bismuth Film Electrodes and Their Application in Electroanalytical Determination of the Biomolecules. J. Solid State Electrochem. 2016, 20, 2241–2250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interferent | A Molar Excess of Interferent | 1 Relative Signal of Quercetin ± SD |
|---|---|---|
| K+ | 500 | 1.04 ± 0.037 |
| Cl− | 500 | 1.03 ± 0.041 |
| Mg2+ | 500 | 1.05 ± 0.038 |
| Ca2+ | 500 | 0.98 ± 0.046 |
| NO3− | 500 | 0.99 ± 0.043 |
| NO2− | 500 | 0.95 ± 0.039 |
| Glucose | 1000 | 1.1 ± 0.036 |
| 10,000 | 0.91 ± 0.045 | |
| Folic acid | 200 | 0.85 ± 0.044 |
| Pyridoxine | 200 | 0.97 ± 0.042 |
| Ascorbic acid | 500 | 0.98 ± 0.035 |
| 10,000 | 1.07 ± 0.046 | |
| Urea | 1000 | 1.02 ± 0.033 |
| Hypoxanthine | 1000 | 0.99 ± 0.042 |
| Sample | Average Measured Content ± SD [mg] | Declared Value [mg] | Recovery ± RSD [%] |
| Pharmaceutical preparation | 60.4 ± 0.5 | 60.0 | 100.7 ± 0.9 |
| Sample | Added [nmol L−1] | Found average content ± SD [nmol L−1] | Recovery ± RSD [%] |
| Urine sample | 5.0 | 5.5 ± 0.2 | 110.0 ± 3.6 |
| 10 | 10.2 ± 0.3 | 102.0 ± 3.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gęca, I.; Korolczuk, M. Adsorptive Stripping Voltammetric Quercetin Determination in Pharmaceuticals and Urine Samples Using a Long Service-Life Array of Carbon Composite Microelectrodes. Molecules 2024, 29, 4464. https://doi.org/10.3390/molecules29184464
Gęca I, Korolczuk M. Adsorptive Stripping Voltammetric Quercetin Determination in Pharmaceuticals and Urine Samples Using a Long Service-Life Array of Carbon Composite Microelectrodes. Molecules. 2024; 29(18):4464. https://doi.org/10.3390/molecules29184464
Chicago/Turabian StyleGęca, Iwona, and Mieczyslaw Korolczuk. 2024. "Adsorptive Stripping Voltammetric Quercetin Determination in Pharmaceuticals and Urine Samples Using a Long Service-Life Array of Carbon Composite Microelectrodes" Molecules 29, no. 18: 4464. https://doi.org/10.3390/molecules29184464
APA StyleGęca, I., & Korolczuk, M. (2024). Adsorptive Stripping Voltammetric Quercetin Determination in Pharmaceuticals and Urine Samples Using a Long Service-Life Array of Carbon Composite Microelectrodes. Molecules, 29(18), 4464. https://doi.org/10.3390/molecules29184464