
Ferroptosis in Ischemic Stroke and Related Traditional Chinese Medicines

 , ,
, ,
Abstract
1. Introduction
2. Ischemic Stroke
2.1. Epidemiology of Stroke
2.2. Etiology and Pathogenesis of Ischemic Stroke
2.3. Clinical Manifestations and Treatment of Ischemic Stroke
3. Ferroptosis and Ischemic Stroke
3.1. Indicators of Ferroptosis
3.1.1. Accumulation of Lipid Peroxides
3.1.2. Iron Accumulation
3.2. The Classic Pathway of Ferroptosis
3.2.1. System Xc−/GPX4 Axis
3.2.2. FSP1/CoQ Axis
3.2.3. GCH1/BH4/DHFR Axis
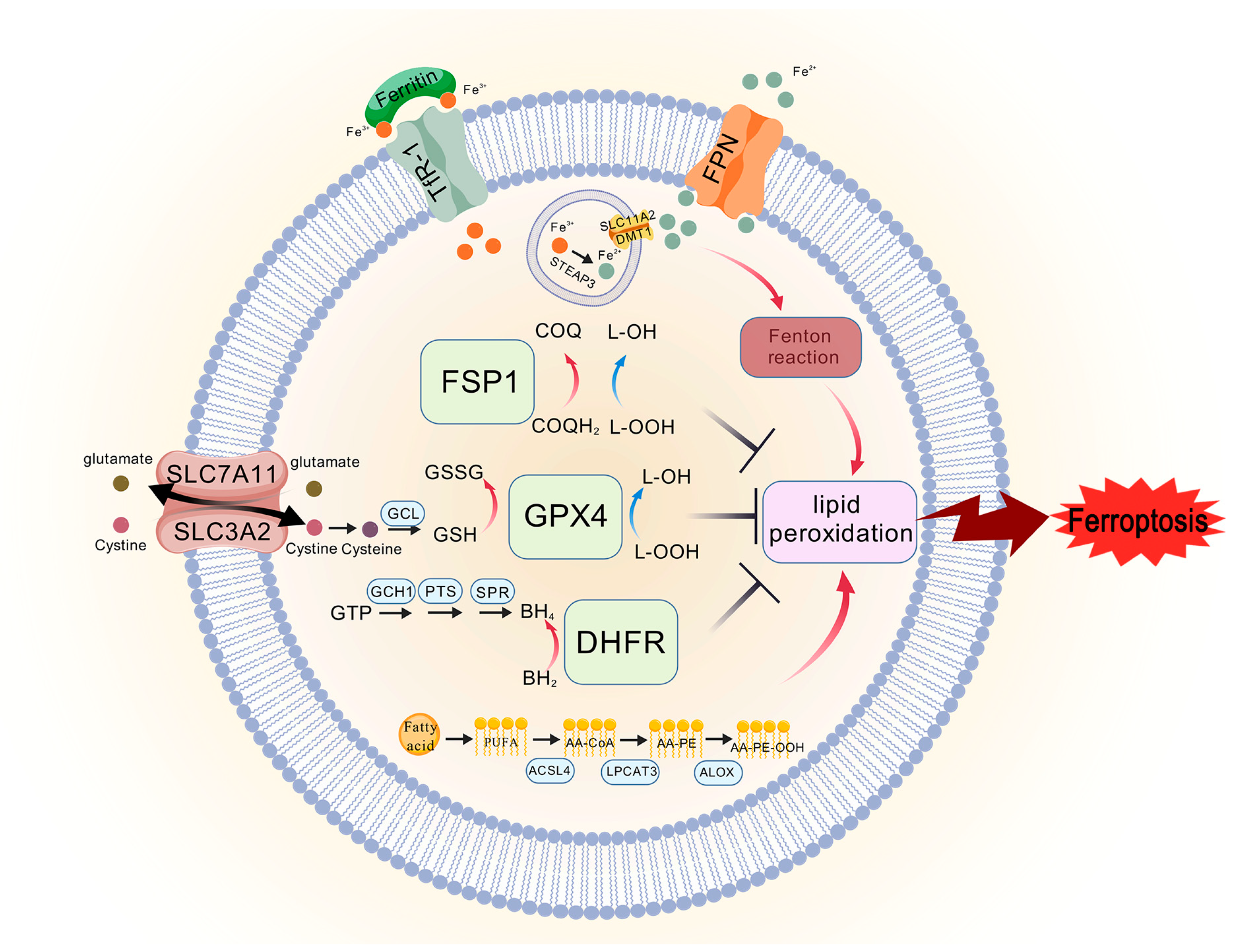
3.3. Inducers and Inhibitors of Ferroptosis
3.3.1. Inducers of Ferroptosis
3.3.2. Inhibitors of Ferroptosis
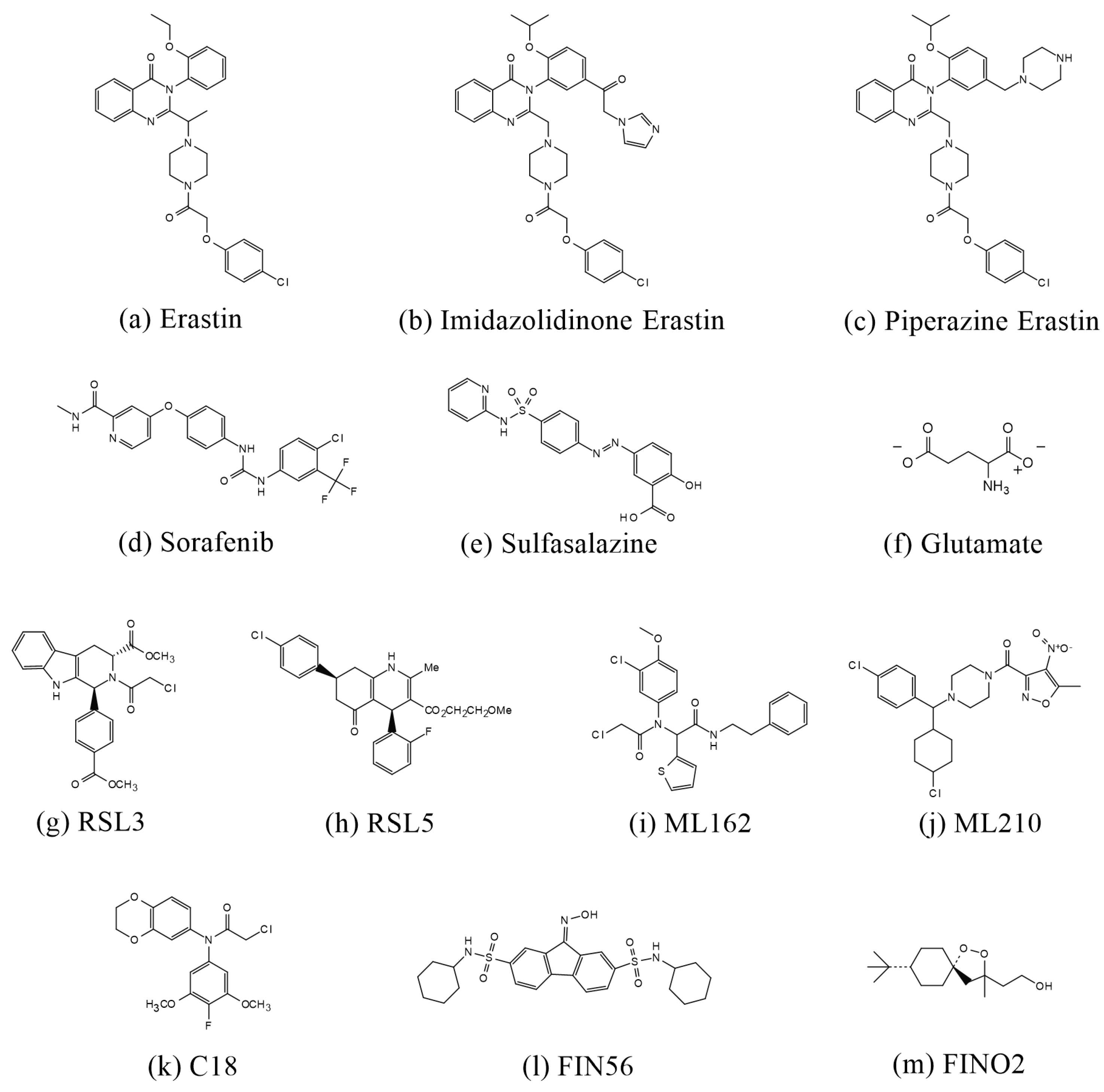
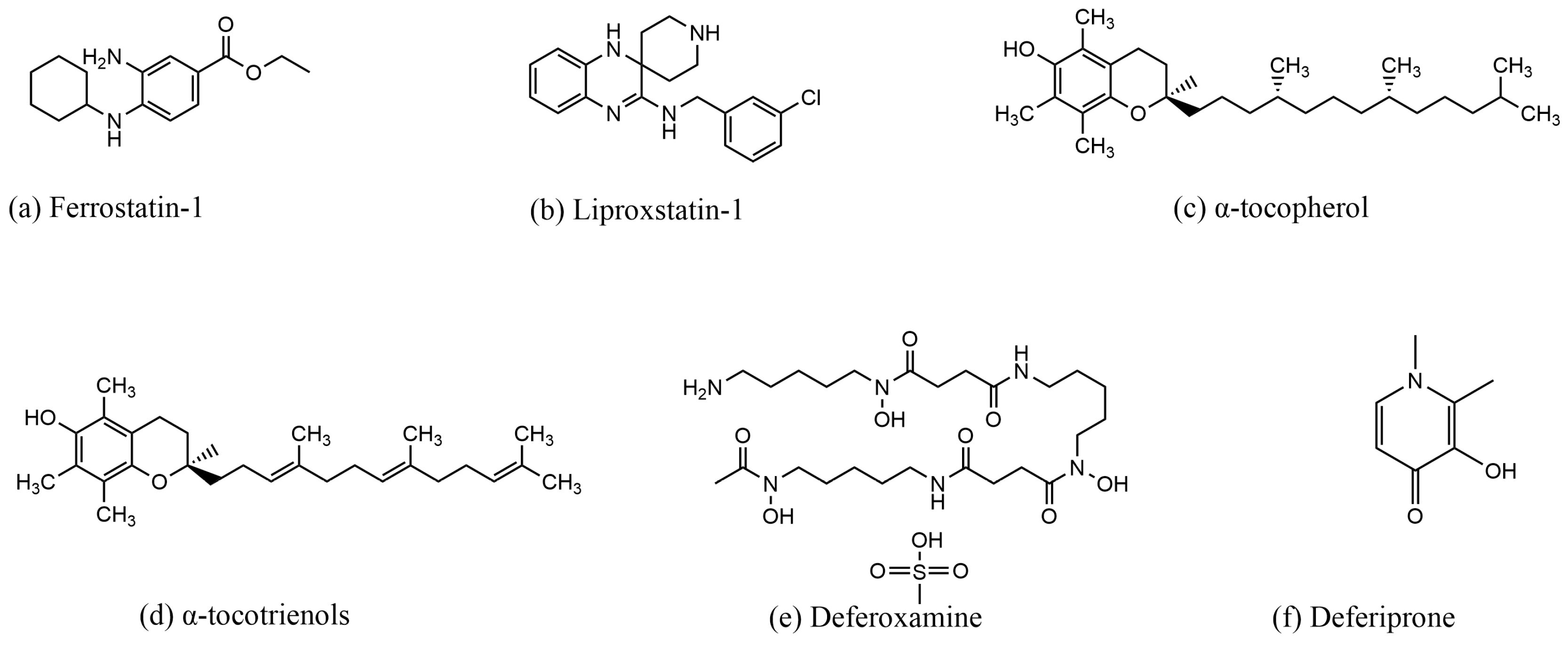
4. Advances in the Reduction in Ferroptosis by Chinese Herbs and Natural Products in the Treatment of Ischemic Stroke
4.1. Regulation of Ferroptosis by Nrf2
4.2. Regulation of Ferroptosis by System Xc−/GPX4 Axis
4.3. Regulation of Ferroptosis through the PI3K/AKT Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Ingredient or Formula | Source | Functional Mechanism | Experimental Models |
|---|---|---|---|
| β-Caryophyllene | lemon, nutmeg, pepper, clove, etc.; | Nrf2/HO-1 | MCAO/R in male SD rats and primary astrocytes treated with OGD/R [85] |
| Vitexin | leaves of Vitex cannabifolia; widely found in Vitex cannabifolia seeds, Vitex cannabifolia leaves, Phyllostachys nigra bamboo leaves, Pennisetum millet, chaste tree, Hawthorn, and Passion Flower, among others | Keap1/Nrf2/HO-1 | MCAO/R in male SD rats and primary cortical neuron cells treated with OGD/R [88] |
| Quercetin | coriander, onion, forsythia, okra, etc. | Nrf2/HO-1 | MCAO in male SD rats; H2O2 or erastin induce HT22 cell ferroptosis [92] |
| Rhein | Rheum palmatum L., Cassia tora L., Polygonum multiflorum Thunb., and Aloe barbadensis Miller | Nrf2/SLC7A11/GPX4 | MCAO in male SD rats and HT22 cells treated with OGD/R [93] |
| Gastrodin | gastrodia elata Blume | Nrf2/Keap1/GPX4, Nrf2/HO-1 | BCCAO to establish vascular dementia models in male SD rats; HT22 cells establish a cell model of hypoxia injury [94,95] |
| Neutral polysaccharide of gastrodia elata Blume | gastrodia elata Blume | Nrf2/HO-1 | MCAO models in C57BL/6 J mice, HT22 cells treated with OGD/R [96] |
| Loureirin C | Dragon’s blood | Nrf2/GPX4 | MCAO/R model in C57BL/6 mice, SH-SY5Y cells treated with OGD/R [97] |
| Icariside II (ICS II) | Herba Epimedii | Nrf2/OXPHOS/NF-κB | MCAO models in C57BL/6 male mice and primary astrocyte treated with OGD/R [98] |
| Astragaloside IV (AST IV) | Astragalus membranaceus Bunge | P62/Keap1/Nrf2 | MCAO/R models in male SD mice and SH-SY5Y cells treated with erastin or OGD/R [101] |
| Rehmannioside A | Rehmannia glutinosa Libosch | PI3K/Akt/Nrf2, SLC7A11/GPX4 | MCAO models in male SD mice; H2O2-induced oxidative stress damage in SH—SY5Y [102] |
| 15,16-Dihydrotanshinone (DHT) | Salvia miltiorrhiza Bunge | Nrf2/GPX4 | PC12 cells and pMCAO models in male SD mice [104] |
| Dihydromyricetin | Ampelopsis grossedentata (Chinese vine tea), Hovenia dulcis (Japanese raisin tree), and some pinus and Cedrus species | Regulating the Expression of GPX4, inhibiting the SPHK1/mTOR signaling pathway | MCAO/R in male SD rats and HT22 cells treated with OGD/R [106] |
| Baicalein | Scutellaria baicalensis Georgi | GPX4/ACSL4/ACSL3 | HT22 cells treated with OGD/R and tMCAO models in C57BL/6 male mice [107] |
| Galangin | Alpinia officinarum Hance | SLC7A11/GPX4 | Using bilateral common carotid artery ligation in male gerbils established a cerebral ischemia model; hippocampal neuron cells treated with OGD [108] |
| Traditional Chinese Medicine | Active Ingredient or Formula | Functional Mechanism | Experimental Models |
|---|---|---|---|
| Naotaifang | Radix Astragali (Huangqi), Rhizoma chuanxiong (Chuangxiong), Pheretima (Dilong), and Bombyx batryticatus (Jiangcan) | TFR1/DMT1, SLC7A1/GPX4 | MCAO model in SD rats [109] |
| Danlou Tablet | Trichosanthes kirilowii Maxim, Salvia miltiorrhiza Bunge, Ligusticum chuanxiong Hort, Allium macrostemon Bunge, Paeonia lactiflora Pall, Pueraria lobata (Willd.) Ohwi, Alisma plantago-aquatica L., Astragalus membranaceus (Fisch.) Bunge, Davallia mariesii T. Moore ex Baker, and Curcuma aeruginosa Roxb | SLC7A11/GPX4 | tMCAO model in male C57BL/6 mice and hy926 cell line treated with OGD/R [110] |
| Xingnaojing Injection | Musk, Borneolum, Radix curcumae, and Fructusgardenia | Upregulating GPX4, FPN, and HO-1 expression and downregulating COX-2, TFR1, and DMT1 expression | MCAO model in male SD rats; SH-SY5Y human neuroblastoma cells establish a hypoxia cell model [111] |
| Danhong injection | Salvia miltiorrhiza (Dan Shen) and Carthamus tinctorius (Hong Hua) | SATB1/SLC7A11/HO-1 | pMCAO model in C57BL/6 mice, and HT22 and primary cortical neuron cells treated with OGD [112] |
| Angong Niuhuang Wan | Calculus bovis, powder of Cornu bubali, Moschus, Margarita, Cinnabaris, Realgar, Coptis chinensis Franch., Scutellaria baicalensis Georgi, Gardenia jasminoides J. Ellis, Curcuma aromatica Salisb., and Borneolum synthcticum | PPAR and PI3K/Akt | MCAO/R and ICH models in SD rats; erastin induces PC 12 cell ferroptosis [115] |
| Paeoniae Radix | Extract of Paeoniae Radix Rubra | PI3K/Akt | MCAO models in male SD mice; H2O2-induced oxidative stress damage in HT22 cells [117] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tu, W.J.; Wang, L.D.; Special Writing Group of China Stroke Surveillance, R. China stroke surveillance report 2021. Mil. Med. Res. 2023, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Bogousslavsky, J.; Caplan, L.R.; Donnan, G.A.; Hennerici, M.G. Classification of stroke subtypes. Cerebrovasc. Dis. 2009, 27, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Guada, L.; Yavagal, D.R. Global Epidemiology of Stroke and Access to Acute Ischemic Stroke Interventions. Neurology 2021, 97 (Suppl. S2), S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Li, Z.X.; Gu, H.Q.; Zhai, Y.; Zhou, Q.; Jiang, Y.; Zhao, X.Q.; Wang, Y.L.; Yang, X.; Wang, C.J.; et al. China Stroke Statistics: An update on the 2019 report from the National Center for Healthcare Quality Management in Neurological Diseases, China National Clinical Research Center for Neurological Diseases, the Chinese Stroke Association, National Center for Chronic and Non-communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention and Institute for Global Neuroscience and Stroke Collaborations. Stroke Vasc. Neurol. 2022, 7, 415–450. [Google Scholar] [PubMed]
- Tuo, Q.Z.; Zhang, S.T.; Lei, P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med. Res. Rev. 2022, 42, 259–305. [Google Scholar] [CrossRef]
- Herpich, F.; Rincon, F. Management of Acute Ischemic Stroke. Crit. Care Med. 2020, 48, 1654–1663. [Google Scholar] [CrossRef]
- Mendelson, S.J.; Prabhakaran, S. Diagnosis and Management of Transient Ischemic Attack and Acute Ischemic Stroke: A Review. JAMA 2021, 325, 1088–1098. [Google Scholar] [CrossRef]
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Chen, X.; Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment (Review). Int. J. Mol. Med. 2022, 49, 15. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J. Exp. Med. 1955, 102, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. Amino acid metabolism in mammalian cell cultures. Science 1959, 130, 432–437. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Wu, S.; Wu, B.; Liu, M.; Chen, Z.; Wang, W.; Anderson, C.S.; Sandercock, P.; Wang, Y.; Huang, Y.; Cui, L.; et al. Stroke in China: Advances and challenges in epidemiology, prevention, and management. Lancet Neurol. 2019, 18, 394–405. [Google Scholar] [CrossRef]
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar]
- Ovbiagele, B.; Goldstein, L.B.; Higashida, R.T.; Howard, V.J.; Johnston, S.C.; Khavjou, O.A.; Lackland, D.T.; Lichtman, J.H.; Mohl, S.; Sacco, R.L.; et al. Forecasting the future of stroke in the United States: A policy statement from the American Heart Association and American Stroke Association. Stroke 2013, 44, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.; Werring, D.J. Stroke: Causes and clinical features. Medicine 2020, 48, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Kamtchum-Tatuene, J.; Jickling, G.C. Blood Biomarkers for Stroke Diagnosis and Management. NeuroMol. Med. 2019, 21, 344–368. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Marotta, C.A. Outcomes validity and reliability of the modified Rankin scale: Implications for stroke clinical trials: A literature review and synthesis. Stroke 2007, 38, 1091–1096. [Google Scholar] [CrossRef]
- Papanagiotou, P.; White, C.J. Endovascular Reperfusion Strategies for Acute Stroke. JACC Cardiovasc. Interv. 2016, 9, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef]
- Guo, J.; Tuo, Q.Z.; Lei, P. Iron, ferroptosis, and ischemic stroke. J. Neurochem. 2023, 165, 487–520. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef]
- Gochenauer, G.E.; Robinson, M.B. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc(-) subunits. J. Neurochem. 2001, 78, 276–286. [Google Scholar] [CrossRef]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remiao, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Belalcazar, A.D.; Ball, J.G.; Frost, L.M.; Valentovic, M.A.; Wilkinson, J. Transsulfuration Is a Significant Source of Sulfur for Glutathione Production in Human Mammary Epithelial Cells. ISRN Biochem. 2014, 2013, 637897. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Wei, X.; Yi, X.; Zhu, X.H.; Jiang, D.S. Posttranslational Modifications in Ferroptosis. Oxidative Med. Cell. Longev. 2020, 2020, 8832043. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Yan, R.; Xie, E.; Li, Y.; Li, J.; Zhang, Y.; Chi, X.; Hu, X.; Xu, L.; Hou, T.; Stockwell, B.R.; et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 2022, 32, 687–690. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e9. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Lv, L.; Lu, T.; Ding, M.; Yu, Z.; Chen, X.; Zhou, X.; Wang, X. alpha-KG inhibits tumor growth of diffuse large B-cell lymphoma by inducing ROS and TP53-mediated ferroptosis. Cell Death Discov. 2023, 9, 182. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Hu, W.; Chen, M.; Wang, W.; Huang, F.; Tian, X.; Xie, L. Pomelo Peel Essential Oil Ameliorates Cerebral Ischemia-Reperfusion Injury through Regulating Redox Homeostasis in Rats and SH-SY5Y Cells. Oxidative Med. Cell Longev. 2022, 2022, 8279851. [Google Scholar] [CrossRef]
- Weiwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Faraji, P.; Borchert, A.; Ahmadian, S.; Kuhn, H. Butylated Hydroxytoluene (BHT) Protects SH-SY5Y Neuroblastoma Cells from Ferroptotic Cell Death: Insights from In Vitro and In Vivo Studies. Antioxidants 2024, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Leng, J.; Tan, J.; Zhao, Y.; Xie, S.; Zhao, S.; Yan, X.; Zhu, L.; Luo, J.; Kong, L.; et al. Discovery of Novel Potent Covalent Glutathione Peroxidase 4 Inhibitors as Highly Selective Ferroptosis Inducers for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2023, 66, 10036–10059. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Sun, Y.; Berleth, N.; Wu, W.; Schlutermann, D.; Deitersen, J.; Stuhldreier, F.; Berning, L.; Friedrich, A.; Akgun, S.; Mendiburo, M.J.; et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 2021, 12, 1028. [Google Scholar] [CrossRef]
- Abrams, R.P.; Carroll, W.L.; Woerpel, K.A. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem. Biol. 2016, 11, 1305–1312. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Chu, J.; Liu, C.X.; Song, R.; Li, Q.L. Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen. Res. 2020, 15, 528–536. [Google Scholar]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.M.; Bishop, K.S. On the Existence of a Hitherto Unrecognized Dietary Factor Essential for Reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Peh, H.Y.; Tan, W.S.; Liao, W.; Wong, W.S. Vitamin E therapy beyond cancer: Tocopherol versus tocotrienol. Pharmacol. Ther. 2016, 162, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Diverse cytoprotective actions of vitamin E isoforms—role as peroxyl radical scavengers and complementary functions with selenoproteins. Free Radic. Biol. Med. 2021, 175, 121–129. [Google Scholar] [CrossRef]
- Atkinson, J.; Harroun, T.; Wassall, S.R.; Stillwell, W.; Katsaras, J. The location and behavior of alpha-tocopherol in membranes. Mol. Nutr. Food Res. 2010, 54, 641–651. [Google Scholar] [CrossRef]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef]
- Hershko, C. Iron chelators in medicine. Mol. Asp. Med. 1992, 13, 113–165. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, X.; Lu, D.; Yin, M.; Shan, M.; Gao, Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021, 36, 951–964. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, J.; Sun, K.; Guo, J.; Yao, X.; Wang, G.; Hou, L.; Xu, J.; Guo, J.; Guo, F. Deferoxamine Alleviates Osteoarthritis by Inhibiting Chondrocyte Ferroptosis and Activating the Nrf2 Pathway. Front. Pharmacol. 2022, 13, 791376. [Google Scholar] [CrossRef]
- Yuan, S.; Wei, C.; Liu, G.; Zhang, L.; Li, J.; Li, L.; Cai, S.; Fang, L. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1alpha/SLC7A11 pathway. Cell Prolif. 2022, 55, e13158. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J. New orally active iron chelators. Lancet 1985, 1, 817. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J. Deferiprone: A Forty-Year-Old Multi-Targeting Drug with Possible Activity against COVID-19 and Diseases of Similar Symptomatology. Int. J. Mol. Sci. 2022, 23, 6735. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Peng, J.; Zhang, E.; Ji, D.; Gao, Z.; Tang, Y.; Yao, X.; Xia, X. Pathologically high intraocular pressure disturbs normal iron homeostasis and leads to retinal ganglion cell ferroptosis in glaucoma. Cell Death Differ. 2023, 30, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Nobuta, H.; Yang, N.; Ng, Y.H.; Marro, S.G.; Sabeur, K.; Chavali, M.; Stockley, J.H.; Killilea, D.W.; Walter, P.B.; Zhao, C.; et al. Oligodendrocyte Death in Pelizaeus-Merzbacher Disease Is Rescued by Iron Chelation. Cell Stem Cell 2019, 25, 531–541.e6. [Google Scholar] [CrossRef]
- Hu, Q.; Zuo, T.; Deng, L.; Chen, S.; Yu, W.; Liu, S.; Liu, J.; Wang, X.; Fan, X.; Dong, Z. beta-Caryophyllene suppresses ferroptosis induced by cerebral ischemia reperfusion via activation of the NRF2/HO-1 signaling pathway in MCAO/R rats. Phytomedicine 2022, 102, 154112. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.A.; Martins, N.M.; Sisti, F.M.; Fernandes, L.S.; Ferreira, R.S.; de Freitas, O.; Santos, A.C. The cannabinoid beta-caryophyllene (BCP) induces neuritogenesis in PC12 cells by a cannabinoid-receptor-independent mechanism. Chem. Biol. Interact. 2017, 261, 86–95. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Yang, Z.B.; Zhang, R.; Luo, X.J.; Peng, J. The Protective Effect of Vitexin Compound B-1 on Rat Cerebral I/R Injury through a Mechanism Involving Modulation of miR-92b/NOX4 Pathway. CNS Neurol. Disord. Drug Targets 2023, 22, 137–147. [Google Scholar]
- Guo, L.; Shi, L. Vitexin Improves Cerebral ischemia—reperfusion Injury by Attenuating Oxidative Injury and Ferroptosis via Keap1/Nrf2/HO-1signaling. Neurochem. Res. 2023, 48, 980–995. [Google Scholar] [CrossRef]
- Javadinia, S.S.; Abbaszadeh-Goudarzi, K.; Mahdian, D.; Hosseini, A.; Ghalenovi, M.; Javan, R. A review of the protective effects of quercetin-rich natural compounds for treating ischemia-reperfusion injury. Biotech. Histochem. 2022, 97, 237–246. [Google Scholar] [CrossRef]
- Yang, R.; Shen, Y.J.; Chen, M.; Zhao, J.Y.; Chen, S.H.; Zhang, W.; Song, J.K.; Li, L.; Du, G.H. Quercetin attenuates ischemia reperfusion injury by protecting the blood-brain barrier through Sirt1 in MCAO rats. J. Asian Nat. Prod. Res. 2022, 24, 278–289. [Google Scholar] [CrossRef]
- Baccan, M.M.; Chiarelli-Neto, O.; Pereira, R.M.; Esposito, B.P. Quercetin as a shuttle for labile iron. J. Inorg. Biochem. 2012, 107, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ai, Q.; Zhao, F.; Li, H.; Sun, Y.; Tang, K.; Yang, Y.; Chen, N.; Liu, F. Quercetin attenuates cerebral ischemic injury by inhibiting ferroptosis via Nrf2/HO-1 signaling pathway. Eur. J. Pharmacol. 2024, 963, 176264. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, T.A.; Zhang, W.Y.; Huang, S.R.; Hu, Y.; Sun, J. Rhein attenuates cerebral ischemia-reperfusion injury via inhibition of ferroptosis through NRF2/SLC7A11/GPX4 pathway. Exp. Neurol. 2023, 369, 114541. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, E.; Yang, H.; Chen, Y.; Tao, L.; Xu, Y.; Chen, T.; Shen, X. Gastrodin Ameliorates Cognitive Dysfunction in Vascular Dementia Rats by Suppressing Ferroptosis via the Regulation of the Nrf2/Keap1-GPx4 Signaling Pathway. Molecules 2022, 27, 6311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Cheng, H.; Su, J.; Wang, X.; Wang, Q.; Chu, J.; Li, Q. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. in Vitro 2020, 62, 104715. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, P.; Zhu, H.; Gu, L.; Li, Y.; Feng, S.; Zeng, Z.; Chen, Q.; Zhou, B.; Xiong, X. Neutral polysaccharide from Gastrodia elata alleviates cerebral ischemia-reperfusion injury by inhibiting ferroptosis-mediated neuroinflammation via the NRF2/HO-1 signaling pathway. CNS Neurosci. Ther. 2024, 30, e14456. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Y.; Wang, Y.; Meng, Q.; Xu, L.; Liu, Y.; Zhou, D.; Wang, Y.; Liang, D.; Li, W.; et al. Loureirin C inhibits ferroptosis after cerebral ischemia reperfusion through regulation of the Nrf2 pathway in mice. Phytomedicine 2023, 113, 154729. [Google Scholar] [CrossRef]
- Gao, J.; Ma, C.; Xia, D.; Chen, N.; Zhang, J.; Xu, F.; Li, F.; He, Y.; Gong, Q. Icariside II preconditioning evokes robust neuroprotection against ischaemic stroke, by targeting Nrf2 and the OXPHOS/NF-kappaB/ferroptosis pathway. Br. J. Pharmacol. 2023, 180, 308–329. [Google Scholar] [CrossRef]
- Feng, L.; Gao, J.; Liu, Y.; Shi, J.; Gong, Q. Icariside II alleviates oxygen-glucose deprivation and reoxygenation-induced PC12 cell oxidative injury by activating Nrf2/SIRT3 signaling pathway. Biomed. Pharmacother. 2018, 103, 9–17. [Google Scholar] [CrossRef]
- Li, L.; Hou, X.; Xu, R.; Liu, C.; Tu, M. Research review on the pharmacological effects of astragaloside IV. Fundam. Clin. Pharmacol. 2017, 31, 17–36. [Google Scholar] [CrossRef]
- Wang, L.; Liu, C.; Wang, L.; Tang, B. Astragaloside IV mitigates cerebral ischaemia-reperfusion injury via inhibition of P62/Keap1/Nrf2 pathway-mediated ferroptosis. Eur. J. Pharmacol. 2023, 944, 175516. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Wu, Y.; Liu, S.; Luo, C.; Lu, Y.; Liu, M.; Wang, L.; Zhang, Y.; Liu, X. Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J. Ethnopharmacol. 2022, 289, 115021. [Google Scholar] [CrossRef] [PubMed]
- Su, C.Y.; Ming, Q.L.; Rahman, K.; Han, T.; Qin, L.P. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 2015, 13, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Duan, F.; Yang, R.; Dai, Y.; Chen, X.; Li, S. 15, 16-Dihydrotanshinone I protects against ischemic stroke by inhibiting ferroptosis via the activation of nuclear factor erythroid 2-related factor 2. Phytomedicine 2023, 114, 154790. [Google Scholar] [CrossRef]
- Dihydromyricetin. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. [Google Scholar]
- Xie, J.; Zhang, T.; Li, P.; Wang, D.; Liu, T.; Xu, S. Dihydromyricetin Attenuates Cerebral Ischemia Reperfusion Injury by Inhibiting SPHK1/mTOR Signaling and Targeting Ferroptosis. Drug Des. Dev. Ther. 2022, 16, 3071–3085. [Google Scholar] [CrossRef]
- Li, M.; Meng, Z.; Yu, S.; Li, J.; Wang, Y.; Yang, W.; Wu, H. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem. Biol. Interact. 2022, 366, 110137. [Google Scholar] [CrossRef]
- Guan, X.; Li, Z.; Zhu, S.; Cheng, M.; Ju, Y.; Ren, L.; Yang, G.; Min, D. Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sci. 2021, 264, 118660. [Google Scholar] [CrossRef]
- Lan, B.; Ge, J.W.; Cheng, S.W.; Zheng, X.L.; Liao, J.; He, C.; Rao, Z.Q.; Wang, G.Z. Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J. Integr. Med. 2020, 18, 344–350. [Google Scholar] [CrossRef]
- Liu, C.; Liu, E.; Li, Z.; Li, W.; Jin, J.; Sui, H.; Chen, G.; Sun, Z.; Xi, H. Danlou tablet attenuates ischemic stroke injury and blood—brain barrier damage by inhibiting ferroptosis. J. Ethnopharmacol. 2024, 322, 117657. [Google Scholar] [CrossRef]
- Liu, H.; An, N.; Wang, L.; Li, Y.; Song, K.; Sun, Y.; Gao, Y. Protective effect of Xingnaojing injection on ferroptosis after cerebral ischemia injury in MCAO rats and SH-SY5Y cells. J. Ethnopharmacol. 2023, 301, 115836. [Google Scholar] [CrossRef]
- Zhan, S.; Liang, J.; Lin, H.; Cai, J.; Yang, X.; Wu, H.; Wei, J.; Wang, S.; Xian, M. SATB1/SLC7A11/HO-1 Axis Ameliorates Ferroptosis in Neuron Cells After Ischemic Stroke by Danhong Injection. Mol. Neurobiol. 2023, 60, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yan, S.; Xu, L.; Zhu, G.; Yu, X.; Tong, X. Use of angong niuhuang in treating central nervous system diseases and related research. Evid. Based Complement. Alternat. Med. 2014, 2014, 346918. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, B.; Chen, X.; Gao, C.; Wang, S.; Yuen, S.C.; Yang, D.; Shen, J. Neuroprotective Effects and Hepatorenal Toxicity of Angong Niuhuang Wan Against Ischemia-Reperfusion Brain Injury in Rats. Front. Pharmacol. 2019, 10, 593. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zheng, E.; Tong, L.; Liu, Y.; Li, X.; Yang, H.; Jiang, J.; Chang, Z.; Yang, H. Angong Niuhuang Wan inhibit ferroptosis on ischemic and hemorrhagic stroke by activating PPARgamma/AKT/GPX4 pathway. J. Ethnopharmacol. 2024, 321, 117438. [Google Scholar] [CrossRef]
- He, D.Y.; Dai, S.M. Anti-inflammatory and immunomodulatory effects of paeonia lactiflora pall. a traditional chinese herbal medicine. Front. Pharmacol. 2011, 2, 10. [Google Scholar] [CrossRef]
- Zhao, F.; Peng, C.; Li, H.; Chen, H.; Yang, Y.; Ai, Q.; Chen, N.; Liu, F. Paeoniae Radix Rubra extract attenuates cerebral ischemia injury by inhibiting ferroptosis and activating autophagy through the PI3K/Akt signalling pathway. J. Ethnopharmacol. 2023, 315, 116567. [Google Scholar] [CrossRef]
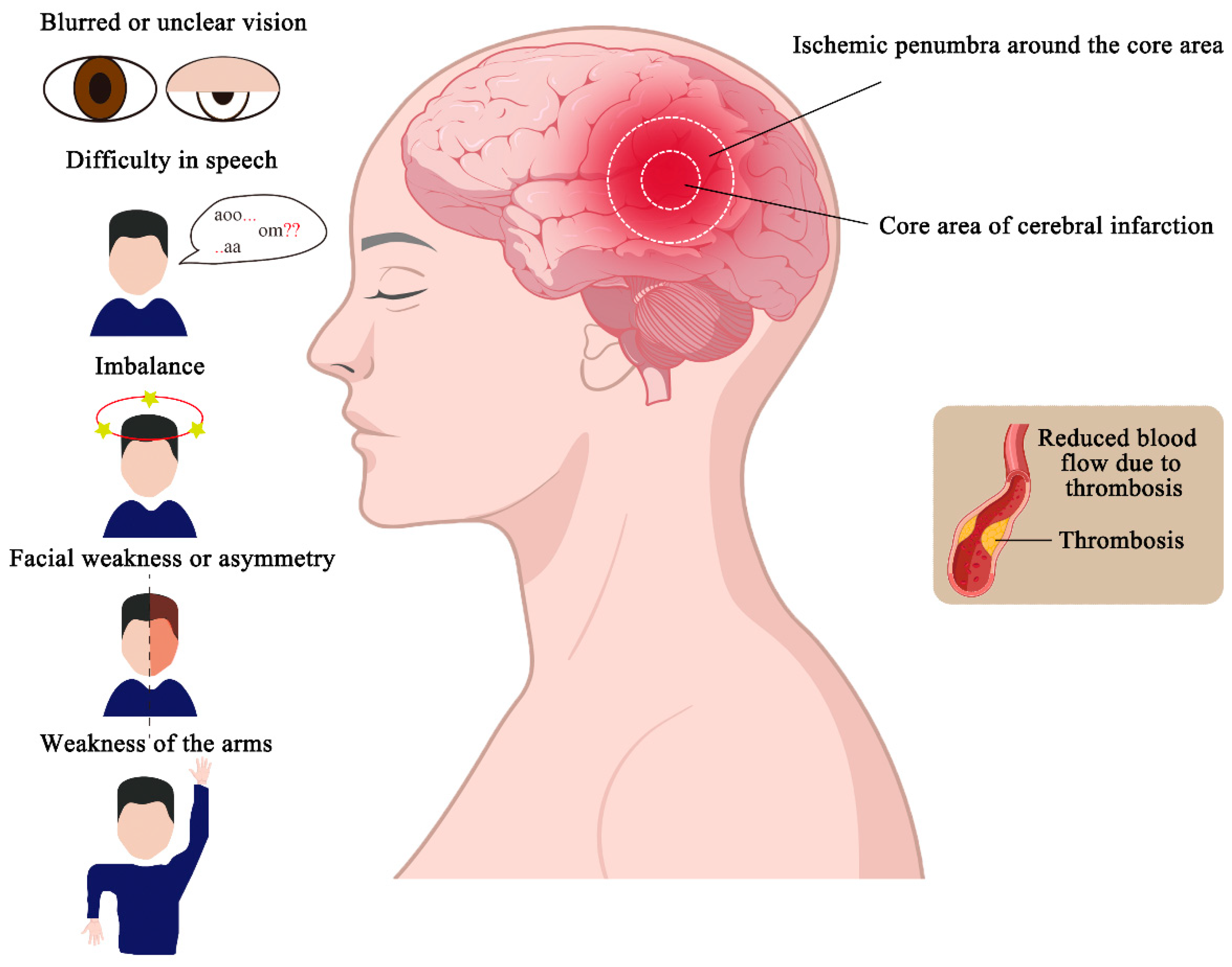
| Classification | Drugs or Compounds | Functional Mechanism |
|---|---|---|
| Inducer | Erastin | Inhibits system Xc−, prevents cystine import, and reduces GSH levels [14,47] |
| Imidazolidinone Erastin | Inhibits system Xc−, prevents cystine import, and reduces GSH levels [47] | |
| Piperazine Erastin | Inhibits system Xc−, prevents cystine import, and reduces GSH levels [47,48,49] | |
| Sorafenib | Inhibits system Xc−, prevents cystine import, and reduces GSH levels [50] | |
| Salazosulfapyridine | Inhibits system Xc−, prevents cystine import, and reduces GSH levels [51,52] | |
| Glutamate | Inhibits system Xc−, prevents cystine import, and reduces GSH levels [14,38,53] | |
| RSL3 | Binding to GPX4 leads to GPX4 inactivation [38,48,57] | |
| RSL5 | Regulates iron accumulation by VADCS and promotes lipid peroxide accumulation [38,48,56] | |
| ML162 and ML210 | Binding to GPX4 leads to GPX4 inactivation [59,60,61] | |
| C18 | Covalently binds to GPX4 and inhibits GPX4 activity [62] | |
| FIN56 | Promotes GPX4 degradation, binds and activates SQS, and depletes coenzyme Q10 [63,64,65] | |
| FINO2 | Oxidizes Fe2+ and promotes iron accumulation [63,66,67] | |
| Inhibitor | Ferrostatin-1 | Inhibits the production of lipid ROS [14,68,69] |
| Liproxstatin-1 | Scavenges lipid peroxides [38,70,71] | |
| α-tocopherol | Scavenges lipid peroxides [73,74,75] | |
| α-tocotrienols | Scavenges lipid peroxides [38,73,74] | |
| Deferoxamine | Chelates Fe3+ and reduces iron concentration [38,76,79,80] | |
| Deferiprone | Chelates Fe3+ and reduces iron concentration [81,82,83,84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, R.; Sun, X.; Liu, Z.; Zhang, J.; Yang, G.; Tian, J.; Wang, Y. Ferroptosis in Ischemic Stroke and Related Traditional Chinese Medicines. Molecules 2024, 29, 4359. https://doi.org/10.3390/molecules29184359
Ma R, Sun X, Liu Z, Zhang J, Yang G, Tian J, Wang Y. Ferroptosis in Ischemic Stroke and Related Traditional Chinese Medicines. Molecules. 2024; 29(18):4359. https://doi.org/10.3390/molecules29184359
Chicago/Turabian StyleMa, Runchen, Xiaohui Sun, Zhaofeng Liu, Jianzhao Zhang, Gangqiang Yang, Jingwei Tian, and Yunjie Wang. 2024. "Ferroptosis in Ischemic Stroke and Related Traditional Chinese Medicines" Molecules 29, no. 18: 4359. https://doi.org/10.3390/molecules29184359
APA StyleMa, R., Sun, X., Liu, Z., Zhang, J., Yang, G., Tian, J., & Wang, Y. (2024). Ferroptosis in Ischemic Stroke and Related Traditional Chinese Medicines. Molecules, 29(18), 4359. https://doi.org/10.3390/molecules29184359