
A Stereoselective Synthesis of a Novel α,β-Unsaturated Imine-Benzodiazepine through Condensation Reaction, Crystal Structure, and DFT Calculations

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
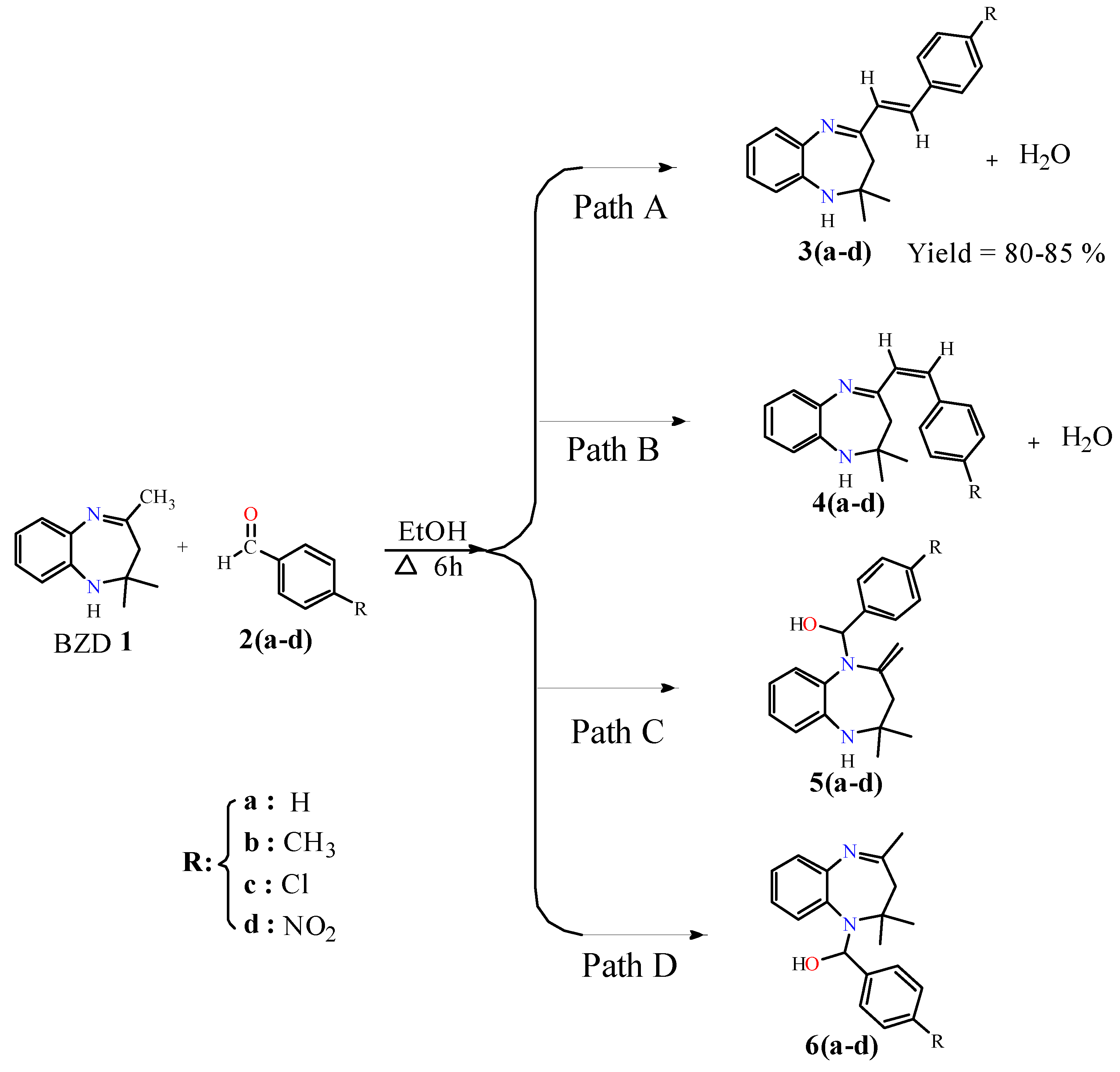
Synthesis
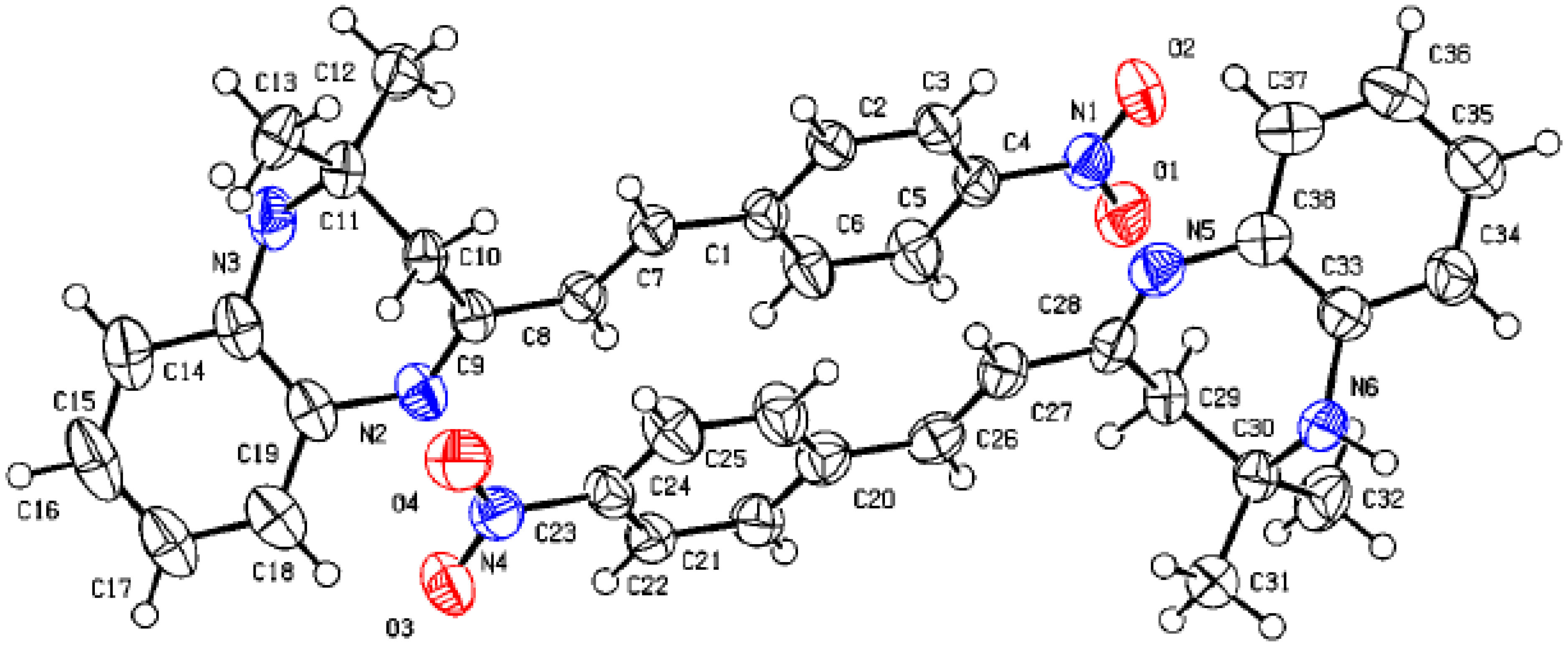
3. Crystal Structure of Compound 3d
4. Theoretical Study
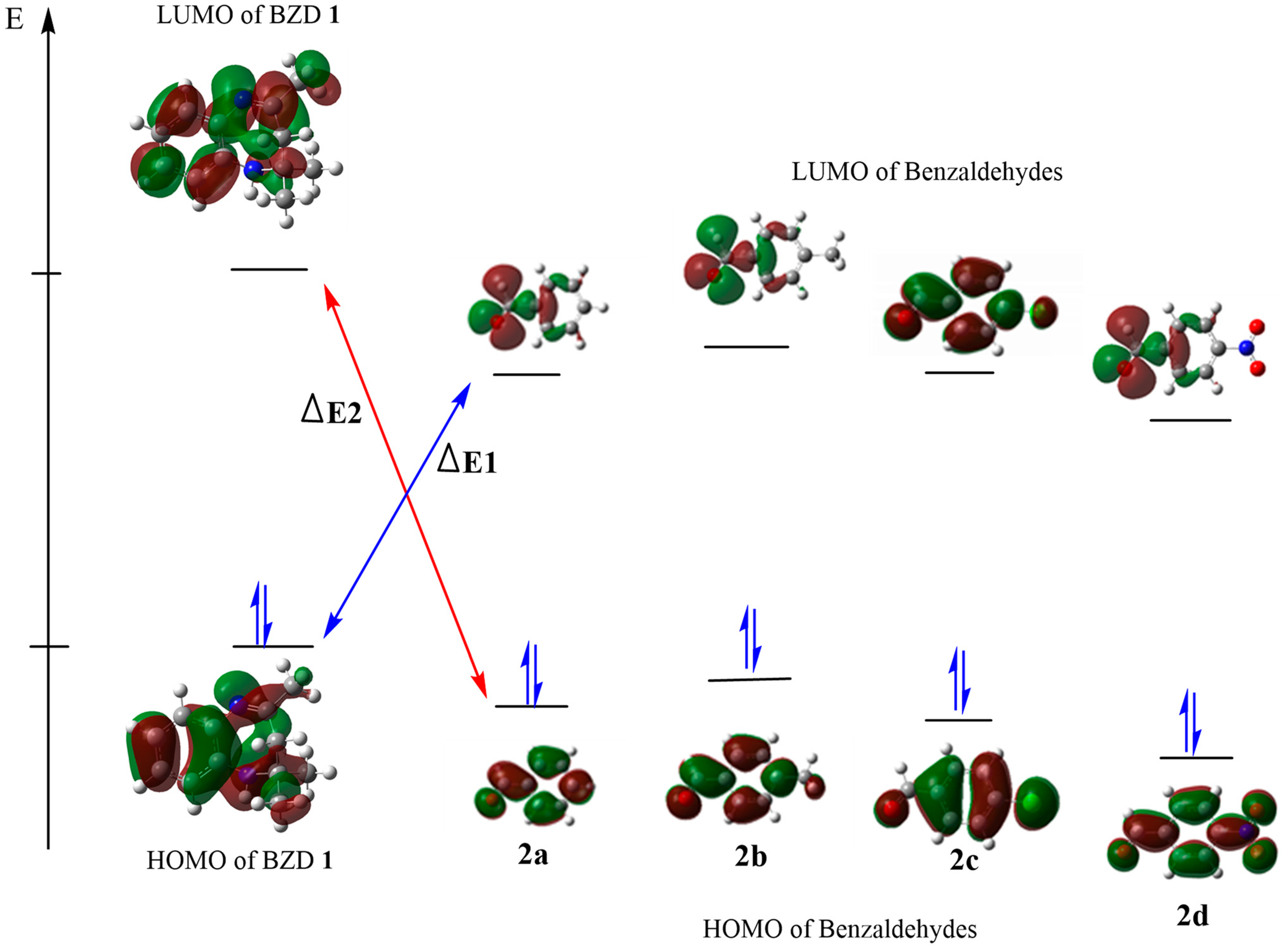
Analysis of the Conceptual DFT Reactivity Indices of the Reagents
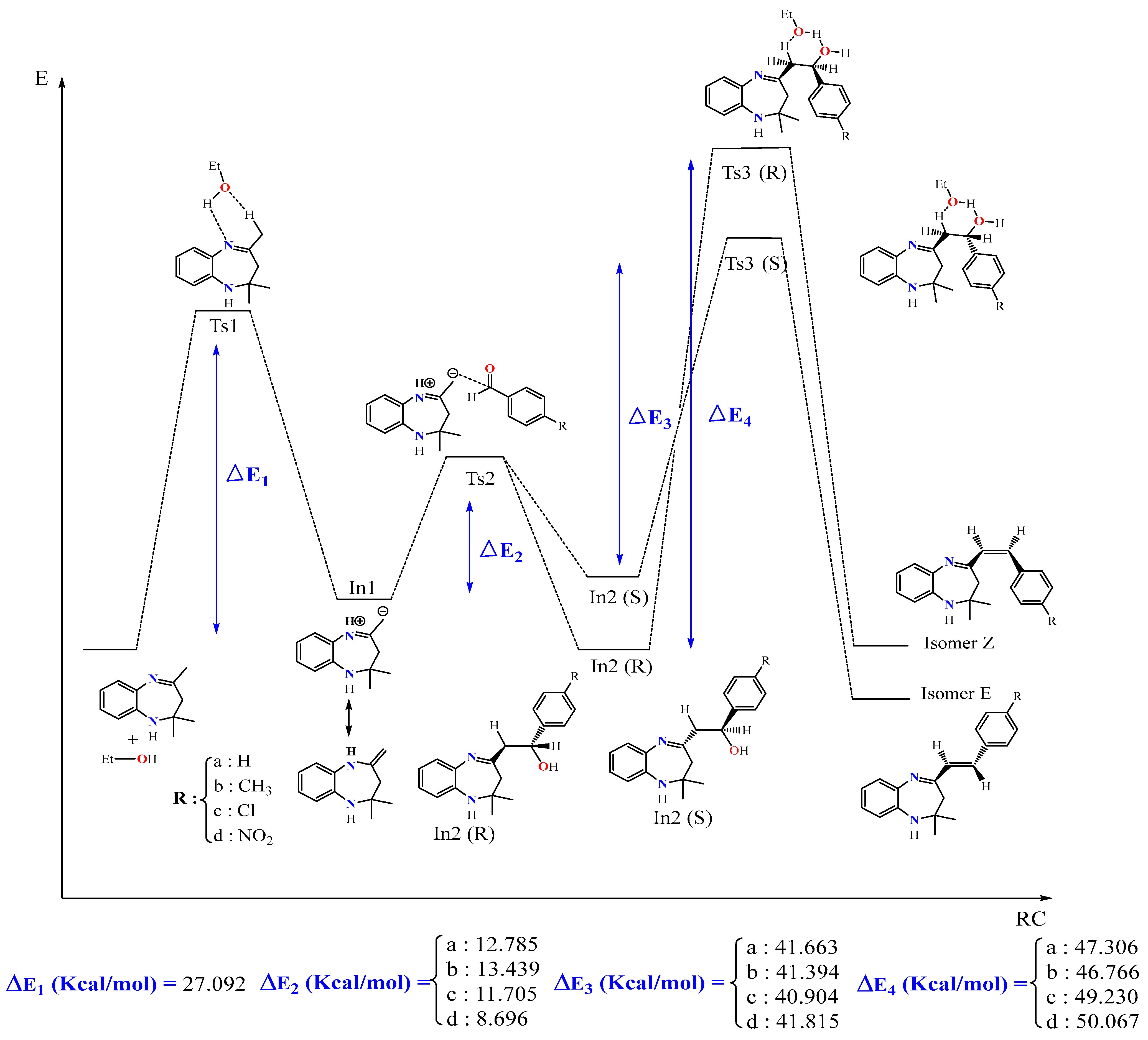
5. Kinetic Study
6. Material and Methods
6.1. Chemistry
6.2. Preparation of 2,2,4-Trimethyl-2.3-Dihydro-1H-1,5-Benzodiazepine 1
6.3. Preparation of 1,5-Benzodiazepines 3(a–d)
- (E)-2,2-Diméthyl-4-styryl-2,3-dihydro-1H-1,5-benzodiazepine 3a
- (E)-2,2-Dimethyl-4-(4-methylstyryl)-2,3-dihydro-1H-1,5-benzodiazepine 3b
- (E)-4-(4-Chlorostyryl)-2,2-diméthyl-2,3-dihydro-1H-1,5-benzodiazepine 3c
- (E)-2,2-Dimethyl-4-(4-nitrostyryl)-2,3-dihydro-1H-1,5-benzodiazepine 3d
6.4. Crystallographic Data Collection, Structure Solution, and Refinement
6.5. Computational Method
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catalani, V.; Botha, M.; Corkery, J.M.; Guirguis, A.; Vento, A.; Schifano, F. Designer Benzodiazepines’ Activity on Opioid Receptors: A Docking Study. Curr. Pharm. Des. 2022, 28, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Tolu-Bolaji, O.; Sojinu, S.O.; Okedere, A.P.; Ajani, O.O. A review on the chemistry and pharmacological properties of benzodiazepine motifs in drug design. Arab J. Basic Appl. Sci. 2022, 29, 287–306. [Google Scholar] [CrossRef]
- Mishra, R.; Sharma, A.K.; Kumar, R.; Baweja, V.; Mothsra, P.; Singh, M.K.; Yadav, S.B. Solid support based synthesis of 1,5-benzodiazepines: A mini review. Synth. Commun. 2022, 52, 481–503. [Google Scholar] [CrossRef]
- Teli, S.; Teli, P.; Soni, S.; Sahiba, N.; Agarwal, S. Synthetic aspects of 1,4- and 1,5-benzodiazepines using o-phenylenediamine: A study of past quinquennial. RSC Adv. 2023, 13, 3694–3714. [Google Scholar] [CrossRef]
- Peng, L.; Morford, K.L.; Levander, X.A. Benzodiazepines and Related Sedatives. Med. Clin. N. Am. 2022, 106, 113–129. [Google Scholar] [CrossRef]
- Mahanna-Gabrielli, E.; Schenning, K.J.; Deiner, S.G.; Whittington, R.A. Pro-Con Debate: Judicious Benzodiazepine Administration for Preoperative Anxiolysis in Older Patients. Anesth. Analg. 2023, 137, 280–288. [Google Scholar] [CrossRef]
- Caiana, E.; De Veras, B.; De Souza, A.; Queiroz, N. Synthesis of Hydroxybenzodiazepines with Potential Antioxidant and Antifungal Action. J. Braz. Chem. Soc. 2021, 32, 626–637. [Google Scholar] [CrossRef]
- Sheikhi-Mohammareh, S.; Mashreghi, M.; Shiri, A. Robust approach leading to novel densely functionalized four-cyclic benzo[e]pyrazolo [5′,1′:2,3]pyrimido[4,5-b][1,4]diazepines with antibacterial activity toward resistant strains. J. Iran. Chem. Soc. 2020, 17, 1555–1566. [Google Scholar] [CrossRef]
- Anil, S.M.; Shobith, R.; Kiran, K.R.; Swaroop, T.R.; Mallesha, N.; Sadashiva, M.P. Facile synthesis of 1,4-benzodiazepine-2,5-diones and quinazolinones from amino acids as anti-tubercular agents. New J. Chem. 2019, 43, 182–187. [Google Scholar] [CrossRef]
- Benzbiria, N.; Thoume, A.; El Caid, Z.A.; Echihi, S.; Elmakssoudi, A.; Zarrouk, A.; Zertoubi, M. An investigation on the utilization of a synthesized benzodiazepine derivative as a corrosion inhibitor for carbon steel in sulfuric solution: Chemical and electrochemical synthesis, surface analysis (SEM/AFM), DFT and MC simulation. Colloids Surf. Physicochem. Eng. Asp. 2024, 681, 132744. [Google Scholar] [CrossRef]
- Benzbiria, N.; Echihi, S.; Belghiti, M.E.; Thoume, A.; Elmakssoudi, A.; Zarrouk, A.; Zertoubi, M.; Azzi, M. Novel synthetized benzodiazepine as efficient corrosion inhibitor for copper in 3.5% NaCl solution. Mater. Today Proc. 2021, 37, 3932–3939. [Google Scholar] [CrossRef]
- Oubaaqa, M.; Rbaa, M.; Ouakki, M.; Idouhli, R.; Maatallah, M.; Jarid, A.; Warad, I.; Abousalem, A.S.; Lakhrissi, B.; Zarrouk, A.; et al. Novel triphenyl imidazole based on 8-hydroxyquinoline as corrosion inhibitor for mild steel in molar hydrochloric acid: Experimental and theoretical investigations. J. Appl. Electrochem. 2022, 52, 413–433. [Google Scholar] [CrossRef]
- Adardour, M.; Lasri, M.; Lahcen, M.A.; Maatallah, M.; Idouhli, R.; Alanazi, M.M.; Lahmidi, S.; Abouelfida, A.; Mague, J.T.; Baouid, A. Exploring the Efficacy of Benzimidazolone Derivative as Corrosion Inhibitors for Copper in a 3.5 wt.% NaCl Solution: A Comprehensive Experimental and Theoretical Investigation. Molecules 2023, 28, 6948. [Google Scholar] [CrossRef] [PubMed]
- Adardour, M.; Boutafda, A.; Hdoufane, I.; Aghraz, A.; Hafidi, M.; Zaballos-García, E.; Cherqaoui, D.; Baouid, A. Efficient and simple synthesis of novel 1,2,3-triazolyl-linked benzimidazolone, molecular docking and evaluation of their antimicrobial activity. Synth. Commun. 2020, 50, 3490–3506. [Google Scholar] [CrossRef]
- Ettahiri, W.; Dalbouha, A.; Baouid, A.; Alsubari, A.; Mague, J.T.; Taleb, M.; Ramli, Y. 5,6,7,8-Tetrahydro-[1,2,4]triazolo[5,1-b]quinazolin-9(4 H)-one. IUCrData 2023, 8, x230409. [Google Scholar] [CrossRef]
- Verma, M.; Sharma, R.; Bharti, R.; Tangri, A. Green one-pot synthesis of N-based heterocycles involving o-phenylenediamine. Mater. Today Proc. 2021, 37, 2321–2328. [Google Scholar] [CrossRef]
- Sarkar, R.; Gupta, A.; Jamatia, R.; Pal, A.K. Reduced graphene oxide supported copper oxide nanocomposites: An efficient heterogeneous and reusable catalyst for the synthesis of ynones, 1,3-diynes and 1,5-benzodiazepines in one-pot under sustainable reaction conditions. Appl. Organomet. Chem. 2020, 34, e5646. [Google Scholar] [CrossRef]
- Wen, T.T.; Wang, L.Z. CoFe2O4@SiO2@APTES@HO-PBA@Cu(OAc)2: A highly efficient and recyclable nanocatalyst for one-pot synthesis of multifunctional 1,5-benzodiazepines via three-component domino reaction. Mater. Today Chem. 2022, 26, 101071. [Google Scholar] [CrossRef]
- Pang, Y.; Lin, H.; Ou, C.; Cao, Y.; An, B.; Yan, J.; Li, X. Design, synthesis, and biological evaluation of novel benzodiazepine derivatives as anticancer agents through inhibition of tubulin polymerization in vitro and in vivo. Eur. J. Med. Chem. 2019, 182, 111670. [Google Scholar] [CrossRef]
- Odame, F.; Madanhire, T.; Hosten, E.; Lobb, K. Crystal structure, Hirshfeld surface analysis and computational studies of (E)-2,2-dimethyl-4-styryl-2,3-dihydro-1H-benzo[b][1,4]diazepine. Moroc. J. Chem. 2023, 11, 579–896. [Google Scholar] [CrossRef]
- Lahcen, M.A.; Adardour, M.; Mortada, S.; Oubahmane, M.; Hmaimou, S.; Loughzail, M.; Hdoufane, I.; Lahmidi, S.; Faouzi, M.E.A.; Cherqaoui, D.; et al. Synthesis, characterization, X-ray, α-glucosidase inhibition and molecular docking study of new triazolic systems based on 1,5-benzodiazepine via 1,3-dipolar cycloaddition reactions. J. Biomol. Struct. Dyn. 2023, 42, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Hmaimou, S.; Adardour, M.; Lahcen, M.A.; Ettahiri, W.; Lahmidi, S.; Mague, J.T.; Taleb, M.; Maatallah, M.; Baouid, A. Synthesis, Characterization, Crystal Structure, and Theoretical Calculations of Novel 1,5-Benzodiazepine Derivatives Obtained via 1,3-Dipolar Cycloaddition Reactions. Polycycl. Aromat. Compd. 2024, 1–19. [Google Scholar] [CrossRef]
- Soumane, Y.B.; Baouid, A.; El Hadrami, E.M.; Idouhli, R.; El Ammari, L.; Tama, A.B.; Maatallah, M.; Berraho, M.; Saadi, M. Synthesis, Characterization, Theoretical and Electrochemical Studies of Pyrazoline Derivatives, Mediterr. J. Chem. 2020, 10, 845. [Google Scholar] [CrossRef]
- Adamovich, S.N.; Vchislo, N.V.; Oborina, E.N.; Ushakov, I.A.; Rozentsveig, I.B. Novel α,β-unsaturated imine derivatives of 3-aminopropylsilatrane. Mendeleev Commun. 2017, 27, 443–445. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Bettigeri, S.V.; Joshi, N.S. Molecular Iodine Catalyzed Highly Rapid Synthesis of 1,5-Benzodiazepine Derivatives Under Mild Conditions. Synth. Commun. 2004, 34, 1447–1453. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Sousa, A.S.; Domingo, L.R. Electrophilicity and nucleophilicity scales at different DFT computational levels. J. Phys. Org. Chem. 2023, 36, e4503. [Google Scholar] [CrossRef]
- Grambow, C.A.; Pattanaik, L.; Green, W.H. Reactants, products, and transition states of elementary chemical reactions based on quantum chemistry. Sci. Data 2020, 7, 137. [Google Scholar] [CrossRef]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; De Proft, F.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual density functional theory: Status, prospects, issues. Theor. Chem. Acc. 2020, 139, 36. [Google Scholar] [CrossRef]
- Vuckovic, S.; Gerolin, A.; Daas, K.J.; Bahmann, H.; Friesecke, G.; Gori-Giorgi, P. Density functionals based on the mathematical structure of the strong-interaction limit of DFT. WIREs Comput. Mol. Sci. 2023, 13, e1634. [Google Scholar] [CrossRef]
- Lotfi, M.; Hamzehloueian, M.; Haghdadi, M. A DFT study on the mechanism and selectivity of [3 + 2] cycloaddition reactions leading to pyrole [2,1-a] phthalazine compounds. Theor. Chem. Acc. 2021, 140, 57. [Google Scholar] [CrossRef]
- Nabih, K.; Maatallah, M.; Baouid, A.; Jarid, A. Theoretical Analysis of the Mechanism of the 1,3-Dipolar Cycloaddition of Benzodiazepine with N-Aryl-C-ethoxycarbonylnitrilimine. J. Chem. 2020, 2020, 8695405. [Google Scholar] [CrossRef]
- Shinde, R.A.; Adole, V.A.S.; Jagdale, B.S.; Bhavsing Pawar, T. Experimental and Theoretical Studies on the Molecular Structure, FT-IR, NMR, HOMO, LUMO, MESP, and Reactivity Descriptors of (E)-1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one. Mater. Sci. Res. India 2020, 17, 54–72. [Google Scholar] [CrossRef]
- Odame, F.; Madanhire, T.; Hosten, E.C.; Lobb, K. Crystal Structure, Hirshfeld Surface Analysis and Computational Studies of Two Benzo[b][1,4]Diazepine Derivatives. J. Struct. Chem. 2023, 64, 2326–2342. [Google Scholar] [CrossRef]
- Fang, Y.; Hou, Y.; Yang, H.; Chen, R.; Li, W.; Ma, J.; Han, D.; Cao, X.; Liu, S.; Shen, Y.; et al. Elucidating Orbital Delocalization Effects on Boosting Electrochemiluminescence Efficiency of Carbon Nitrides. Adv. Opt. Mater. 2022, 10, 2201017. [Google Scholar] [CrossRef]
- Sánchez-Márquez, J.; García, V.; Zorrilla, D.; Fernández, M. On Electronegativity, Hardness, and Reactivity Descriptors: A New Property-Oriented Basis Set. J. Phys. Chem. A 2020, 124, 4700–4711. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Guo, C.; Li, M.; Rong, C.; Liu, S. Finite difference representation of information-theoretic approach in density functional theory. Chemistry 2023, 142, 83. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Maatallah, M.; Cherqaoui, D.; Jarid, A.; Liebman, J.F. Large gallanes and the PSEPT theory: A theoretical study of GanHn+2 clusters (n = 7–9). J. Mol. Model. 2012, 18, 3321–3328. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 3d |
|---|---|
| Formula | C19H19N3O2 |
| Dcalc./g cm−3 | 1.244 |
| m/mm−1 | 0.664 |
| Formula weight | 321.37 |
| Color | red |
| Shape | Prism-shaped |
| Size/mm3 | 0.19 × 0.17 × 0.13 |
| T/K | 150.00(10) |
| Crystal system | Orthorhombic |
| Flack parameter | 0.5 |
| Space group | Pna21 |
| a; b; c (Å) | 16.2480(2); 10.08130(10); 20.9539(3) |
| a; b;c (°) | 90; 90; 90 |
| V/Å3 | 3432.27(7) |
| Z | 8 |
| Z’ | 2 |
| Wavelength/Å | 1.54184 |
| Radiation type | Cu Ka |
| Qmax; Qmin/° | 67.075; 4.220 |
| Measured Refl’s. | 23,523 |
| Indep’t Refl’s | 5130 |
| Refl’s I ≥ 2 s(I) | 4794 |
| Rint | 0.0410 |
| Parameters | 445 |
| Restraints | 1 |
| Largest peak | 0.362 |
| Deepest hole | −0.176 |
| GooF | 1.064 |
| wR2 (all data) | 0.1136 |
| wR2 | 0.1111 |
| R1 (all data) | 0.0439 |
| R1 | 0.0413 |
| H | ΔE | CH3 | ΔE | Cl | ΔE | NO2 | ΔE | |
|---|---|---|---|---|---|---|---|---|
| 3 | −922.01575 | 0.000 | −961.33426 | 0.000 | −1381.61211 | 0.000 | −1126.52168 | 0.000 |
| 4 | −922.00365 | −7.595 | −961.32198 | −7.703 | −1381.59989 | −7.673 | −1126.50901 | −7.950 |
| 5 | −921.98564 | −18.895 | −961.30379 | −19.117 | −1381.58245 | −18.617 | −1126.49182 | −18.741 |
| 6 | −921.97577 | −25.088 | −961.29387 | −25.345 | −1381.57249 | −24.865 | −1126.47276 | −30.700 |
| HOMO | LUMO | A | I | µ | ᵡ | η | σ | ω | N | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −5.265 | −0.545 | 0.545 | 5.265 | 2.905 | −2.905 | 4.720 | 0.211 | 0.896 | 4.103 |
| 2a | −6.941 | −1.712 | 1.712 | 6.941 | −2.614 | 2.614 | −5.229 | −0.191 | −0.653 | 2.427 |
| 2b | −6.839 | −1.597 | 1.597 | 6.839 | −2.621 | 2.621 | −5.242 | −0.190 | −0.655 | 2.529 |
| 2c | −7.148 | −1.987 | 1.987 | 7.148 | −2.580 | 2.580 | −5.161 | −0.193 | −0.645 | 2.220 |
| 2d | −7.571 | −3.116 | 3.116 | 7.571 | −2.227 | 2.227 | −4.455 | −0.224 | −0.556 | 1.797 |
| R | Para-H | Para-CH3 | Para-Cl | Para-NO2 |
|---|---|---|---|---|
| Ts1 | 27.092 | 27.092 | 27.092 | 27.092 |
| In1 | 18.410 | 18.410 | 18.410 | 18.410 |
| Ts2 | 12.785 | 13.439 | 11.705 | 8.696 |
| In2 (Z) | 17.519 | 17.678 | 16.835 | 15.911 |
| In2 (E) | 24.416 | 24.415 | 24.229 | 23.922 |
| Ts3 (Z) | 41.662 | 41.394 | 40.904 | 41.815 |
| Ts3 (E) | 47.306 | 46.766 | 49.230 | 50.067 |
| Product (Z) | 26.038 | 25.925 | 25.232 | 26.237 |
| Product (E) | 31.936 | 31.746 | 33.402 | 33.547 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hmaimou, S.; Ait Lahcen, M.; Adardour, M.; Alanazi, M.M.; Kabra, A.; Maatallah, M.; Baouid, A. A Stereoselective Synthesis of a Novel α,β-Unsaturated Imine-Benzodiazepine through Condensation Reaction, Crystal Structure, and DFT Calculations. Molecules 2024, 29, 4323. https://doi.org/10.3390/molecules29184323
Hmaimou S, Ait Lahcen M, Adardour M, Alanazi MM, Kabra A, Maatallah M, Baouid A. A Stereoselective Synthesis of a Novel α,β-Unsaturated Imine-Benzodiazepine through Condensation Reaction, Crystal Structure, and DFT Calculations. Molecules. 2024; 29(18):4323. https://doi.org/10.3390/molecules29184323
Chicago/Turabian StyleHmaimou, Samir, Marouane Ait Lahcen, Mohamed Adardour, Mohammed M. Alanazi, Atul Kabra, Mohamed Maatallah, and Abdesselam Baouid. 2024. "A Stereoselective Synthesis of a Novel α,β-Unsaturated Imine-Benzodiazepine through Condensation Reaction, Crystal Structure, and DFT Calculations" Molecules 29, no. 18: 4323. https://doi.org/10.3390/molecules29184323
APA StyleHmaimou, S., Ait Lahcen, M., Adardour, M., Alanazi, M. M., Kabra, A., Maatallah, M., & Baouid, A. (2024). A Stereoselective Synthesis of a Novel α,β-Unsaturated Imine-Benzodiazepine through Condensation Reaction, Crystal Structure, and DFT Calculations. Molecules, 29(18), 4323. https://doi.org/10.3390/molecules29184323