
Structural Insights into Endostatin–Heparan Sulfate Interactions Using Modeling Approaches

 and
and
Abstract
1. Introduction
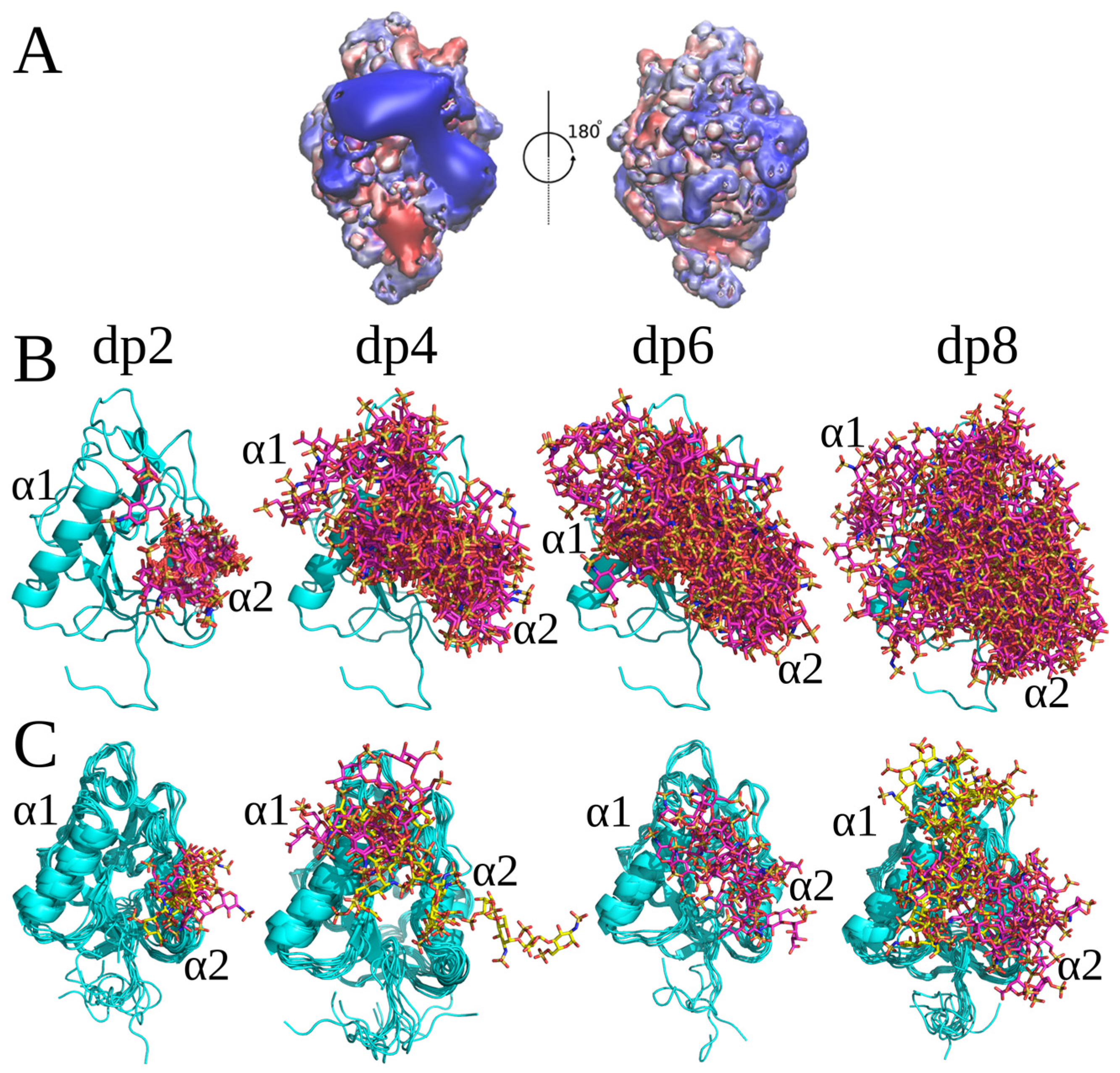
2. Results
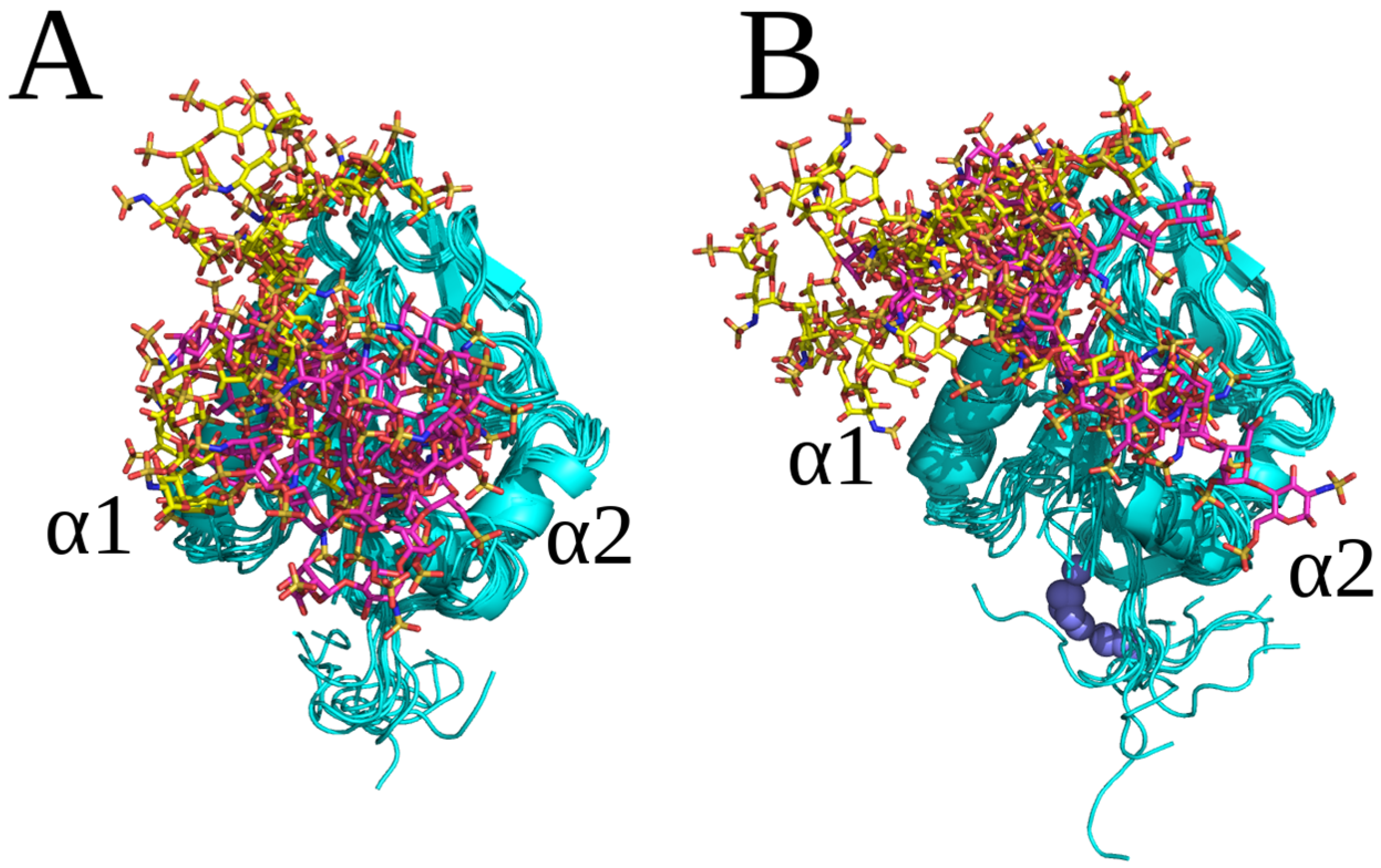
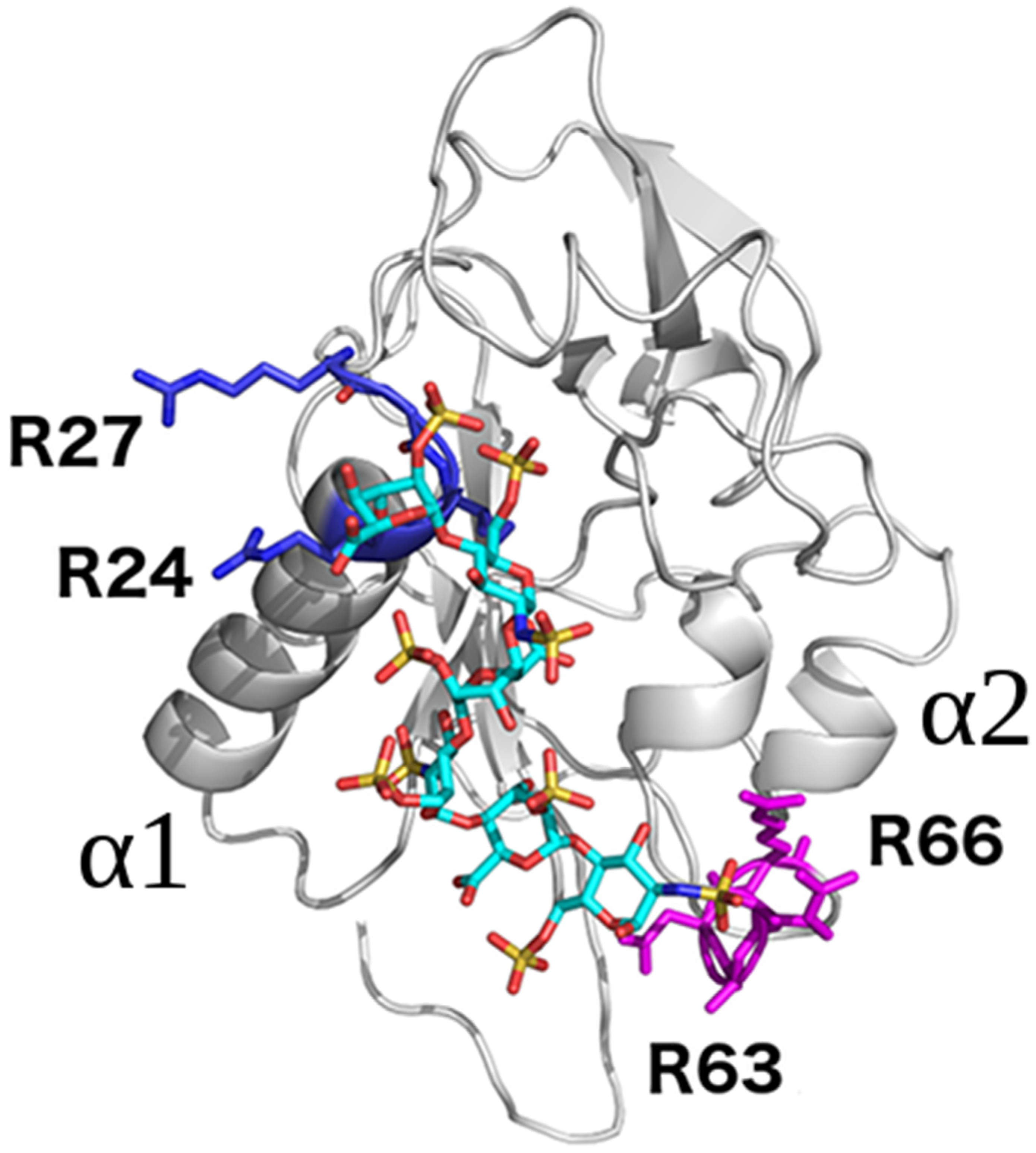
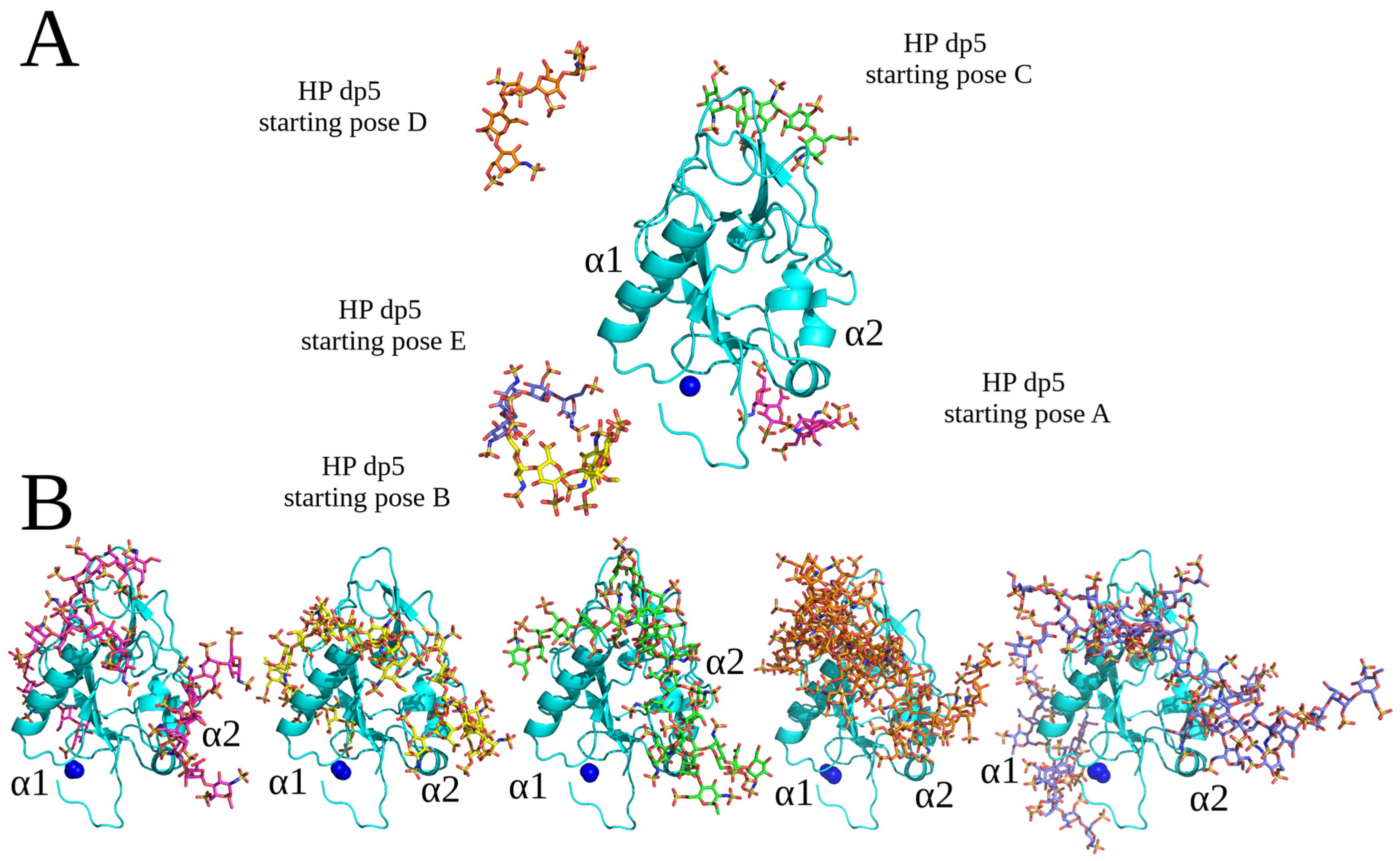
2.1. Conventional Molecular Docking and MD Simulations
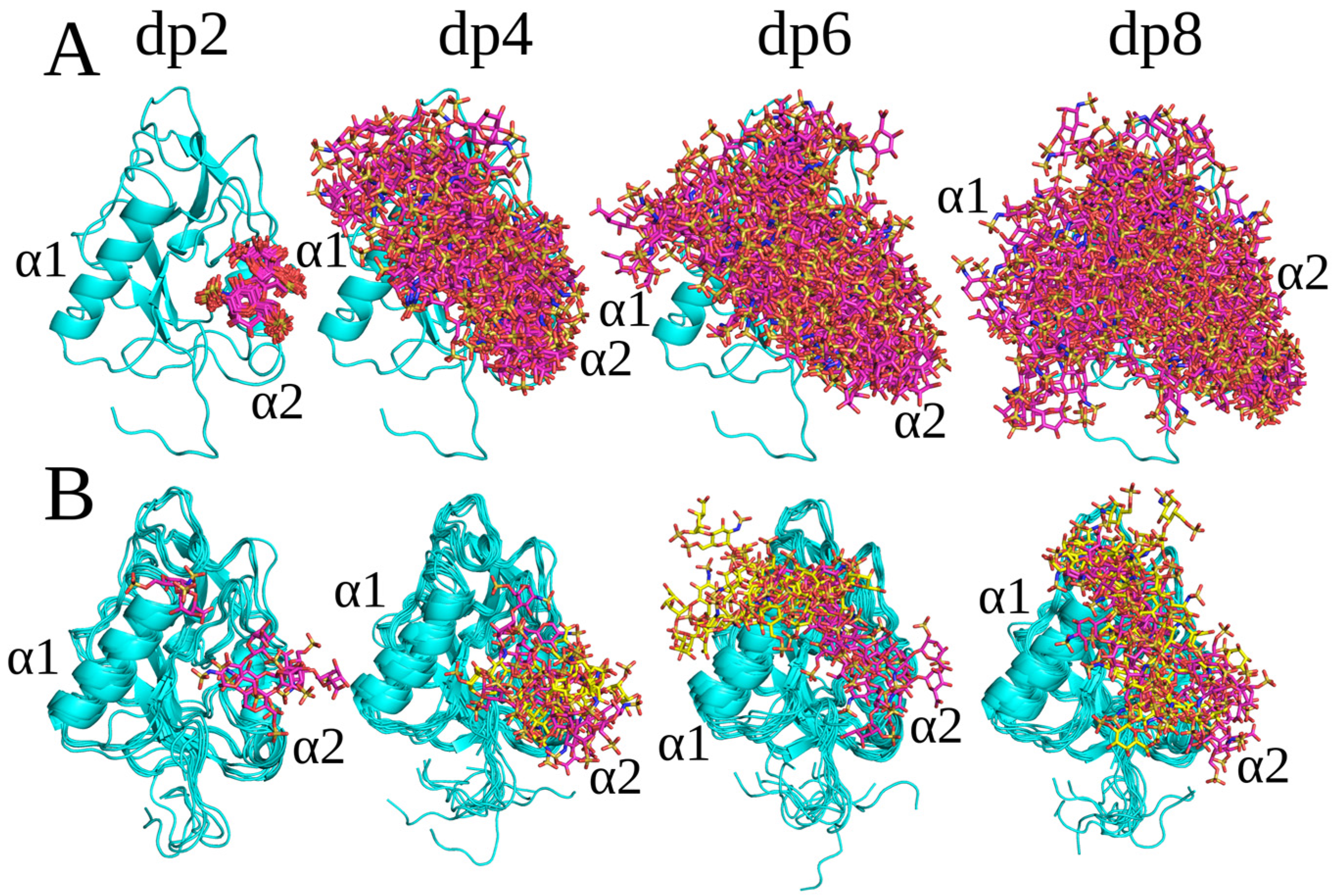
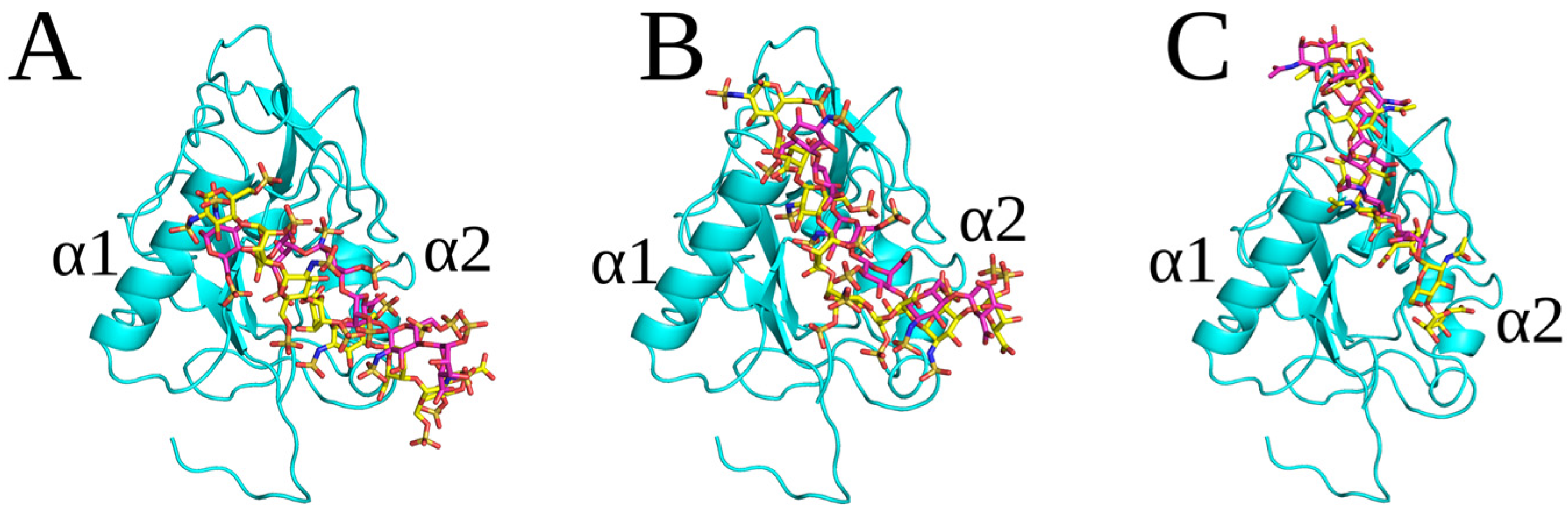
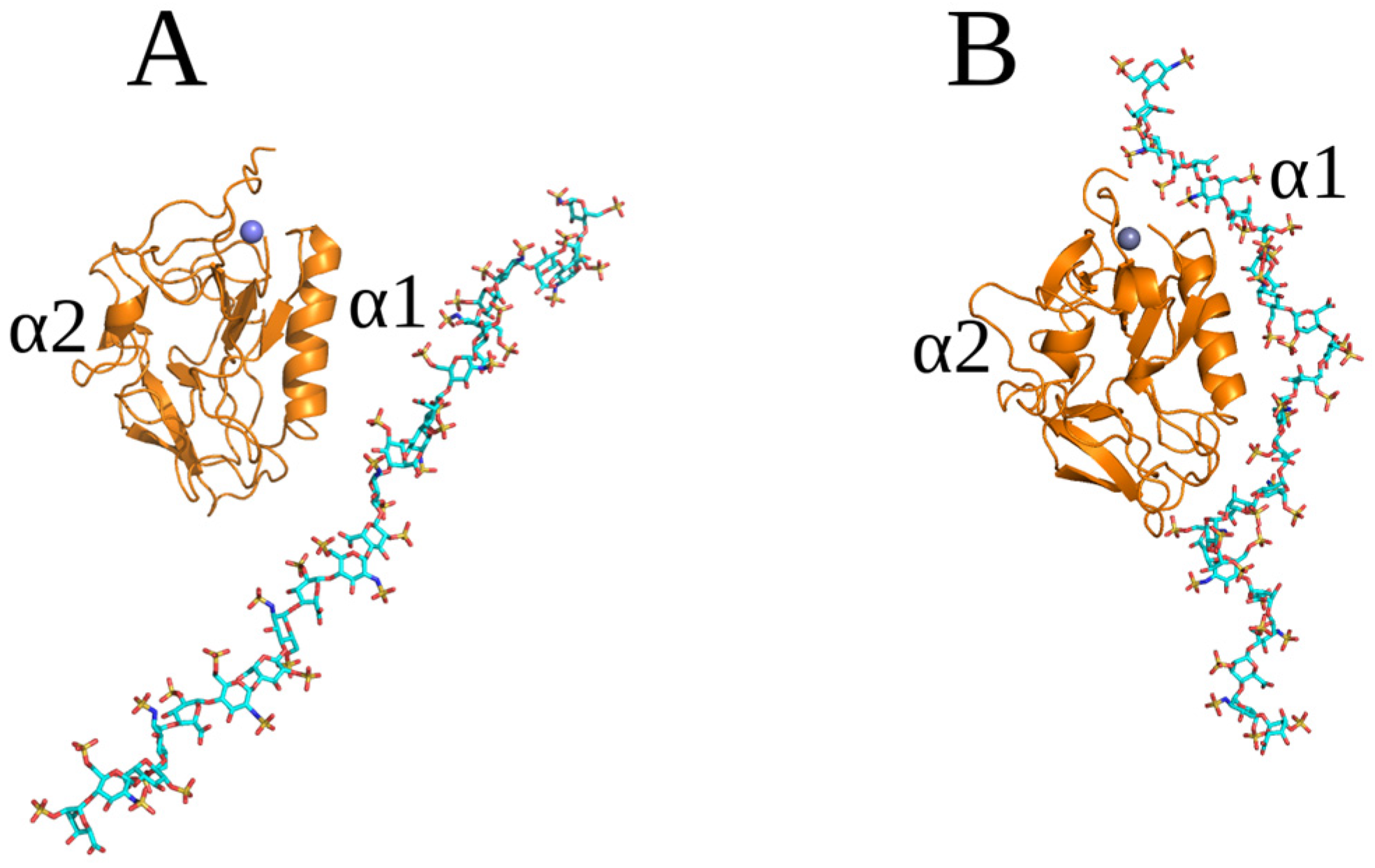
2.2. RS-REMD Analysis
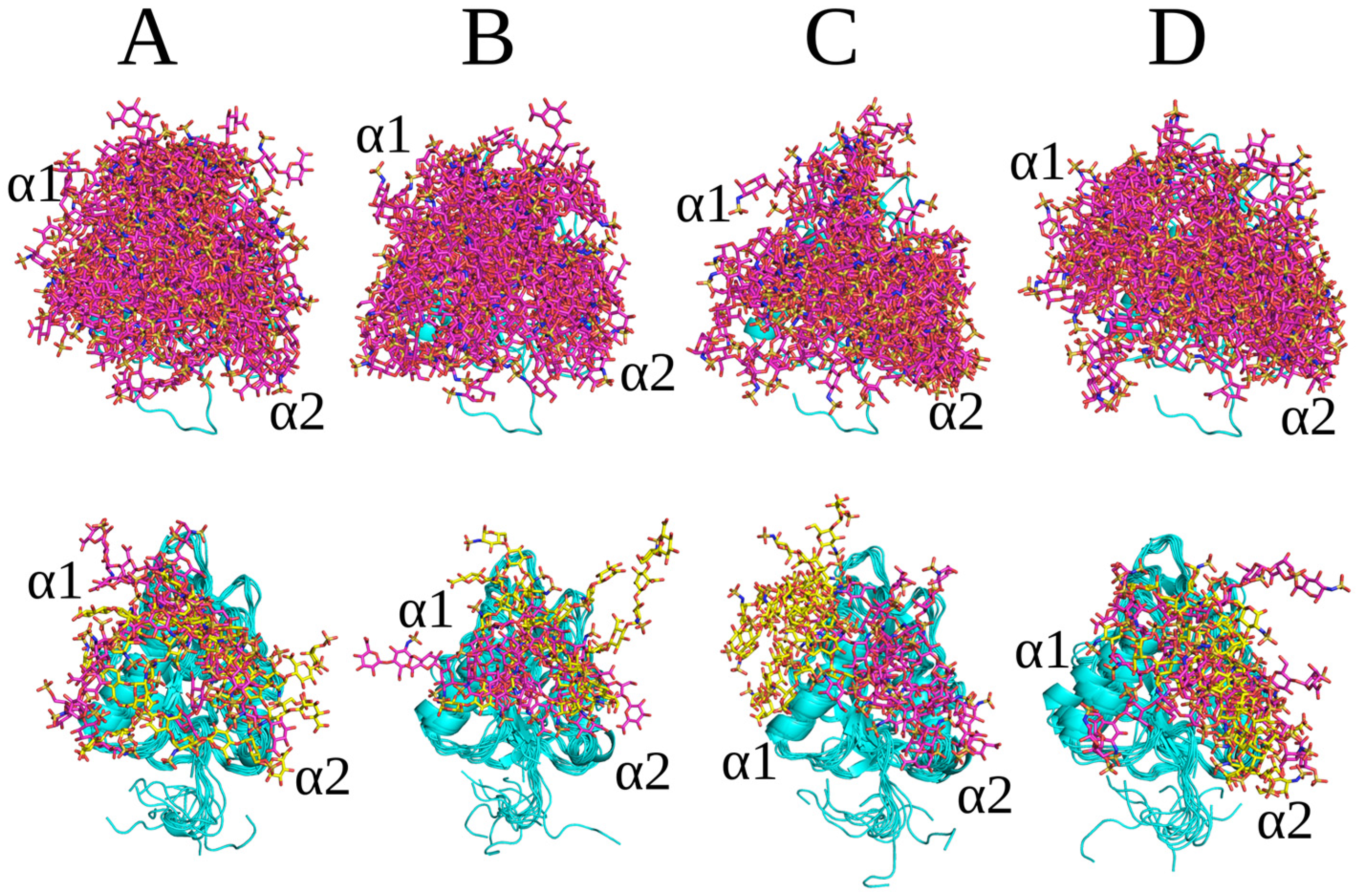
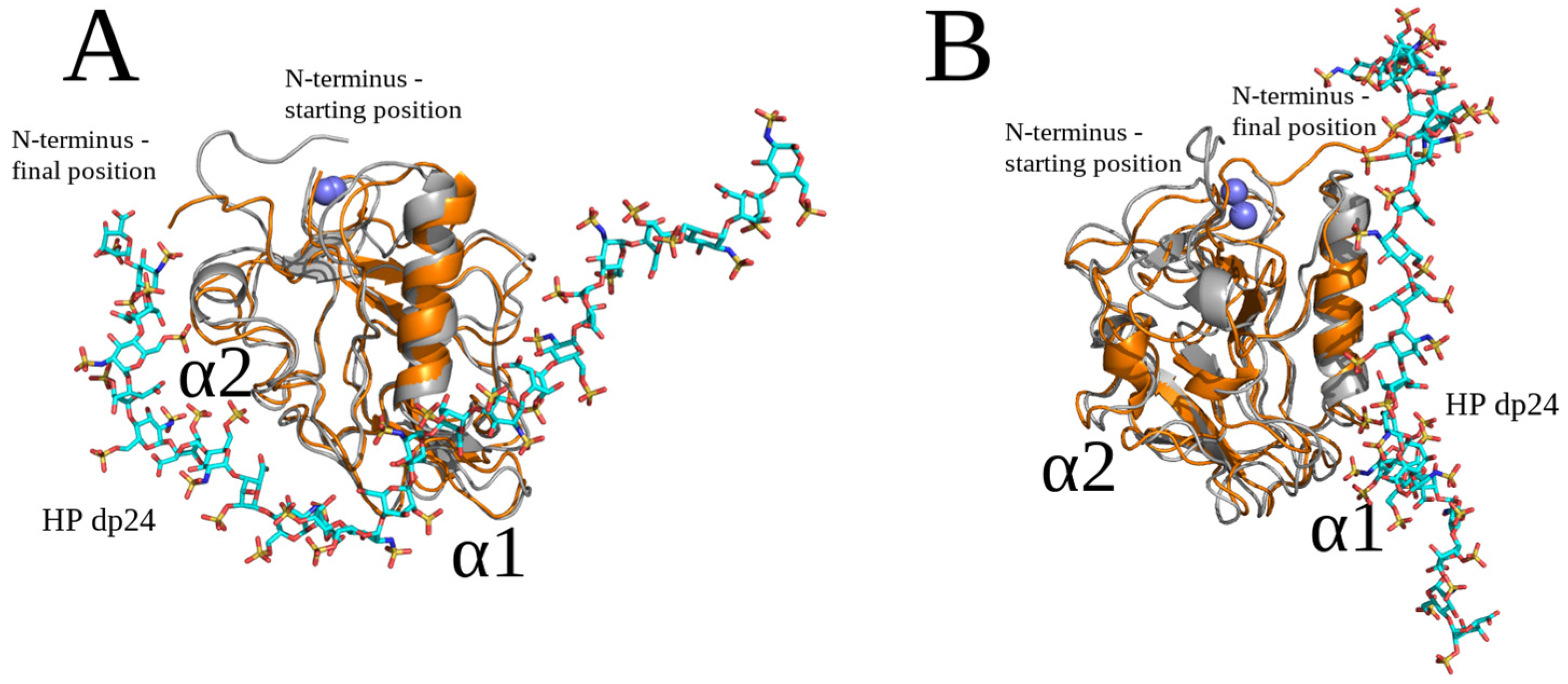
2.3. Unbiased MD Simulation Analysis
2.4. Endostatin Mutations
3. Discussion
4. Materials and Methods
4.1. Endostatin’s Structure
4.2. Electrostatic Potential Calculations
4.3. Conventional Molecular Docking
4.4. Molecular Dynamics and MM-GBSA Calculations
4.5. Repulsive Scaling–Replica Exchange Molecular Dynamics (RS-REMD)
4.5.1. Molecular Dynamics Simulation
4.5.2. Binding Free Energy Calculations/Scoring
4.5.3. Refinement
4.5.4. MD Data Analysis
4.6. Unbiased MD Simulation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Vallet, S.D.; Clerc, O.; Ricard-Blum, S. Glycosaminoglycan–protein interactions: The first draft of the glycosaminoglycan Interactome. J. Histochem. Cytochem. 2021, 69, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.D.; Berthollier, C.; Ricard-Blum, S. The glycosaminoglycan interactome 2.0. Am. J. Physiol.-Cell Physiol. 2022, 322, C1271–C1278. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Perez, S. Glycosaminoglycan interaction networks and databases. Curr. Opin. Struct. Biol. 2022, 74, 102355. [Google Scholar] [CrossRef]
- Paiardi, G.; Milanesi, M.; Wade, R.C.; D’Ursi, P.; Rusnati, M. A bittersweet computational journey among glycosaminoglycans. Biomolecules 2021, 11, 739. [Google Scholar] [CrossRef]
- Bojarski, K.K.; Sage, J.; Lalmanach, G.; Lecaille, F.; Samsonov, S.A. In silico and in vitro mapping of specificity patterns of glycosaminoglycans towards cysteine cathepsins B, L, K, S and V. J. Mol. Graph. Model. 2022, 113, 108153. [Google Scholar] [CrossRef]
- Pérez, S.; Bonnardel, F.; Lisacek, F.; Imberty, A.; Ricard Blum, S.; Makshakova, O. GAG-DB, the new interface of the three-dimensional landscape of glycosaminoglycans. Biomolecules 2020, 10, 1660. [Google Scholar] [CrossRef]
- Clerc, O.; Mariethoz, J.; Rivet, A.; Lisacek, F.; Pérez, S.; Ricard-Blum, S. A pipeline to translate glycosaminoglycan sequences into 3D models. Application to the exploration of glycosaminoglycan conformational space. Glycobiology 2019, 29, 36–44. [Google Scholar] [CrossRef]
- Griffith, A.R.; Rogers, C.J.; Miller, G.M.; Abrol, R.; Hsieh-Wilson, L.C.; Goddard, W.A., III. Predicting glycosaminoglycan surface protein interactions and implications for studying axonal growth. Proc. Natl. Acad. Sci. USA 2017, 114, 13697–13702. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Pisabarro, M.T. Computational analysis of interactions in structurally available protein–glycosaminoglycan complexes. Glycobiology 2016, 26, 850–861. [Google Scholar] [CrossRef]
- Künze, G.; Huster, D.; Samsonov, S.A. Investigation of the structure of regulatory proteins interacting with glycosaminoglycans by combining NMR spectroscopy and molecular modeling–the beginning of a wonderful friendship. Biol. Chem. 2021, 402, 1337–1355. [Google Scholar] [CrossRef]
- Hintze, V.; Samsonov, S.A.; Anselmi, M.; Moeller, S.; Becher, J.; Schnabelrauch, M.; Scharnweber, D.; Pisabarro, M.T. Sulfated glycosaminoglycans exploit the conformational plasticity of bone morphogenetic protein-2 (BMP-2) and alter the interaction profile with its receptor. Biomacromolecules 2014, 15, 3083–3092. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Samsonov, S.A.; Pichert, A.; Lemmnitzer, K.; Schiller, J.; Huster, D.; Pisabarro, M.T.; von Bergen, M.; Kalkhof, S. Structural analysis of the interleukin-8/glycosaminoglycan interactions by amide hydrogen/deuterium exchange mass spectrometry. Methods 2015, 89, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Makshakova, O.; Angulo, J.; Bedini, E.; Bisio, A.; de Paz, J.L.; Fadda, E.; Guerrini, M.; Hricovini, M.; Hricovini, M.; et al. Glycosaminoglycans: What remains to be deciphered? JACS Au 2023, 3, 628–656. [Google Scholar] [CrossRef]
- Poluzzi, C.; Iozzo, R.V.; Schaefer, L. Endostatin and endorepellin: A common route of action for similar angiostatic cancer avengers. Adv. Drug Deliv. Rev. 2016, 97, 156–173. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Vallet, S.D. Matricryptins network with matricellular receptors at the surface of endothelial and tumor cells. Front. Pharmacol. 2016, 7, 11. [Google Scholar] [CrossRef]
- Li, K.; Shi, M.; Qin, S. Current status and study progress of recombinant human endostatin in cancer treatment. Oncol. Ther. 2018, 6, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Féraud, O.; Lortat-Jacob, H.; Rencurosi, A.; Fukai, N.; Dkhissi, F.; Vittet, D.; Imberty, A.; Olsen, B.R.; van Der Rest, M. Characterization of endostatin binding to heparin and heparan sulfate by surface plasmon resonance and molecular modeling: Role of divalent cations. J. Biol. Chem. 2004, 279, 2927–2936. [Google Scholar] [CrossRef]
- Boehm, T.; O’Reilly, M.S.; Keough, K.; Shiloach, J.; Shapiro, R.; Folkman, J. Zinc-binding of endostatin is essential for its antiangiogenic activity. Biochem. Biophys. Res. Commun. 1998, 252, 190–194. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Anand-Apte, B.; Lee, M.; Sasaki, T.; Fukai, N.; Shapiro, R.; Que, I.; Lowik, C.; Timpl, R.; Olsen, B.R. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999, 18, 4414–4423. [Google Scholar] [CrossRef]
- Woodhead, N.E.; Long, W.F.; Williamson, F.B. Binding of zinc ions to heparin. Analysis by equilibrium dialysis suggests the occurrence of two, entropy-driven, processes. Biochem. J. 1986, 237, 281. [Google Scholar] [CrossRef]
- Ding, Y.H.; Javaherian, K.; Lo, K.M.; Chopra, R.; Boehm, T.; Lanciotti, J.; Harris, B.A.; Li, Y.; Shapiro, R.; Hohenester, E.; et al. Zinc-dependent dimers observed in crystals of human endostatin. Proc. Natl. Acad. Sci. USA 1998, 95, 10443–10448. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Sasaki, T.; Mann, K.; Timpl, R. Variable zinc coordination in endostatin. J. Mol. Biol. 2000, 297, 1–6. [Google Scholar] [CrossRef]
- Sasaki, T.; Larsson, H.; Kreuger, J.; Salmivirta, M.; Claesson-Welsh, L.; Lindahl, U.; Hohenester, E.; Timpl, R. Structural basis and potential role of heparin/heparan sulfate binding to the angiogenesis inhibitor endostatin. EMBO J. 1999, 18, 6240–6248. [Google Scholar] [CrossRef]
- Siebenmorgen, T.; Engelhard, M.; Zacharias, M. Prediction of protein–protein complexes using replica exchange with repulsive scaling. J. Comput. Chem. 2020, 41, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Maszota-Zieleniak, M.; Marcisz, M.; Kogut, M.M.; Siebenmorgen, T.; Zacharias, M.; Samsonov, S.A. Evaluation of replica exchange with repulsive scaling approach for docking glycosaminoglycans. J. Comput. Chem. 2021, 42, 1040–1053. [Google Scholar] [CrossRef]
- Marcisz, M.; Zacharias, M.; Samsonov, S.A. Modeling protein–glycosaminoglycan complexes: Does the size matter? J. Chem. Inf. Model. 2021, 61, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Samsonov, S.A.; Pisabarro, M.T. Importance of IdoA and IdoA (2S) ring conformations in computational studies of glycosaminoglycan–protein interactions. Carbohydr. Res. 2013, 381, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Ricard-Blum, S.; Moschcovich, L.; Wineman, E.; Mesilaty, S.; Kessler, E. Binding of procollagen C-proteinase enhancer-1 (PCPE-1) to heparin/heparan sulfate: Properties and role in PCPE-1 interaction with cells. J. Biol. Chem. 2010, 285, 33867–33874. [Google Scholar] [CrossRef]
- Penk, A.; Baumann, L.; Huster, D.; Samsonov, S.A. NMR and molecular modeling reveal specificity of the interactions between CXCL14 and glycosaminoglycans. Glycobiology 2019, 29, 715–725. [Google Scholar] [CrossRef]
- Bojarski, K.K.; Samsonov, S.A. Role of Oligosaccharide Chain Polarity in Protein–Glycosaminoglycan Interactions. J. Chem. Inf. Model. 2020, 61, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Fu, Y.; Zhou, H.; He, Y.; Luo, Y. Contributions of Zn (II)-binding to the structural stability of endostatin. FEBS Lett. 2007, 581, 3027–3032. [Google Scholar] [CrossRef] [PubMed]
- Marcisz, M.; Maszota-Zieleniak, M.; Huard, B.; Samsonov, S.A. Advanced molecular dynamics approaches to model a tertiary complex APRIL/TACI with long glycosaminoglycans. Biomolecules 2021, 11, 1349. [Google Scholar] [CrossRef]
- Wang, N.; Silver, D.L.; Thiele, C.; Tall, A.R. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J. Biol. Chem. 2001, 276, 23742–23747. [Google Scholar] [CrossRef]
- Tersariol, I.L.; Dietrich, C.P.; Nader, H.B. Interaction of heparin with myosin ATPase: Possible involvement with the hemorrhagic activity and a correlation with antithrombin III high affinity-heparin molecules. Thromb. Res. 1992, 68, 247–258. [Google Scholar] [CrossRef]
- Felix, C.F.; Oliveira, V.H.; Moreira, O.C.; Mignaco, J.A.; Barrabin, H.; Scofano, H.M. Inhibition of plasma membrane Ca2+-ATPase by heparin is modulated by potassium. Int. J. Biochem. Cell Biol. 2007, 39, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Conformational Analysis Tools. Available online: http://www.md-simulations.de/CAT/index.html (accessed on 10 January 2024).
- Sattelle, B.M.; Hansen, S.U.; Gardiner, J.; Almond, A. Free energy landscapes of iduronic acid and related monosaccharides. JACS 2010, 132, 13132–13134. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER16; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993, 293, 849–858. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96). kdd 1996, 96, 226–231. [Google Scholar]
- Samsonov, S.A.; Gehrcke, J.P.; Pisabarro, M.T. Flexibility and explicit solvent in molecular-dynamics-based docking of protein–glycosaminoglycan systems. J. Chem. Inf. Model. 2014, 54, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of chemical theory and computation 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. Free energy calculations of glycosaminoglycan–protein interactions. Glycobiology 2009, 19, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Marcisz, M.; Gaardløs, M.; Bojarski, K.K.; Siebenmorgen, T.; Zacharias, M.; Samsonov, S.A. Explicit Solvent Repulsive Scaling Replica Exchange Molecular Dynamics (RS-REMD) in Molecular Modeling of Protein-Glycosaminoglycan Complexes. J. Comp. Chem. 2022, 43, 1633–1640. [Google Scholar] [CrossRef]
- Marcisz, M.; MAszota-Zieleniak, M.; Samsonov, S.A. Repulsive Scaling Replica Exchange Molecular Dynamics in Modeling Protein-Glycosaminoglycan Complexes. Proteoglycans Methods Protoc. Methods Mol. Biol. 2023, 2619, 153–167. [Google Scholar]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.2r3pre; Schrödinger, LLC: New York, NY, USA, 2010.
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔG [kcal/mol] | GAG |
|---|---|
| −30.8 ± 8.5 | HP (GlcNS(6S)-IdoA(2S)) dp2, 1C4 |
| −45.6 ± 7.7 | HP (GlcNS(6S)-IdoA(2S)) dp4, 1C4 |
| −50.3 ± 11.0 | HP (GlcNS(6S)-IdoA(2S)) dp6, 1C4 |
| −53.8 ± 17.9 | HP (GlcNS(6S)-IdoA(2S)) dp8, 1C4 |
| −33.6 ± 7.2 | HP (GlcNS(6S)-IdoA(2S)) dp2, 2S0 |
| −31.1 ± 5.7 | HP (GlcNS(6S)-IdoA(2S)) dp4, 2S0 |
| −68.5 ± 14.4 | HP (GlcNS(6S)-IdoA(2S)) dp6, 2S0 |
| −70.5 ± 14.9 | HP (GlcNS(6S)-IdoA(2S)) dp8, 2S0 |
| −18.8 ± 4.2 | desulfated HS (GlcNAc-IdoA) dp2 |
| −25.7 ± 4.2 | desulfated HS (GlcNAc-IdoA) dp4 |
| −43.4 ± 1.8 | desulfated HS (GlcNAc-IdoA) dp6 |
| −48 ± 13.6 | desulfated HS (GlcNAc-IdoA) dp8 |
| −48.2 ± 8.8 | HS (GlcNS(6S)-GlcA) dp6 |
| −47.7 ± 10.7 | HS (GlcNS-GlcA) dp6 |
| −48.0 ± 12.1 | HS (GlcNS-IdoA(2S)) dp6, 1C4 |
| −56.9 ± 11.1 | HS (GlcNS-IdoA(2S)) dp6, 2S0 |
| ΔG (kcal/mol) | GAG |
|---|---|
| −62.5 ± 14.5 | HP (GlcNS(6S)-IdoA(2S))4 1C4 |
| −51.8 ± 22.1 | (GlcNS(6S)-GlcA)4 |
| −42.9 ± 13.3 | (GlcNS-GlcA)4 |
| −49.2 ± 13.6 | (GlcNS-IdoA(2S))4 2S0 |
| −51.9 ± 13.4 | (GlcNS-IdoA(2S))4 1C4 |
| ΔG (kcal/mol) | Complex (n°) |
|---|---|
| −91.0 | 1 |
| −106.7 | 2 |
| −65.7 | 3 |
| −71.4 | 4 |
| −80.6 | 5 |
| −87.3 | 6 |
| −92.0 | 7 |
| −88.9 | 8 |
| −56.4 | 9 |
| −79.3 | 10 |
| −101.1 | 11 |
| −94.1 | 12 |
| −78.8 | 13 |
| −67.4 | 14 |
| −99.8 | 15 |
| −52.9 | 16 |
| −105.1 | 17 |
| −84.0 | 18 |
| −65.7 | 19 |
| −79.5 | 20 |
| ΔG [kcal/mol] | Amino Acid Residue |
|---|---|
| −3.9 | His1 |
| −4.6 | Arg4 |
| −8.0 | Arg24 |
| −12.9 | Arg27 |
| −5.9 | Arg38 |
| −5.6 | Arg47 |
| −10.8 | Arg53 |
| −5.8 | Arg62 |
| −7.8 | Arg63 |
| −4.6 | Arg66 |
| −2.8 | Lys75 |
| −2.3 | Arg99 |
| −2.9 | Lys106 |
| −2.9 | Arg110 |
| −2.4 | Lys117 |
| −3.9 | Arg128 |
| −4.5 | Arg129 |
| −11.2 | Arg139 |
| −2.4 | Arg156 |
| ΔG [kcal/mol] | Mutant |
|---|---|
| −79.3 ± 14.5 | D30R |
| −81.6 ± 15.5 | D56R |
| −64.1 ± 10.3 | D65R |
| −107.2 ± 12.7 | D30R, D56R |
| −111.4 ± 19.8 | D30R, D56R, D65R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uciechowska-Kaczmarzyk, U.; Frank, M.; Samsonov, S.A.; Maszota-Zieleniak, M. Structural Insights into Endostatin–Heparan Sulfate Interactions Using Modeling Approaches. Molecules 2024, 29, 4040. https://doi.org/10.3390/molecules29174040
Uciechowska-Kaczmarzyk U, Frank M, Samsonov SA, Maszota-Zieleniak M. Structural Insights into Endostatin–Heparan Sulfate Interactions Using Modeling Approaches. Molecules. 2024; 29(17):4040. https://doi.org/10.3390/molecules29174040
Chicago/Turabian StyleUciechowska-Kaczmarzyk, Urszula, Martin Frank, Sergey A. Samsonov, and Martyna Maszota-Zieleniak. 2024. "Structural Insights into Endostatin–Heparan Sulfate Interactions Using Modeling Approaches" Molecules 29, no. 17: 4040. https://doi.org/10.3390/molecules29174040
APA StyleUciechowska-Kaczmarzyk, U., Frank, M., Samsonov, S. A., & Maszota-Zieleniak, M. (2024). Structural Insights into Endostatin–Heparan Sulfate Interactions Using Modeling Approaches. Molecules, 29(17), 4040. https://doi.org/10.3390/molecules29174040