3.2. Clustering Results
In this section, we present the clustering results obtained from applying four different deep learning methodologies: Deep Belief Network (DBN), Convolutional Autoencoder (CAE), Variational Autoencoder (VAE), and Adversarial Autoencoder (AAE). For each of these deep learning architectures, we employed k-means clustering with cluster sizes of 16, 32, and 64. The results showcase the effectiveness of each method in grouping the molecules based on their spectral geometric properties. By comparing these results, we aim to identify the most suitable deep learning approach and cluster size for capturing the inherent structural patterns in the molecular dataset. Detailed interpretations of the clustering outcomes will provide insights into the performance and robustness of each methodology in the context of molecular clustering.
Table 1 highlights the clustering performance for DBNs with different partitions.
The Calinski–Harabasz index is used to evaluate the quality of clustering by measuring the ratio of between-cluster dispersion to within-cluster dispersion. Higher values indicate better-defined clusters. For the DBN + k-means results, we observe that with clusters, the index is 1532.07, indicating a moderate level of cluster separation. As the number of clusters increases to , the index rises to 2175.96, suggesting an improvement in the separation and definition of the clusters. With clusters, the index significantly increases to 14,471.6, indicating that the clusters are exceptionally well-defined and separated. This trend demonstrates that increasing the number of clusters leads to better-defined clusters as measured by the Calinski–Harabasz index.
The Davies–Bouldin index assesses the average similarity ratio of each cluster with its most similar cluster, with lower values indicating better clustering. For clusters, the index is 0.44316, suggesting that the clusters are relatively compact and well separated. When the number of clusters is increased to , the index decreases to 0.35608, indicating improved compactness and separation of clusters. At , the index further decreases to 0.19196, showing that the clusters are even more compact and well separated. This decreasing trend highlights the effectiveness of using a higher number of clusters for achieving better clustering results.
The Dunn index measures the ratio of the minimum inter-cluster distance to the maximum intra-cluster distance, with higher values indicating better clustering. For clusters, the index is 0.04986, reflecting a relatively low separation between clusters. As the number of clusters increases to , the index improves to 0.112182, indicating better cluster separation. With , the index significantly improves to 0.4838, suggesting a high degree of separation between clusters. This substantial improvement with more clusters indicates that higher cluster counts contribute to better-defined and more separated clusters.
R-Squared measures the proportion of variance explained by the clustering, with higher values indicating better clustering. For clusters, the R-Squared value is 0.94382, suggesting that a large portion of the variance in the data is explained by the clustering. When the number of clusters is increased to , the R-Squared value rises to 0.9737, showing an improved explanation of the data variance. At , the R-Squared value further increases to 0.99523, indicating that nearly all the variance in the data is captured by the clustering. This trend demonstrates that a higher number of clusters leads to a more comprehensive explanation of the data variance.
The Silhouette Score evaluates how similar a point is to its own cluster compared to other clusters, with values ranging from −1 to 1 and higher values indicating better clustering. For clusters, the score is 0.52044, suggesting good clustering quality. As the number of clusters increases to , the score slightly decreases to 0.4989, indicating a slight decline in clustering quality. At , the score further decreases to 0.43674, suggesting a more noticeable decline in clustering quality with a higher number of clusters. This decreasing trend indicates that while more clusters may provide better-defined clusters, the overall cohesion and separation measured by the Silhouette Score may slightly decline.
The Standard Deviation measures the spread of points within each cluster, with lower values indicating more compact clusters. For clusters, the standard deviation is 1.8814, suggesting a relatively high spread within clusters. As the number of clusters increases to , the standard deviation decreases to 1.0811, indicating more compact clusters. At , the standard deviation significantly decreases to 0.28871, showing highly compact clusters. This trend demonstrates that increasing the number of clusters leads to more compact clustering.
Overall, the clustering results for DBN + k-means indicate that increasing the number of clusters generally improves cluster separation and definition as measured by the Calinski–Harabasz, Davies–Bouldin, Dunn, and R-Squared indices. However, the Silhouette Score suggests a slight decline in clustering quality with a higher number of clusters. The standard deviation shows that clusters become more compact with an increasing number of clusters.
The clustering performance for CAE with different partitions is highlighted in
Table 2.
The CAE + k-means findings show that with clusters, the index is 1465.42, which suggests a moderate level of cluster separation. When the number of clusters is raised to , the index climbs to 1984.89, indicating enhanced cluster delineation. When using 64 clusters, the index experiences a substantial increase to 11,659.3, suggesting the presence of highly distinct and well-defined clusters. The observed pattern indicates that a higher number of clusters generally improves the distinction across clusters, as quantified by the Calinski–Harabasz index.
With 16 clusters, the index is 0.43703, indicating that the clusters are relatively compact and well separated. When the number of clusters is raised to , the index lowers slightly to 0.3888, suggesting an enhancement in the compactness and separation of the clusters. When , the index lowers to 0.253, indicating that the clusters become more compact and distinct with 64 clusters. The declining pattern emphasizes the efficacy of employing a larger number of clusters to obtain superior clustering outcomes.
With 16 clusters, the index is 0.08787, indicating a rather low level of separation between the clusters. As the number of clusters increases to , the index lowers to 0.03924, suggesting a decrease in the separation between clusters. When , the index is 0.0264, indicating that there is still room for more reduction in cluster separation. This trend suggests that as the number of clusters rises, their compactness increases while their distance from each other decreases.
With 16 clusters, the R-Squared value is 0.9446, indicating that a significant amount of the variability in the data can be accounted for by the grouping. By increasing the number of clusters to , the R-Squared value increases to 0.9728, indicating a higher level of explanation for the variance in the data. When , the R-Squared value increases even more to 0.995, which suggests that almost all of the variability in the data is accounted for by the clustering. This trend illustrates that an increased number of clusters results in a more thorough elucidation of the variance in the data.
The clustering quality is considered good with a score of 0.5893 for 16 clusters. When the number of clusters is increased to , the score lowers slightly to 0.5549, suggesting a minor decrease in the quality of clustering. When , the score lowers even further to 0.3656, indicating a significant decrease in the quality of clustering when there are more clusters. This declining trend suggests that increasing the number of clusters may result in more distinct clusters, but it may also lead to a decrease in the overall cohesiveness and separation as measured by the Silhouette Score.
With clusters, the standard deviation is 2.1218, indicating a significant dispersion among the clusters. As the number of clusters increases to , the standard deviation drops to 1.2206, suggesting a higher level of cluster compactness. When , the standard deviation reduces significantly to 0.3887, indicating the presence of highly compact clusters. This pattern illustrates that augmenting the quantity of clusters results in clustering that is more condensed.
To summarize, the clustering outcomes for CAE + k-means demonstrate that augmenting the number of clusters typically enhances the distinction and precision of the clusters, as assessed by the Calinski–Harabasz and Davies–Bouldin indices. Nevertheless, the Dunn index indicates that as the number of clusters increases, there is a reduction in the spacing between the clusters. The R-Squared values exhibit a positive trend, suggesting a more effective explanation of the variability in the data as the number of clusters increases. The Silhouette Score demonstrates a negative correlation between clustering quality and the number of clusters, whereas the standard deviation suggests an increase in cluster compactness.
Table 3 highlights the clustering performance for VAE with different partitions.
The VAE + k-means results, with clusters, yield an index of 1432.01, indicating a considerable degree of cluster separation. When the number of clusters is increased to , the index shows a considerable increase to 3807.64, suggesting a notable enhancement in the definition of the clusters. When the number of clusters is increased to , the index rises to 5630.58, indicating that the clusters become more distinct and separated with a larger number of clusters. The observed pattern indicates that augmenting the number of clusters improves the distinction between clusters, as quantified by the Calinski–Harabasz index.
With 16 clusters, the index is 0.446, suggesting that the clusters are relatively compact and well separated. As the number of clusters increases to , the index lowers to 0.3958, indicating enhanced cluster separation and compactness. When , the index lowers to 0.2166, indicating that the clusters become more compact and distinct from each other. The declining pattern emphasizes the efficacy of employing a larger number of clusters to attain superior clustering outcomes.
With 16 clusters, the index is 0.0219, indicating a rather low level of separation between the clusters. As the number of clusters increases to , the index improves to 0.0614, suggesting enhanced cluster separation. When , the index experiences a minor fall to 0.0271, indicating a decrease in the spacing between clusters. This pattern suggests that although having more clusters may result in improved early differentiation, it becomes increasingly difficult to sustain this differentiation as the number of clusters continues to grow.
With 16 clusters, the R-Squared value is 0.9444, indicating that a significant proportion of the variability in the data can be accounted for by the grouping. By increasing the number of clusters to , the R-Squared value increases to 0.9785, indicating a higher level of explanation for the variance in the data. When , the R-Squared value increases even more to 0.9932, suggesting that almost all of the variability in the data is accounted for by the clustering. This trend illustrates that an increased number of clusters results in a more thorough elucidation of the variance in the data.
The clustering quality is considered good, with a score of 0.576 for clusters. When the number of clusters is increased to , the score lowers slightly to 0.5267, suggesting a minor decrease in the quality of clustering. When , the score reduces to 0.3951, indicating a significant decrease in the quality of clustering as the number of clusters increases. This declining trend suggests that increasing the number of clusters may result in more distinct clusters, but it may also lead to a decrease in the overall cohesiveness and separation as measured by the Silhouette Score.
With clusters, the standard deviation is 1.5746, indicating a significant dispersion among the clusters. As the number of clusters increases to , the standard deviation reduces to 0.663, suggesting a higher level of cluster compactness. When , the standard deviation reduces significantly to 0.371, indicating the presence of highly compact clusters. This pattern illustrates that augmenting the quantity of clusters results in clustering that is more condensed.
In general, the clustering results for VAE + k-means suggest that increasing the number of clusters enhances the separation and definition of the clusters, as evaluated by the Calinski–Harabasz and Davies–Bouldin indices. The Dunn index demonstrates an initial enhancement in the distinction between clusters but experiences a little deterioration as the number of clusters becomes excessively high. The R-Squared values exhibit a positive trend, suggesting a more effective explanation of the variability in the data as the number of clusters increases. The Silhouette Score demonstrates a negative correlation between clustering quality and the number of clusters, whereas the standard deviation suggests an increase in cluster compactness.
The clustering performance for AAE with different partitions is highlighted in
Table 4.
The AAE + k-means results show a moderate level of cluster separation with an index of 1090.47 for clusters. The index climbs to 2744.92 when the number of clusters is increased to , indicating a significant improvement in cluster definition. Clusters with show exceptionally well-defined and distinct features, as the index surges to 52,322.5. This pattern suggests that the Calinski–Harabasz index measures cluster separation better as the number of clusters increases.
The index for clusters with K = 16 is 0.3801, suggesting that the clusters are moderately compact and well separated. A drop in the index to 0.2897 at indicates better cluster separation and compactness as the number of clusters is raised. The clusters become increasingly more compact and well separated when the index drops to 0.1157 at . It is clear from this declining trend that increasing the number of clusters yields better clustering outcomes.
As a result, the index for clusters with K = 16 is 0.1761, which indicates a modest level of separation between them. With a higher number of clusters (K = 32), the index improves to 0.2129, suggesting that the clusters are better separated. A high level of cluster separation is shown by the index’s considerable increase to 0.5673 at . This pattern shows that more clusters result in more distinct and well-defined clusters.
The clustering explains a significant amount of the data variation, as indicated by the R-Squared value of 0.9343 for clusters. An enhanced explanation of the data variance is demonstrated by an R-Squared value of 0.9774 when the number of clusters is raised to . The R-Squared value rises to 0.9978 at , suggesting that clustering accounts for almost all of the data variance. As we can see from this pattern, adding more clusters helps to explain the data variance more thoroughly.
The score of 0.5026 indicates acceptable clustering quality for clusters with K = 16. With an increase in the number of clusters to 32, the score drops to 0.5033, suggesting that the clustering quality remains consistent. The score drops even lower to 0.4793 for , indicating that clustering quality slightly declines as the number of clusters increases. Although a larger number of clusters may result in more clearly defined clusters, this trend suggests that the Silhouette Score’s overall measure of group cohesiveness and separation may slightly decrease.
The standard deviation of 2.0263 for clusters with K = 16 indicates a reasonably high spread within clusters. With an increase in the number of clusters to 32, the standard deviation drops to 0.9135, suggesting that the clusters are becoming more compact. Highly compact clusters are indicated by a considerable decrease in the standard deviation to 0.1435 at . Increasing the number of clusters causes clustering to become more compact, as shown by this pattern.
In general, the Calinski–Harabasz, Davies–Bouldin, and Dunn indices show that increasing the number of clusters generally enhances cluster separation and definition, according to the clustering results for AAE + k-means. An upward trend in the R-Squared values suggests that more clusters provide a more satisfactory explanation of the data variation. Standard deviation demonstrates that clusters become denser as cluster size increases, while Silhouette Score shows stable clustering quality with a small drop at maximum cluster size.
3.3. Nonparametric Tests
To comprehensively evaluate the impact of topological descriptors on the clustering results, we will employ nonparametric statistical tests. These tests are particularly suitable given the nature of our data, which may not adhere to normal distribution assumptions. By applying nonparametric tests, we aim to rigorously assess whether the topological indices significantly influence the formation and quality of clusters across different clustering methodologies. Specifically, we will analyze the effects of topological descriptors on the clusters generated by Deep Belief Network (DBN), Convolutional Autoencoder (CAE), Variational Autoencoder (VAE), and Adversarial Autoencoder (AAE) combined with k-means clustering. The test findings yield vital insights into the structural factors that determine molecular clustering. Additionally, they will emphasize the strength and dependability of each clustering approach in accurately representing the underlying topological features of the molecules.
We present the nonparametric test results in
Table 5 for clusters determined by the DBN.
Table 5 presents the nonparametric test results for the effect of topological descriptors on clusters formed by the Deep Belief Network (DBN) combined with k-means clustering. The tests performed include the Kruskal–Wallis test, the Conover post hoc test, and the Log-Rank test. These tests evaluate the significance of the differences in topological indices across clusters for different numbers of clusters (K = 16, 32, 64).
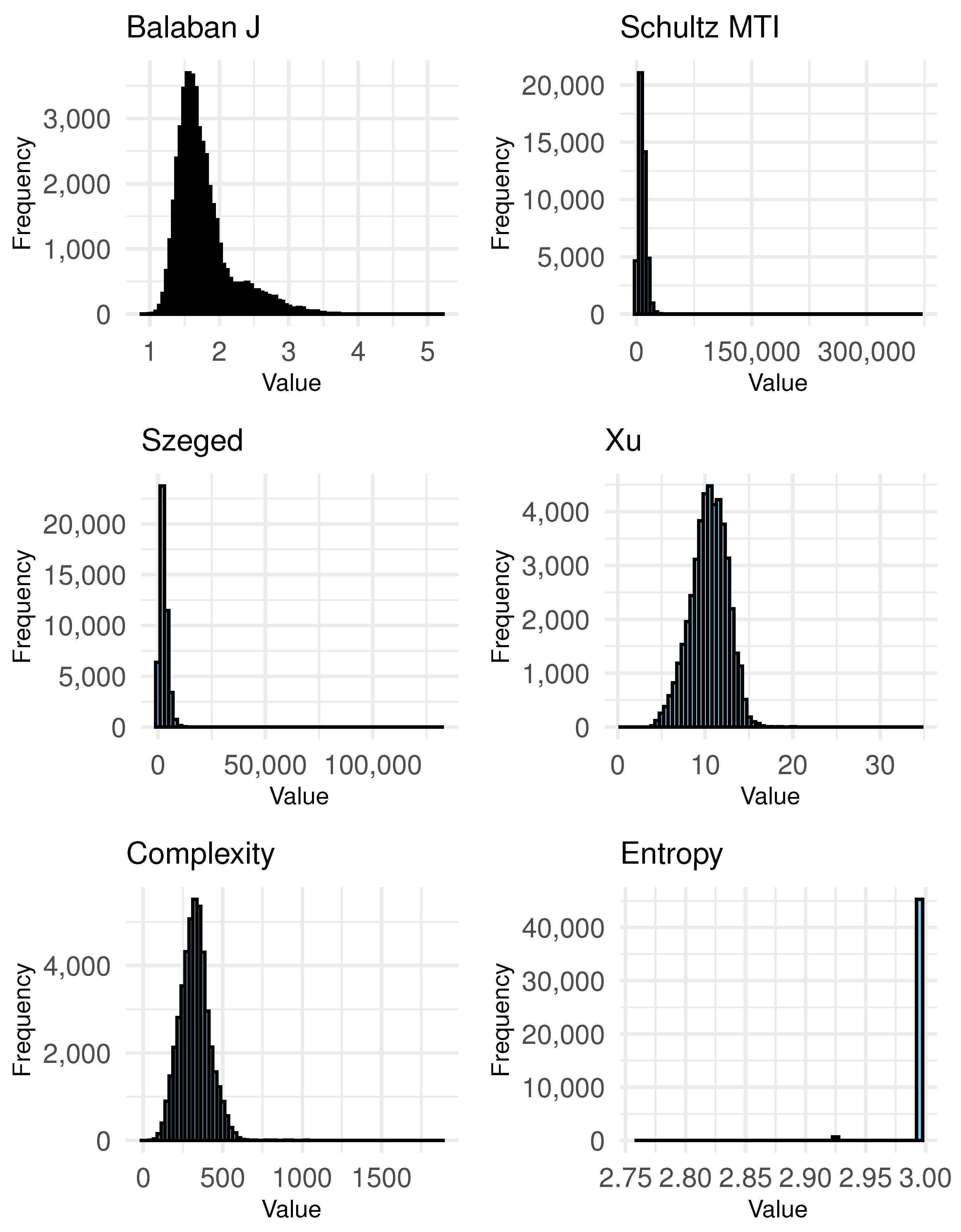
For clusters, the Kruskal–Wallis test shows significant results for Balaban J, indicating that there are differences in this descriptor across clusters. The Conover post hoc test also shows significance for Balaban J. The Log-Rank test supports these findings with a p-value. Other descriptors such as Schultz MTI, Szeged, Xu, Complexity, and Entropy of AC do not show significant differences across clusters, as indicated by higher p-values.
When the number of clusters increases to , the Kruskal–Wallis test indicates significant differences for Schultz MTI, while other descriptors do not show significant results. The Conover test reveals some significance for Schultz MTI and Szeged, indicating that these descriptors have significant differences across clusters. The Log-Rank test supports the significance for Schultz MTI, confirming that this descriptor varies significantly across clusters.
For clusters, the Kruskal–Wallis test does not show significant results for any of the topological descriptors, as all p-values are well above 0.05. This indicates that there are no statistically significant differences in the distribution of these descriptors across the clusters. The Conover and Log-Rank tests similarly do not show significant results for most descriptors, confirming the findings of the Kruskal–Wallis test. Overall, for clusters, the nonparametric tests indicate that there are no significant differences in the topological descriptors across clusters. This suggests that, with a high number of clusters, the impact of topological descriptors on clustering is not statistically significant according to these tests. This result highlights the complexity and potential limitations of using very high numbers of clusters to discern the impact of topological indices on clustering results.
Comparing the results across different numbers of clusters, it is evident that for clusters, only Balaban J shows significant differences across clusters, indicating its impact on clustering. For clusters, multiple descriptors such as Schultz MTI, Szeged, and Complexity show significant differences, suggesting a stronger impact of these descriptors on clustering as the number of clusters increases. However, for clusters, none of the descriptors show significant differences across clusters, indicating that the high number of clusters may dilute the impact of individual topological descriptors on the clustering results.
The nonparametric test findings for clusters defined by CEA are displayed in
Table 6.
For clusters, the Kruskal–Wallis test shows no significant results for any of the topological descriptors, as indicated by high p-values (e.g., Balaban J: , Schultz MTI: , Szeged: ). The Conover and Log-Rank tests also confirm these findings with high p-values, indicating that the topological descriptors do not significantly impact the clustering results for this number of clusters.
When the number of clusters increases to , the Kruskal–Wallis test again shows no significant results for any of the descriptors, with high p-values (e.g., Balaban J: , Schultz MTI: , Complexity: ). The Conover test reveals lower p-values for Schultz MTI, but they are still not significant. The Log-Rank test similarly shows high p-values for all descriptors, indicating that the impact of topological descriptors remains limited for clusters.
For clusters, the Kruskal–Wallis test does not show significant results for any of the topological descriptors, as all p-values are high. The Conover test confirms these findings with high p-values for all descriptors, such as Balaban J, Schultz MTI, and Complexity. The Log-Rank test also supports these results with high p-values, indicating no significant impact of the topological descriptors on the clustering results when there are 64 clusters.
Comparing the results across different numbers of clusters, it is evident that for clusters, none of the topological descriptors show significant differences across clusters, indicating that the clustering structure is not strongly influenced by topological features. For clusters, there are still no significant differences in any of the descriptors. For clusters, none of the descriptors show significant differences across clusters, suggesting that the high number of clusters does not reveal significant impacts of topological descriptors.
The nonparametric test results for CAE + k-means clustering reveal that topological descriptors such as Balaban J, Schultz MTI, Szeged, Xu, Complexity, and Entropy of AC do not significantly influence the clustering results for , , and clusters. These findings suggest that the clustering structure is not significantly affected by the topological descriptors used in this analysis, highlighting the complexity and potential limitations of using these descriptors to discern the impact on clustering results.
We present the nonparametric test results in
Table 7 for clusters determined by VAE.
Based on the high p-values (e.g., Balaban J: , Schultz MTI: , Szeged: ), the Kruskal–Wallis test does not yield any significant results for any of the topological descriptors in the clusters. These findings are also confirmed by the Conover and Log-Rank tests, which yield high p-values. This suggests that the topological descriptors do not have a substantial influence on the clustering results for this number of clusters.
The Kruskal–Wallis test yields no significant results for any of the descriptors when the number of clusters reaches , despite the high p-values. The p-values for Complexity are lower in the Conover test (), but they are still not statistically significant. The Log-Rank test also yields high p-values for all descriptors, suggesting that the influence of topological descriptors is restricted to clusters.
The Kruskal–Wallis test does not yield significant results for any of the topological descriptors in the clusters, as all p-values are high (e.g., Balaban J: , Schultz MTI: , Szeged: , Xu: ). These findings are corroborated by the Conover test, which yields high p-values for all descriptors, including Balaban J (), Schultz MTI (), and Complexity (). The Log-Rank test also supports these results with high p-values, suggesting that the topological descriptors do not have a significant impact on the clustering results when there are 64 clusters.
It is clear that the clustering structure is not significantly influenced by topological features when comparing the results across different numbers of clusters, as none of the topological descriptors exhibit significant differences across clusters for . There are no significant differences in any of the descriptors for clusters. None of the descriptors exhibit significant disparities across clusters for clusters, indicating that the high number of clusters does not reveal significant impacts of topological descriptors.
The clustering results for the , , and clusters are not significantly influenced by topological descriptors such as Balaban J, Schultz MTI, Szeged, Xu, Complexity, and Entropy of AC, as confirmed by the nonparametric test results for VAE + k-means clustering. These results indicate that the topological descriptors employed in this analysis do not have a substantial impact on the clustering structure, underscoring the potential limitations and complexity of utilizing these descriptors to determine their impact on clustering results.
The nonparametric test findings for clusters defined by AAE are displayed in
Table 8.
For clusters, the Kruskal–Wallis test shows no significant results for any of the topological descriptors, as indicated by high p-values (e.g., Balaban J: , Schultz MTI: , Szeged: ). The Conover and Log-Rank tests also confirm these findings with high p-values, indicating that the topological descriptors do not significantly impact the clustering results for this number of clusters.
When the number of clusters increases to , the Kruskal–Wallis test shows no significant results for any of the descriptors, with high p-values (e.g., Balaban J: , Schultz MTI: , Szeged: ). The Conover test reveals lower p-values for Balaban J (), but they are still not significant. The Log-Rank test similarly shows high p-values for all descriptors, indicating that the impact of topological descriptors remains limited for clusters.
For clusters, the Kruskal–Wallis test does not show significant results for any of the topological descriptors, as all p-values are high (e.g., Balaban J: , Schultz MTI: , Szeged: , Xu: ). The Conover test, however, shows significant results for Balaban J (), indicating some impact of this descriptor on the clustering results. The Log-Rank test does not support significant results for most descriptors, confirming the high p-values for Balaban J (), Schultz MTI (), and Szeged ().
Comparing the results across different numbers of clusters, it is evident that for clusters, none of the topological descriptors show significant differences across clusters, indicating that the clustering structure is not strongly influenced by topological features. For clusters, there are still no significant differences in any of the descriptors. For clusters, only Balaban J shows some significance according to the Conover test (), but other tests do not confirm this significance. This suggests that the high number of clusters may reveal some impact of individual topological descriptors on the clustering results, though the overall impact remains limited.
The nonparametric test results for AAE + k-means clustering reveal that topological descriptors such as Balaban J, Schultz MTI, Szeged, Xu, Complexity, and Entropy of AC do not significantly influence the clustering results for and clusters. For clusters, the Conover test indicates some significance for Balaban J, but other tests do not confirm this. These findings suggest that the clustering structure is generally not significantly affected by the topological descriptors used in this analysis.







{kind=link}
{kind=link}
{kind=link}
{kind=link}