
Slowing Down the “Magic Bullet”: Encapsulation of Imatinib in Fe-MOF for Cardiotoxicity Reduction and Improvement in Anticancer Activity

Abstract

1. Introduction
2. Results and Discussion
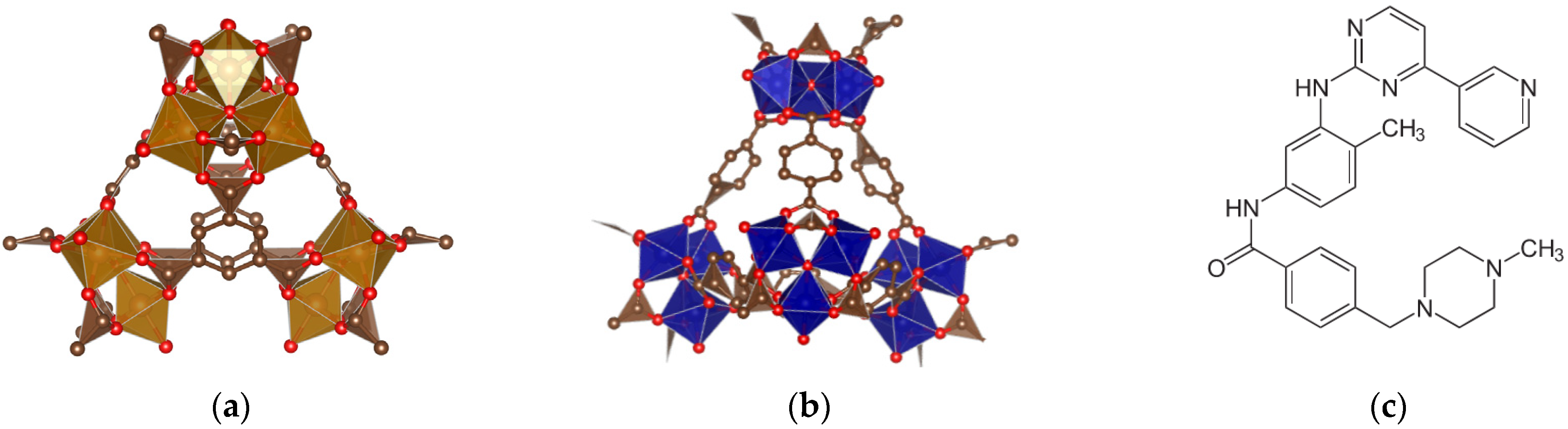
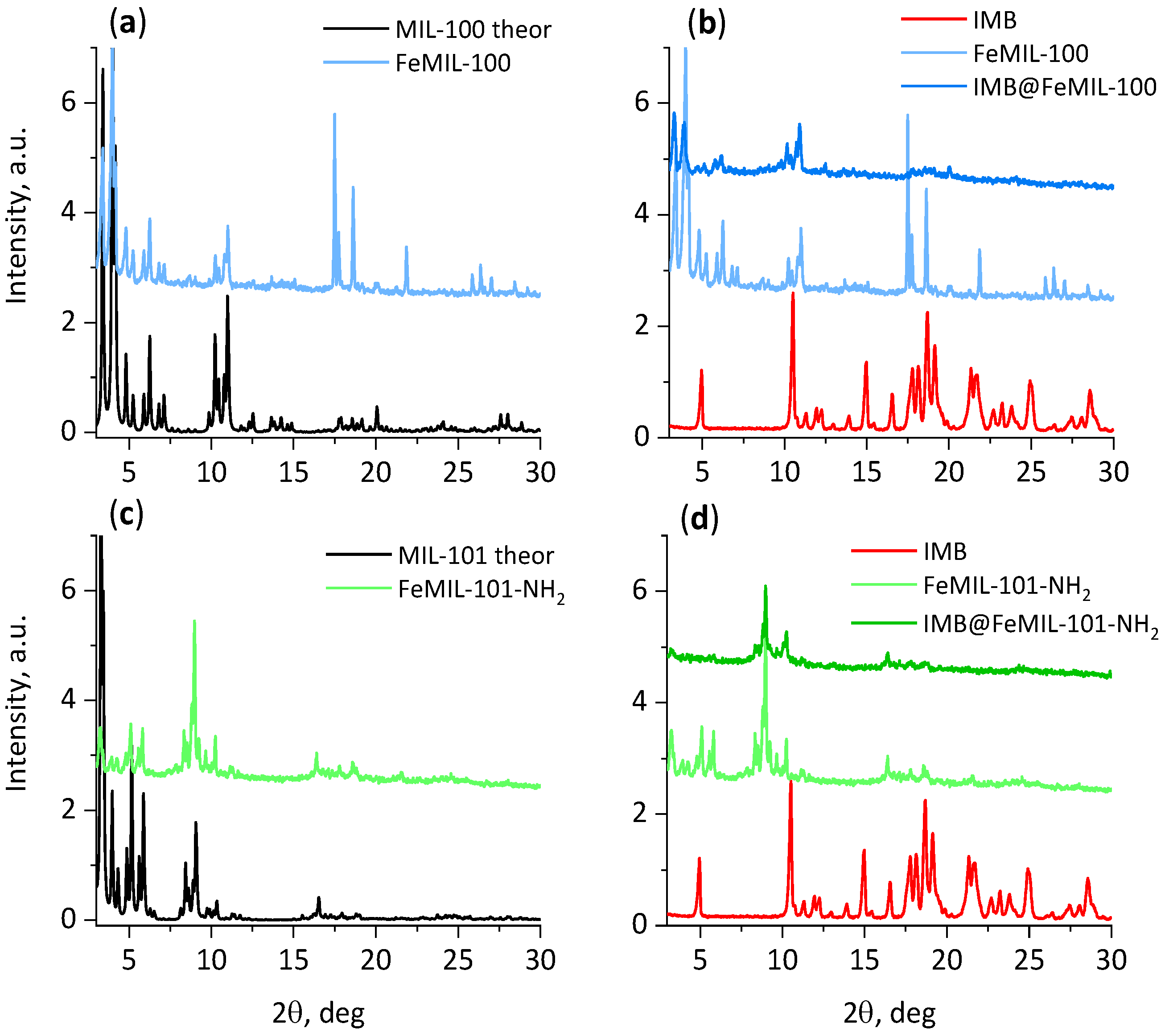
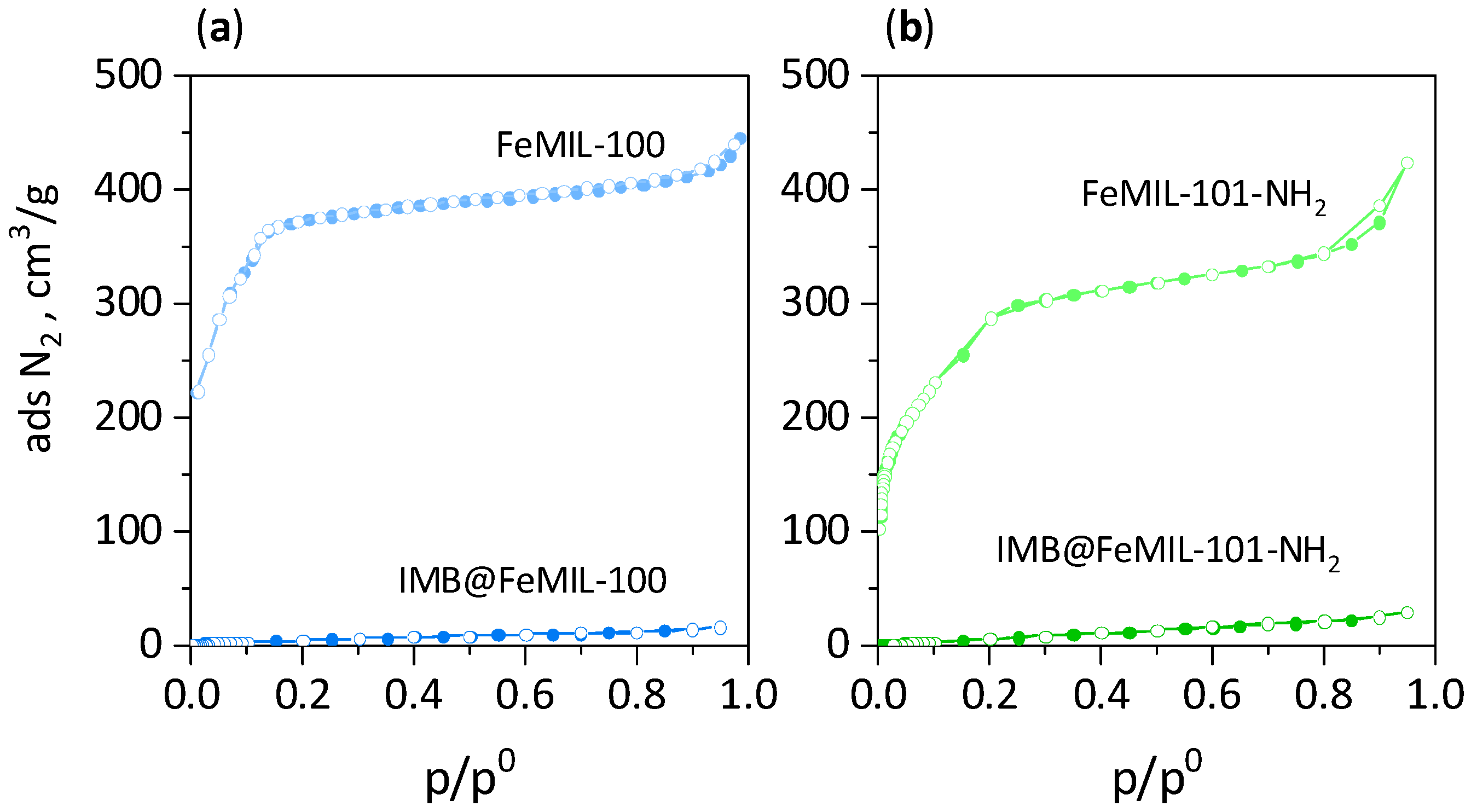
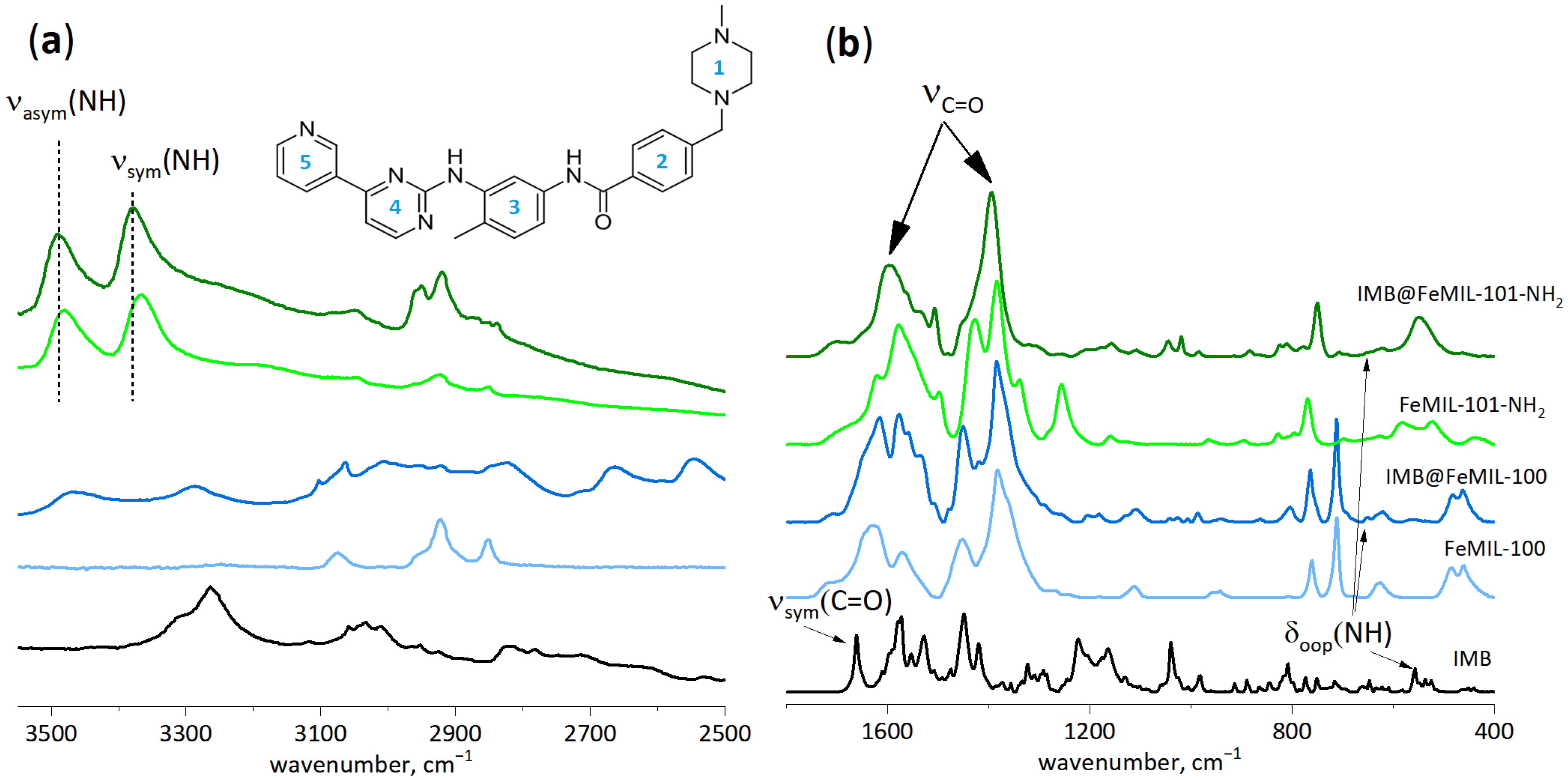
2.1. Preparation and Characterization of IMB@FeMIL-100 and IMB@FeMIL-101-NH2 Composites
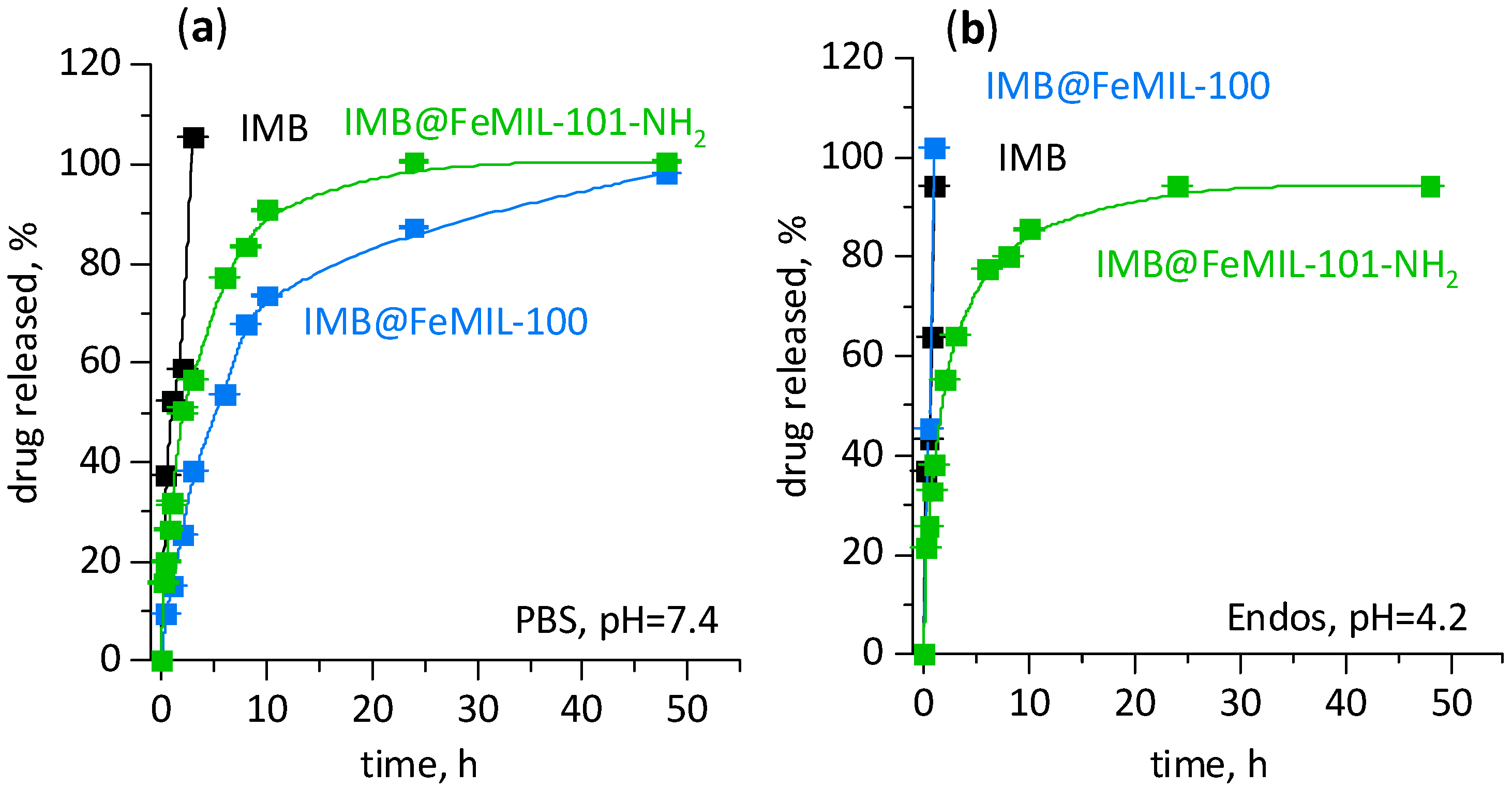
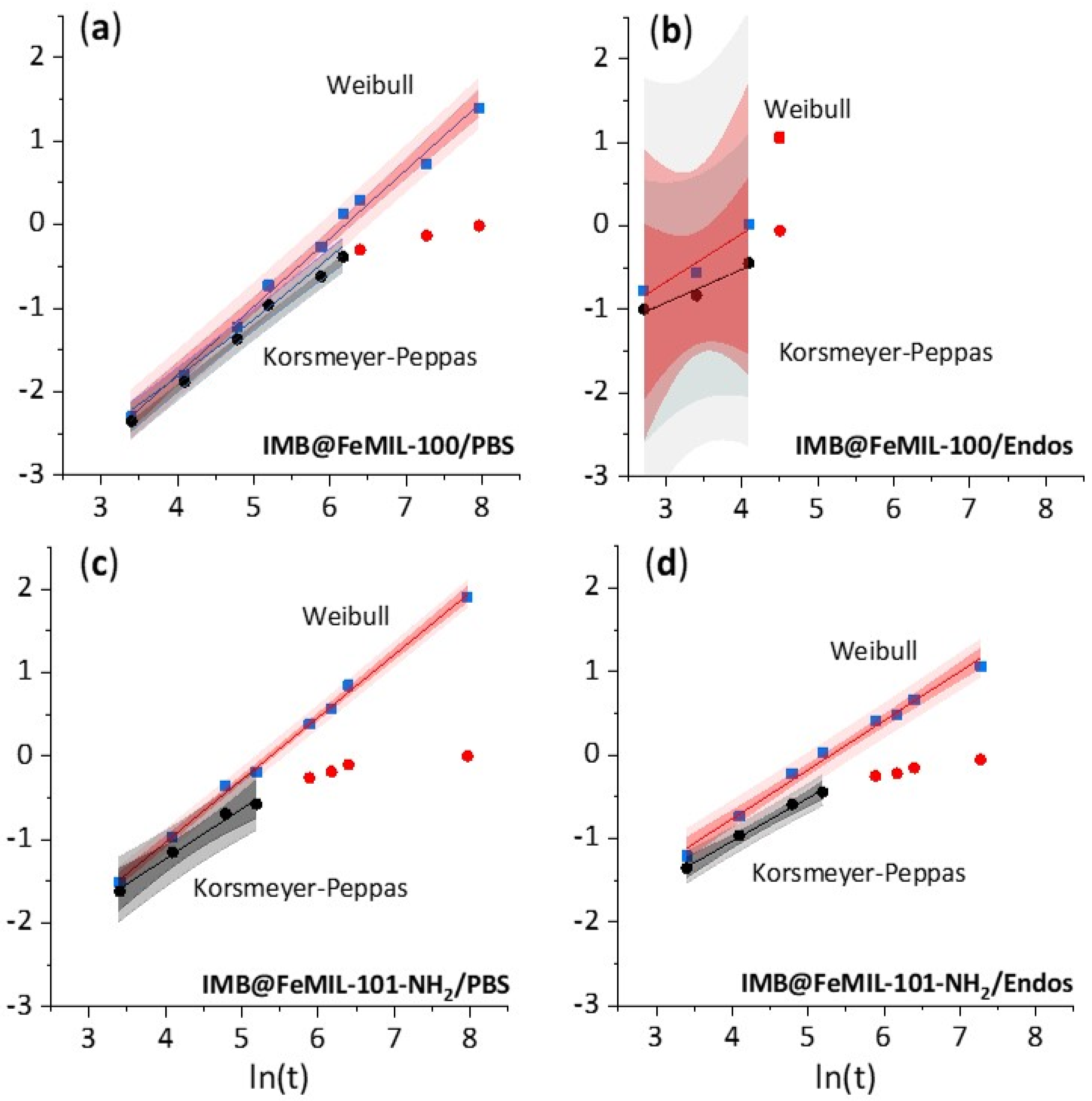
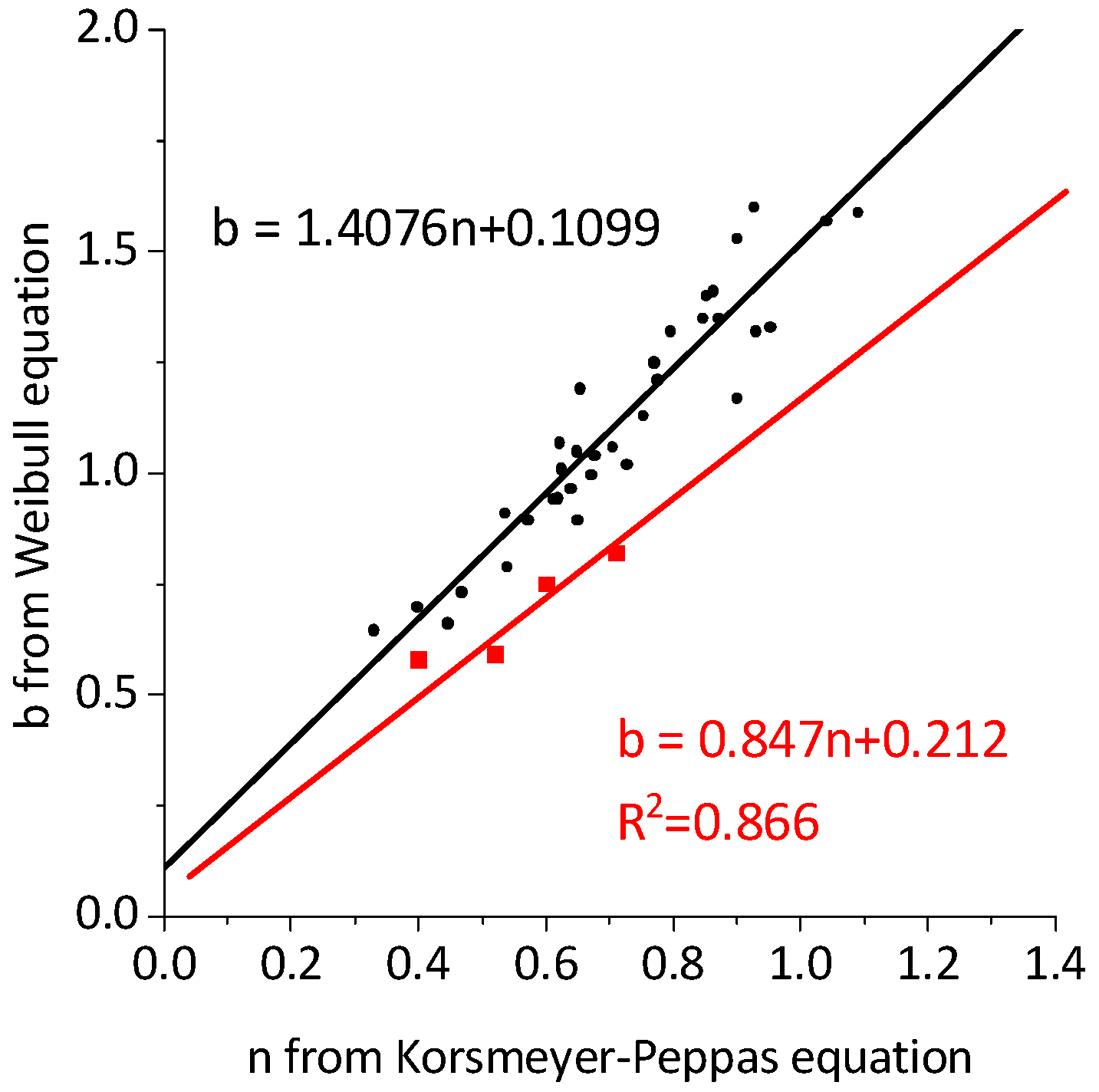
2.2. Drug Release
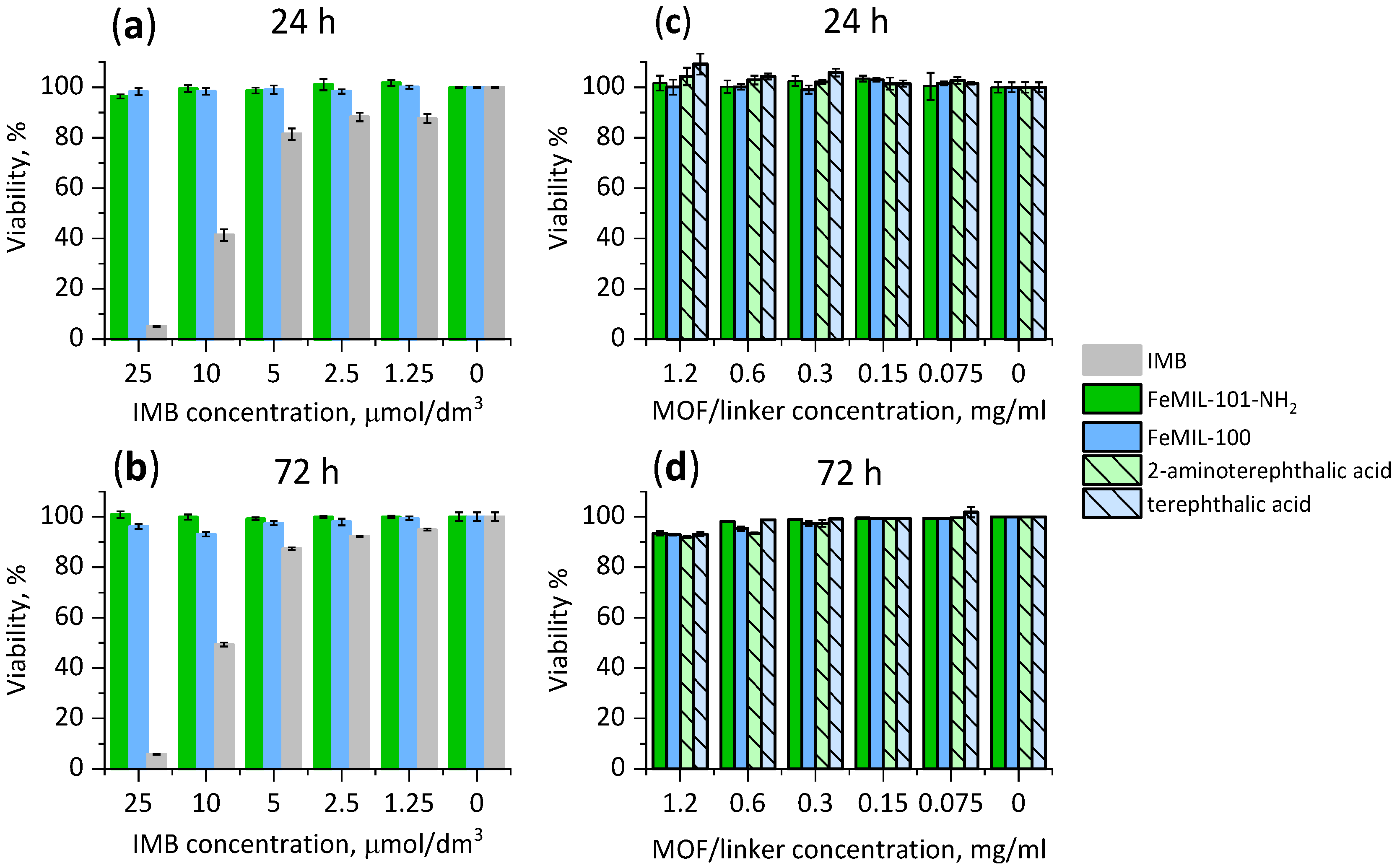
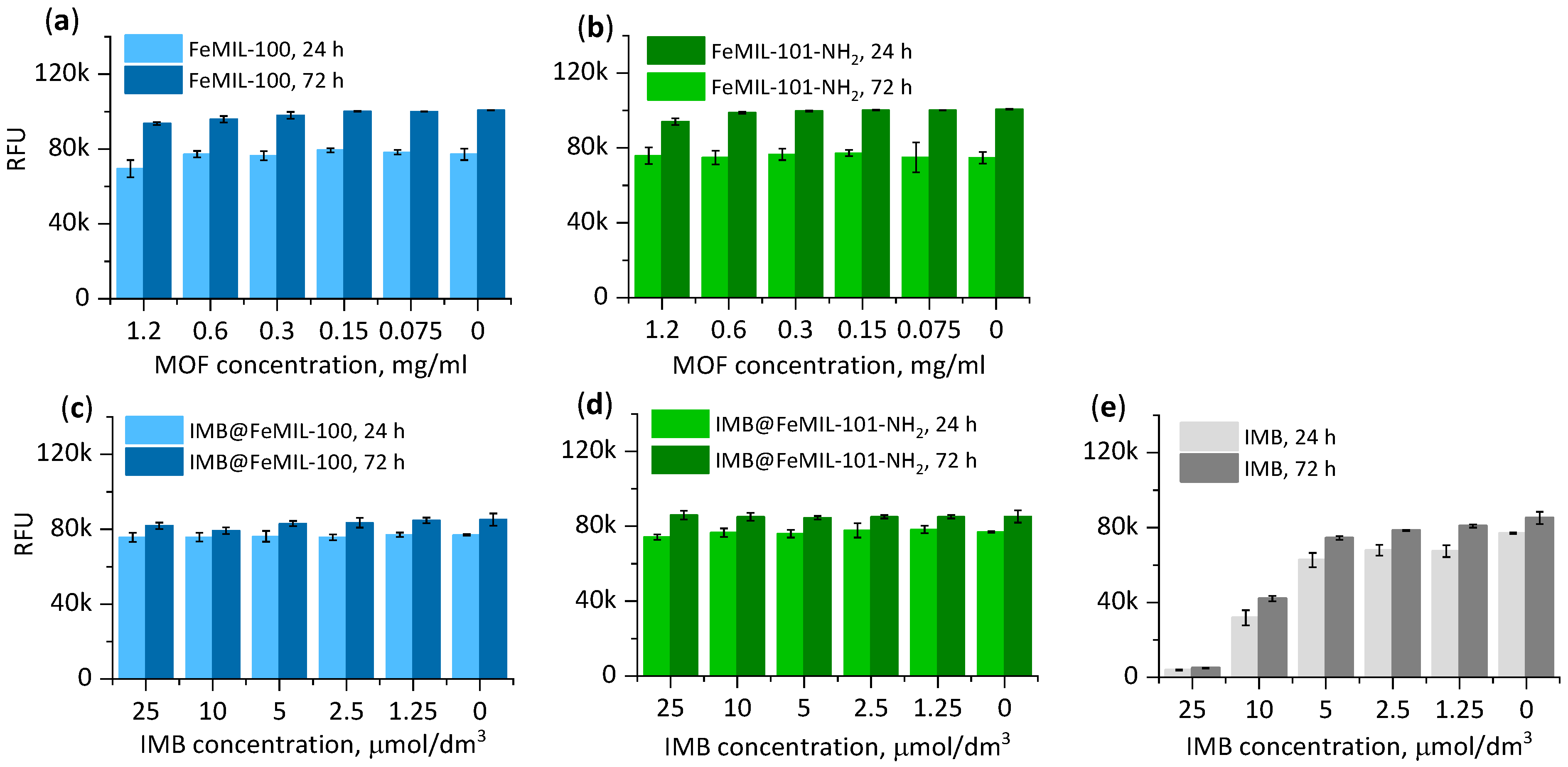
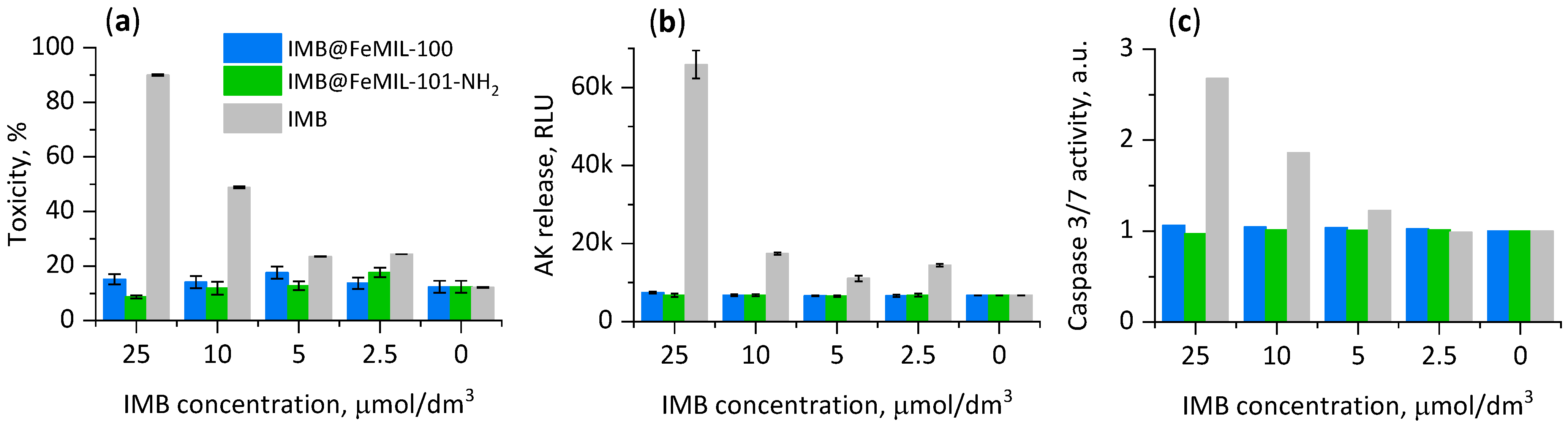
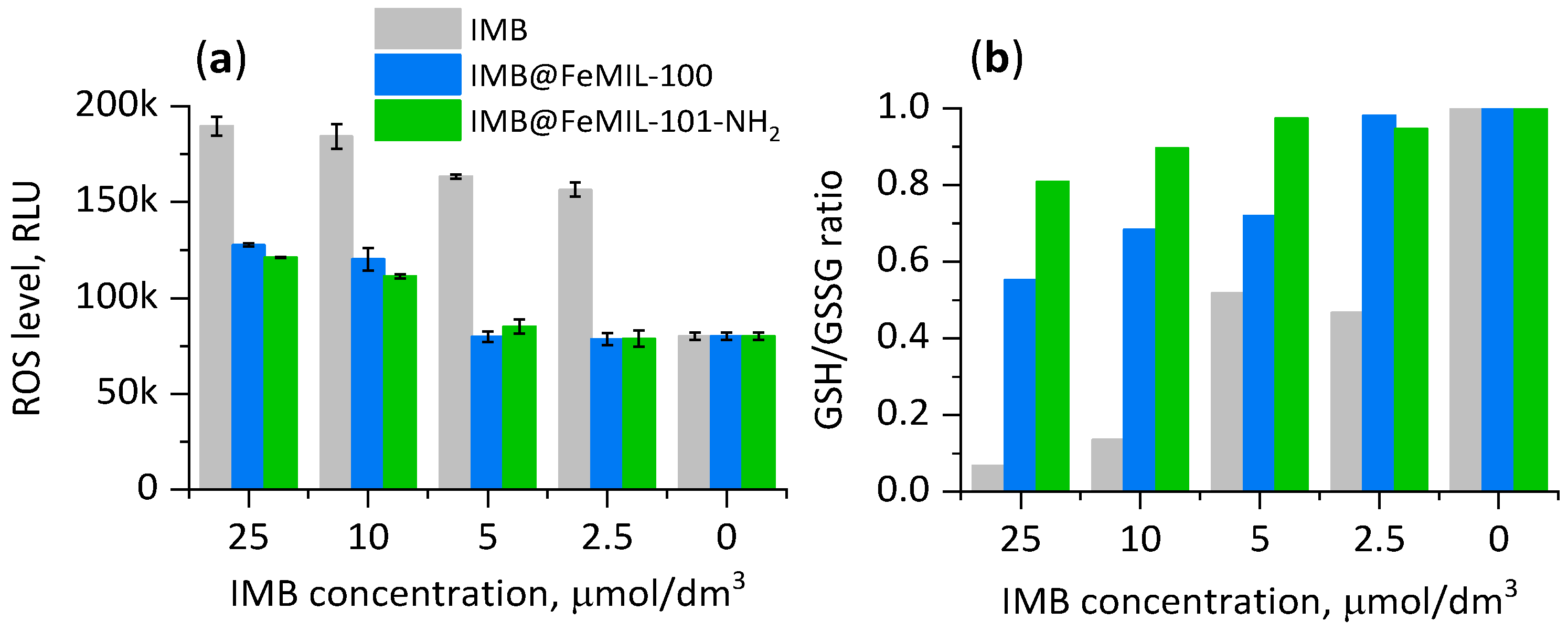
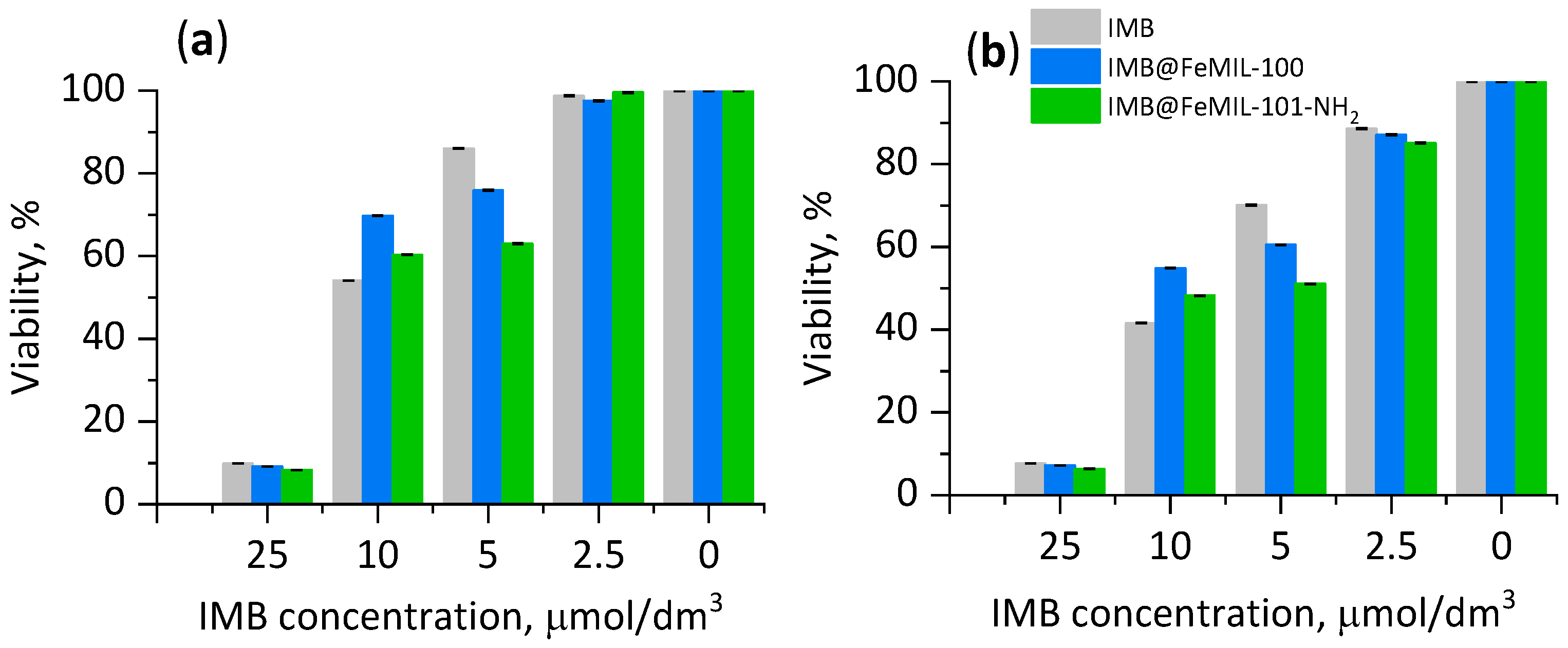
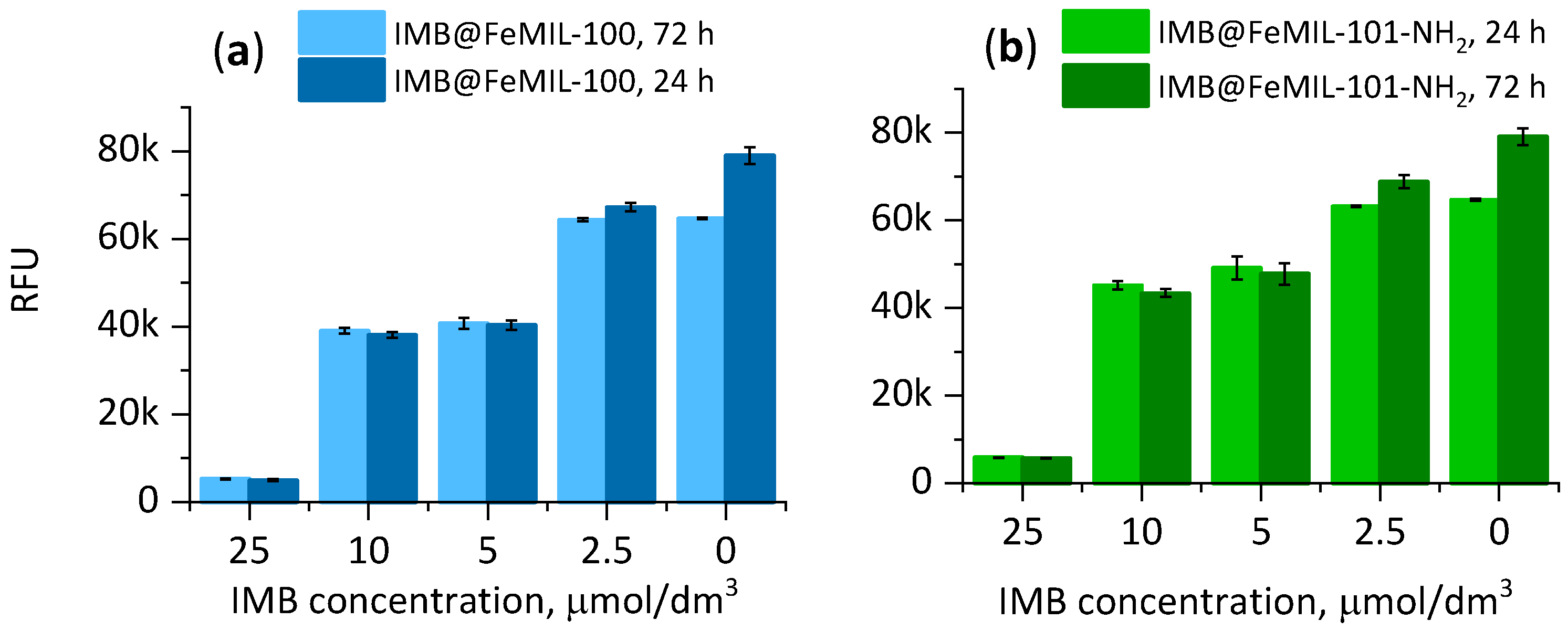
2.3. Cellular Cytotoxicity Studies
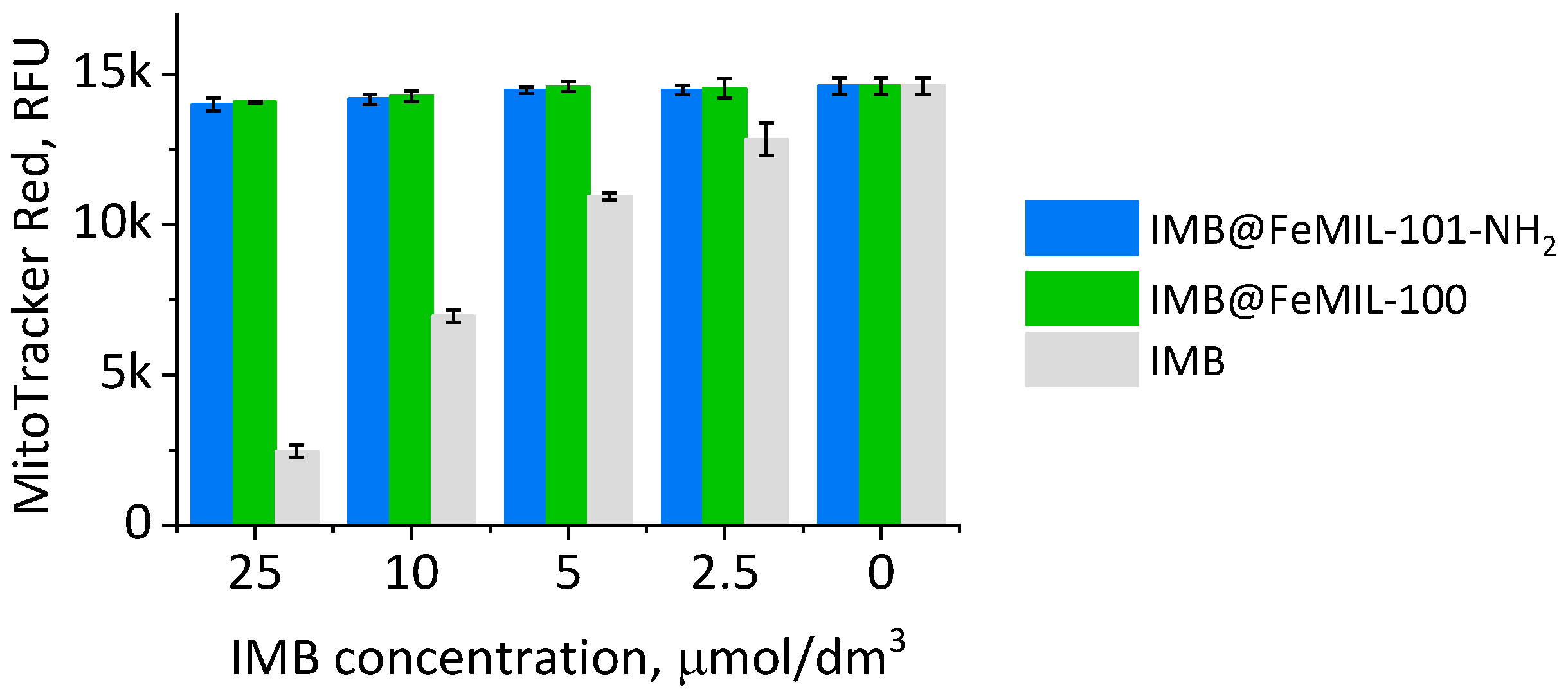
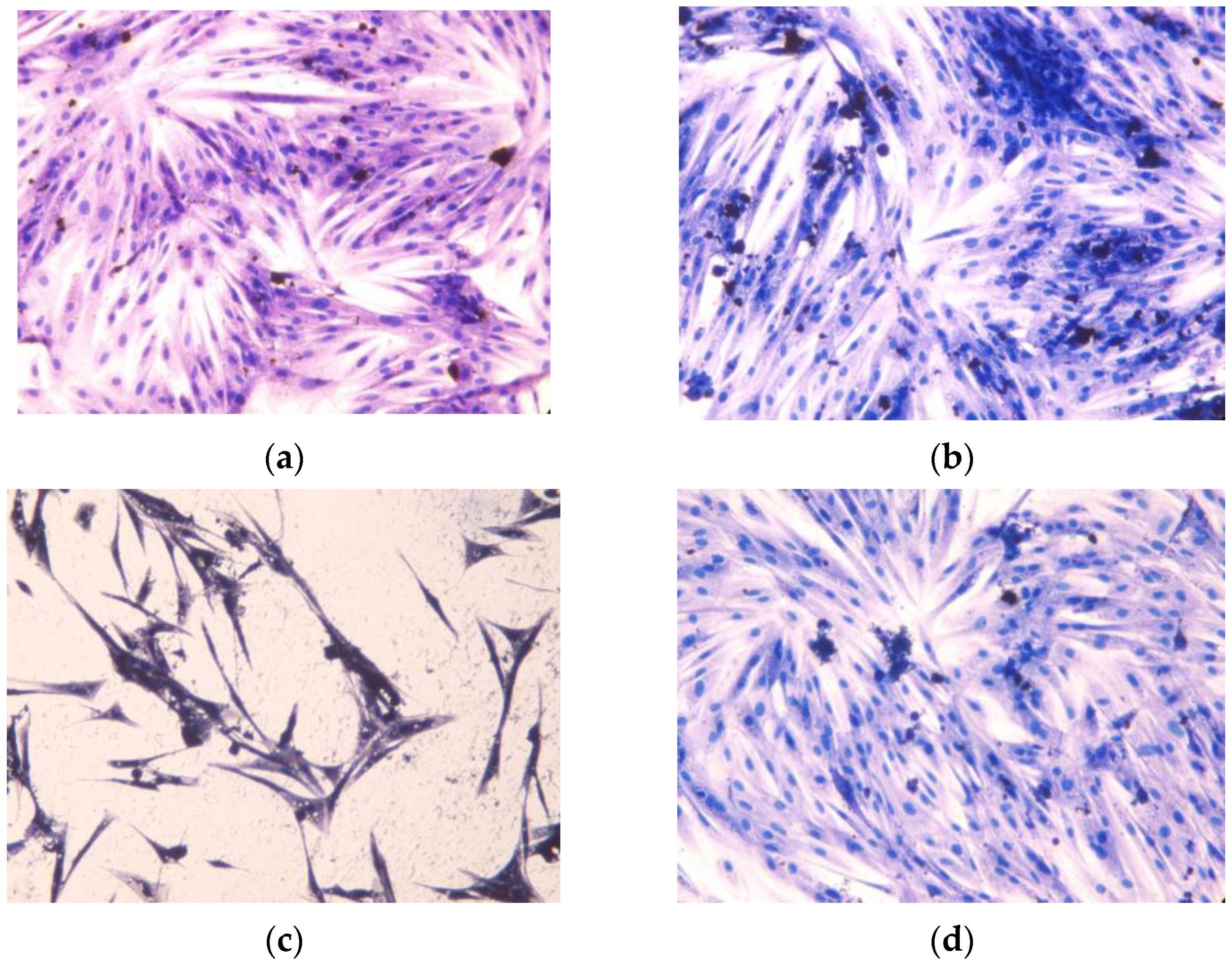
2.3.1. Toxicity for Cardiomyocytes
2.3.2. Toxicity for HL60 Human Leukemia Cells
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Synthesis of FeMOFs
4.3. Preparation of IMB@MOF Composites
4.4. Characterization
4.5. In Vitro IMB Release Profiles
4.6. Cell Cultures and Viability Assay
4.6.1. Cell Cultures
4.6.2. Cell Viability and Cytotoxicity
4.6.3. Mitochondrial Membrane Potential
4.6.4. Cell Morphology
4.6.5. Oxidative Stress and Superoxide Production
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Toruner, G.A.; Tang, G.L.; Yin, C.C.; Wang, W.; Hu, S.M.; Thakral, B.; Wang, S.A.; Miranda, R.N.; Khoury, J.D.; et al. Chronic myeloid leukemia with insertion-derived BCR-ABL1 fusion: Redefining complex chromosomal abnormalities by correlation of FISH and karyotype predicts prognosis (vol 85, pg 631, 2020). Mod. Pathol. 2020, 33, 2088. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Matulonis, U.; Salgia, R.; Kanakura, Y.; Druker, B.; Griffin, J.D. Factor independence of human myeloid-leukemia cell-lines is associated with increased phosphorylation of the protooncogene RAF-1. Exp. Hematol. 1994, 22, 1111–1117. [Google Scholar] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Fontana, D.; Zambrotta, G.; Scannella, A.; Piazza, R.; Gambacorti-Passerini, C. Late relapse of chronic myeloid leukemia after allogeneic bone marrow transplantation points to KANSARL (KANSL1::ARL17A) alteration: A case report with insights on the molecular landscape. Ann. Hematol. 2024, 103, 1561–1568. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.; Johnson, J.R.; Duan, J.; Gobburu, J.; Rahman, A.; Benson, K.; Leighton, J.; Kim, S.K.; Wood, R.; et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2002, 8, 935–942. [Google Scholar]
- Kerkela, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.W.; Distler, O. Cardiotoxicity of imatinib mesylate: An extremely rare phenomenon or a major side effect? Ann. Rheum. Dis. 2007, 66, 836. [Google Scholar] [CrossRef]
- Garraway, L.A.; Jänne, P.A. Circumventing Cancer Drug Resistance in the Era of Personalized Medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef]
- Jain, K.K. An Overview of Drug Delivery Systems. In Drug Delivery Systems; Jain, K.K., Ed.; Springer: New York, NY, USA, 2020; pp. 1–54. [Google Scholar]
- Sheeba, C.J.; Marslin, G.; Revina, A.M.; Khandelwal, V.; Balakumar, K.; Prakash, J.; Franklin, G. Delivery as nanoparticles reduces imatinib mesylate-induced cardiotoxicity and improves anticancer activity. Int. J. Nanomed. 2015, 10, 3163–3170. [Google Scholar] [CrossRef]
- Karimi, M.; Karimian, K.; Heli, H. A nanoemulsion-based delivery system for imatinib and in vitro anticancer efficacy. Braz. J. Pharm. Sci. 2020, 56, e18973. [Google Scholar] [CrossRef]
- Karal-Yilmaz, O.; Ozkan, A.; Akgun, E.; Kukut, M.; Baysal, K.; Avsar, T.; Kilic, T. Controlled release of imatinib mesylate from PLGA microspheres inhibit craniopharyngioma mediated angiogenesis. J. Mater. Sci. Mater. Med. 2013, 24, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.; Bahadur, A.; Saeed, A.; Rahman, M.S.u.; Naseer, M.M. Biocompatible, pH-responsive, and biodegradable polyurethanes as smart anti-cancer drug delivery carriers. React. Funct. Polym. 2018, 127, 153–160. [Google Scholar] [CrossRef]
- Labala, S.; Mandapalli, P.K.; Kurumaddali, A.; Venuganti, V.V.K. Layer-by-Layer Polymer Coated Gold Nanoparticles for Topical Delivery of Imatinib Mesylate To Treat Melanoma. Mol. Pharm. 2015, 12, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Torrik, A.; Zaerin, S.; Zarif, M. Doxorubicin and Imatinib co-drug delivery using non-covalently functionalized carbon nanotube: Molecular dynamics study. J. Mol. Liq. 2022, 362, 119789. [Google Scholar] [CrossRef]
- Panigrahi, G.; Medhi, H.; Wasnik, K.; Patra, S.; Gupta, P.; Pareek, D.; Maity, S.; Mandey, M.; Paik, P. Hollow mesoporous SiO2-ZnO nanocapsules and effective in vitro delivery of anticancer drugs against different cancers with low doses of drugs. Mater. Chem. Phys. 2022, 287, 126287. [Google Scholar] [CrossRef]
- Kimura, S.; Egashira, K.; Nakano, K.; Iwata, E.; Miyagawa, M.; Tsujimoto, H.; Hara, K.; Kawashima, Y.; Tominaga, R.; Sunagawa, K. Local Delivery of Imatinib Mesylate (STI571)-Incorporated Nanoparticle Ex Vivo Suppresses Vein Graft Neointima Formation. Circulation 2008, 118, S65–S70. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.-E.; Guo, M.; Deng, Y.; Pan, Y.; Wang, X.; Maduraiveeran, G.; Liu, J.; Lu, C. MOF-Based Platform for Kidney Diseases: Advances, Challenges, and Prospects. Pharmaceutics 2024, 16, 793. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lu, H.; Chen, J.; Jiang, C.; Jiang, G.; Maduraiveeran, G.; Pan, Y.; Liu, J.; Deng, L.-E. Advances in drug delivery-based therapeutic strategies for renal fibrosis treatment. J. Mater. Chem. B 2024, 12, 6532–6549. [Google Scholar] [CrossRef]
- Jiang, C.; Zhou, L.; Hu, W.; Luo, J.; Nezamzadeh-Ejhieh, A.; Ouyang, J.; Liu, X.; Pan, Y.; Cui, S.; Liu, J. Recent advances in NO-triggered gas therapy by metal-organic frameworks. Mater. Today Chem. 2024, 36, 101964. [Google Scholar] [CrossRef]
- Nguyen, N.T.T.; Nguyen, T.T.T.; Ge, S.; Liew, R.K.; Nguyen, D.T.C.; Tran, T.V. Recent progress and challenges of MOF-based nanocomposites in bioimaging, biosensing and biocarriers for drug delivery. Nanoscale Adv. 2024, 6, 1800–1821. [Google Scholar] [CrossRef] [PubMed]
- Moharramnejad, M.; Malekshah, R.E.; Ehsani, A.; Gharanli, S.; Shahi, M.; Alvan, S.A.; Salariyeh, Z.; Azadani, M.N.; Haribabu, J.; Basmenj, Z.S.; et al. A review of recent developments of metal–organic frameworks as combined biomedical platforms over the past decade. Adv. Colloid Interface Sci. 2023, 316, 102908. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dai, X. Current status of controlled onco-therapies based on metal organic frameworks. RSC Adv. 2024, 14, 12817–12828. [Google Scholar] [CrossRef] [PubMed]
- Arabbaghi, E.K.; Mokhtari, J.; Naimi-Jamal, M.R.; Khosravi, A. Zn-MOF: An efficient drug delivery platform for the encapsulation and releasing of Imatinib Mesylate. J. Porous Mater. 2021, 28, 641–649. [Google Scholar] [CrossRef]
- Abbasi, A.R.; Rizvandi, M.; Azadbakht, A.; Rostamnia, S. Controlled uptake and release of imatinib from ultrasound nanoparticles Cu-3(BTC)(2) metal-organic framework in comparison with bulk structure. J. Colloid Interface Sci. 2016, 471, 112–117. [Google Scholar] [CrossRef]
- Shahin, R.; Yousefi, M.; Ziyadi, H.; Bikhof, M.; Hekmati, M. pH-Responsive and magnetic Fe3O4@UiO-66-NH2@PEI nanocomposite as drug nanocarrier: Loading and release study of Imatinib. Inorg. Chem. Commun. 2023, 147, 110186. [Google Scholar] [CrossRef]
- Afshar, E.; Taher, M.; Hashisho, Z.; Karimi-Maleh, H.; Rajendran, S.; Vasseghian, Y. Determination and measurement of Imatinib by a fluorescence quenching sensor based on halloysite nanotubes modified by zirconium metal organic framework. Mater. Chem. Phys. 2023, 295, 127157. [Google Scholar] [CrossRef]
- Mir, A.; Shabani-Nooshabadi, M. Application of Binary Metal-Organic Framework in an Electrochemical Sensor Decorated With Gold Nanoparticles for the Determination of Anticancer Drug Imatinib in Plasma. IEEE Sens. J. 2023, 23, 12124–12132. [Google Scholar] [CrossRef]
- Ghorbani, M.; Saghafi, A.; Pakseresht, M.; Shams, A.; Keshavarzi, M.; Asghari, S. Crafting an innovative bimetallic MOF-on-MOF/TiO2 composite for effective removal of Imatinib anticancer agent through adsorption and photodegradation. Sep. Purif. Technol. 2024, 336, 126227. [Google Scholar] [CrossRef]
- Férey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F. Crystallized Frameworks with Giant Pores: Are There Limits to the Possible? Acc. Chem. Res. 2005, 38, 217–225. [Google Scholar] [CrossRef]
- Horcajada, P.; Surblé, S.; Serre, C.; Hong, D.-Y.; Seo, Y.-K.; Chang, J.-S.; Grenèche, J.-M.; Margiolaki, I.; Férey, G. Synthesis and catalytic properties of MIL-100(Fe), an iron(III) carboxylate with large pores. Chem. Commun. 2007, 27, 2820–2822. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Qian, Q.; Mizrahi Rodriguez, K.; Smith, Z.P. Hydrothermal Synthesis of Sub-20 nm Amine-Functionalized MIL-101(Cr) Nanoparticles with High Surface Area and Enhanced CO2 Uptake. Ind. Eng. Chem. Res. 2020, 59, 7888–7900. [Google Scholar] [CrossRef]
- Quijia, C.R.; Lima, C.; Silva, C.; Alves, R.C.; Frem, R.; Chorilli, M. Application of MIL-100(Fe) in drug delivery and biomedicine. J. Drug Deliv. Sci. Technol. 2021, 61, 102217. [Google Scholar] [CrossRef]
- Ma, X.; Lepoitevin, M.; Serre, C. Metal–organic frameworks towards bio-medical applications. Mater. Chem. Front. 2021, 5, 5573–5594. [Google Scholar] [CrossRef]
- Lin, Z.; Liao, D.; Jiang, C.; Nezamzadeh-Ejhieh, A.; Zheng, M.; Yuan, H.; Liu, J.; Song, H.; Lu, C. Current status and prospects of MIL-based MOF materials for biomedicine applications. RSC Med. Chem. 2023, 14, 1914–1933. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhai, L.X.; Li, C.X.; Li, Y.J.; Guo, X.Q.; Zhao, Y.B.; Wu, C.L. Exploring and Exploiting Dynamic Noncovalent Chemistry for Effective Surface Modification of Nanoscale Metal-Organic Frameworks. ACS Appl. Mater. Interfaces 2014, 6, 5404–5412. [Google Scholar] [CrossRef] [PubMed]
- Baati, T.; Njim, L.; Neffati, F.; Kerkeni, A.; Bouttemi, M.; Gref, R.; Najjar, M.F.; Zakhama, A.; Couvreur, P.; Serre, C.; et al. In depth analysis of the in vivo toxicity of nanoparticles of porous iron(III) metal-organic frameworks. Chem. Sci. 2013, 4, 1597–1607. [Google Scholar] [CrossRef]
- Wyszogrodzka, G.; Dorozynski, P.; Gil, B.; Roth, W.J.; Strzempek, M.; Marszalek, B.; Weglarz, W.P.; Menaszek, E.; Strzempek, W.; Kulinowski, P. Iron-Based Metal-Organic Frameworks as a Theranostic Carrier for Local Tuberculosis Therapy. Pharm. Res. 2018, 35, 144. [Google Scholar] [CrossRef] [PubMed]
- Wyszogrodzka-Gawel, G.; Dorozynski, P.; Giovagnoli, S.; Strzempek, W.; Pesta, E.; Weglarz, W.P.; Gil, B.; Menaszek, E.; Kulinowski, P. An Inhalable Theranostic System for Local Tuberculosis Treatment Containing an Isoniazid Loaded Metal Organic Framework Fe-MIL-101-NH2—From Raw MOF to Drug Delivery System. Pharmaceutics 2019, 11, 687. [Google Scholar] [CrossRef]
- Hafizovic, J.; Bjørgen, M.; Olsbye, U.; Dietzel, P.D.C.; Bordiga, S.; Prestipino, C.; Lamberti, C.; Lillerud, K.P. The Inconsistency in Adsorption Properties and Powder XRD Data of MOF-5 Is Rationalized by Framework Interpenetration and the Presence of Organic and Inorganic Species in the Nanocavities. J. Am. Chem. Soc. 2007, 129, 3612–3620. [Google Scholar] [CrossRef]
- Kimitsuka, Y.; Hosono, E.; Ueno, S.; Zhou, H.; Fujihara, S. Fabrication of Porous Cubic Architecture of ZnO Using Zn-terephthalate MOFs with Characteristic Microstructures. Inorg. Chem. 2013, 52, 14028–14033. [Google Scholar] [CrossRef]
- Chao, M.-Y.; Zhang, W.-H.; Lang, J.-P. Co2 and Co3 Mixed Cluster Secondary Building Unit Approach toward a Three-Dimensional Metal-Organic Framework with Permanent Porosity. Molecules 2018, 23, 755. [Google Scholar] [CrossRef]
- Monshi, A.; Foroughi, M.R.; Monshi, M.R. Modified Scherrer Equation to Estimate More Accurately Nano-Crystallite Size Using XRD. World J. Nano Sci. Eng. 2012, 2, 154–160. [Google Scholar] [CrossRef]
- Bauer, S.; Serre, C.; Devic, T.; Horcajada, P.; Marrot, J.; Ferey, G.; Stock, N. High-throughput assisted rationalization of the formation of metal organic frameworks in the iron(III) aminoterephthalate solvothermal system. Inorg. Chem. 2008, 47, 7568–7576. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, H.; Vimont, A.; Lavalley, J.-C.; Daturi, M.; Wiersum, A.D.; Llwellyn, P.L.; Horcajada, P.; Férey, G.; Serre, C. Infrared study of the influence of reducible iron(iii) metal sites on the adsorption of CO, CO2, propane, propene and propyne in the mesoporous metal–organic framework MIL-100. Phys. Chem. Chem. Phys. 2011, 13, 11748. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Joshi, B.D.; Tandon, P.; Ayala, A.P.; Bansal, A.K.; Grillo, D. Study of polymorphism in imatinib mesylate: A quantum chemical approach using electronic and vibrational spectra. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 103, 325–332. [Google Scholar] [CrossRef]
- Hemmer, K.; Cokoja, M.; Fischer, R.A. Exploitation of Intrinsic Confinement Effects of MOFs in Catalysis. ChemCatChem 2021, 13, 1683–1691. [Google Scholar] [CrossRef]
- Dai, J.; Zhang, H. Recent Advances in Catalytic Confinement Effect within Micro/Meso Porous Crystalline Materials. Small 2021, 17, 2005334. [Google Scholar] [CrossRef]
- Pandey, P. Evidence of blue-shifting N–H⋯N hydrogen bonding despite elongation of the N–H bond. RSC Adv. 2015, 5, 79661–79664. [Google Scholar] [CrossRef]
- Mishra, S.; Nguyen, H.-Q.; Huang, Q.-R.; Lin, C.-K.; Kuo, J.-L.; Patwari, G.N. Vibrational spectroscopic signatures of hydrogen bond induced NH stretch–bend Fermi-resonance in amines: The methylamine clusters and other N–H⋯N hydrogen-bonded complexes. J. Chem. Phys. 2020, 153, 194301. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, Y.; Gillies, R.J. Tumor pH and Its Measurement. J. Nucl. Med. 2010, 51, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.L. 5-Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Bruschi, M.L., Ed.; Woodhead Publishing: Sawston, UK, 2015; pp. 63–86. [Google Scholar]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N.A.; Moynihan, H.J. Solute diffusion in swollen membranes. IV. Theories for moderately swollen networks. J. Appl. Polym. Sci. 1985, 30, 2589–2606. [Google Scholar] [CrossRef]
- Channab, B.-E.; El Ouardi, M.; Ait Layachi, O.; Marrane, S.E.; El Idrissi, A.; Baqais, A.; Ait Ahsaine, H. Recent trends on MIL-Fe metal–organic frameworks: Synthesis approaches, structural insights, and applications in organic pollutant adsorption and photocatalytic degradation. Environ. Sci. Nano 2023, 10, 2957–2988. [Google Scholar] [CrossRef]
- Bezverkhyy, I.; Weber, G.; Bellat, J.-P. Degradation of fluoride-free MIL-100(Fe) and MIL-53(Fe) in water: Effect of temperature and pH. Microporous Mesoporous Mater. 2016, 219, 117–124. [Google Scholar] [CrossRef]
- Souza, B.E.; Möslein, A.F.; Titov, K.; Taylor, J.D.; Rudić, S.; Tan, J.-C. Green Reconstruction of MIL-100 (Fe) in Water for High Crystallinity and Enhanced Guest Encapsulation. ACS Sustain. Chem. Eng. 2020, 8, 8247–8255. [Google Scholar] [CrossRef]
- Kuznicki, A.; Lorzing, G.R.; Bloch, E.D. Tuning water adsorption, stability, and phase in Fe-MIL-101 and Fe-MIL-88 analogs with amide functionalization. Chem. Commun. 2021, 57, 8312–8315. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Danışman Kalındemirtaş, F.; Birman, H.; Candöken, E.; Bilgiş Gazioğlu, S.; Melikoğlu, G.; Kuruca, S. Cytotoxic Effects of Some Flavonoids and Imatinib on the K562 Chronic Myeloid Leukemia Cell Line: Data Analysis Using the Combination Index Method. Balk. Med. J. 2019, 36, 96–105. [Google Scholar] [CrossRef]
- Gundogdu, E.; Demir, E.-S.; Ekinci, M.; Ozgenc, E.; Ilem-Ozdemir, D.; Senyigit, Z.; Gonzalez-Alvarez, I.; Bermejo, M. An Innovative Formulation Based on Nanostructured Lipid Carriers for Imatinib Delivery: Pre-Formulation, Cellular Uptake and Cytotoxicity Studies. Nanomaterials 2022, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Gupta, M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021. [Google Scholar] [CrossRef]
- Strzempek, W.; Menaszek, E.; Gil, B. Fe-MIL-100 as drug delivery system for asthma and chronic obstructive pulmonary disease treatment and diagnosis. Microporous Mesoporous Mater. 2019, 280, 264–270. [Google Scholar] [CrossRef]
- Bouitbir, J.; Panajatovic, M.V.; Krähenbühl, S. Mitochondrial Toxicity Associated with Imatinib and Sorafenib in Isolated Rat Heart Fibers and the Cardiomyoblast H9c2 Cell Line. Int. J. Mol. Sci. 2022, 23, 2282. [Google Scholar] [CrossRef] [PubMed]
- Kobara, M.; Nessa, N.; Toba, H.; Nakata, T. Induction of autophagy has protective roles in imatinib-induced cardiotoxicity. Toxicol. Rep. 2021, 8, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Liu, M.; Liang, F.; Zhao, L.; Gao, C.; Jiang, X.; Zhang, X.; Zhan, H.; Hu, H.; Zhao, Z. Cardiotoxicity of sorafenib is mediated through elevation of ROS level and CaMKII activity and dysregulation of calcium homoeostasis. Basic Clin. Pharmacol. Toxicol. 2020, 126, 166–180. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Druker, B.J. Perspectives on the development of imatinib and the future of cancer research. Nat. Med. 2009, 15, 1149–1152. [Google Scholar] [CrossRef]
- Yang, J.; Wang, H.; Liu, J.; Ding, M.; Xie, X.; Yang, X.; Peng, Y.; Zhou, S.; Ouyang, R.; Miao, Y. Recent advances in nanosized metal organic frameworks for drug delivery and tumor therapy. RSC Adv. 2021, 11, 3241–3263. [Google Scholar] [CrossRef]
- Guesh, K.; Caiuby, C.A.D.; Mayoral, A.; Diaz-Garcia, M.; Diaz, I.; Sanchez-Sanchez, M. Sustainable Preparation of MIL-100(Fe) and Its Photocatalytic Behavior in the Degradation of Methyl Orange in Water. Cryst. Growth Des. 2017, 17, 1806–1813. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m3/g) | Vmicro (cm3/g) |
|---|---|---|
| FeMIL-100 | 1413 | 0.54 |
| IMB@FeMIL-100 | 50 | 0 |
| FeMIL-101-NH2 | 969 | 0.38 |
| IMB@FeMIL-101-NH2 | 23 | 0 |
| Parameter | IMB | IMB@FeMIL-100 | IMB@FeMIL-101-NH2 | ||||
|---|---|---|---|---|---|---|---|
| Solution | PBS | Endos | PBS | Endos | PBS | Endos | |
| b value | W | 0.59 ± 0.06 | 0.47 ± 0.19 | 0.82 ± 0.03 | 0.58 ± 0.15 | 0.75 ± 0.01 | 0.59 ± 0.02 |
| n value km value | KP | 0.42 ± 0.05 7.76 × 10−3 | 0.68 ± 0.30 2.00 × 10−3 | 0.71 ± 0.03 1.70 × 10−5 | 0.40 ± 0.09 7.59 × 10−3 | 0.60 ± 0.05 2.34 × 10−4 | 0.52 ± 0.03 8.13 × 10−4 |
| Pearson’s r coefficient | W | 0.995 | 0.916 | 0.996 | 0.966 | 0.999 | 0.995 |
| KP | 0.992 | 0.928 | 0.997 | 0.973 | 0.993 | 0.998 | |
| RSS | W | 0.002 | 0.055 | 0.083 | 0.023 | 0.019 | 0.039 |
| KP | 0.001 | 0.022 | 0.016 | 0.008 | 0.009 | 0.002 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzempek, W.; Menaszek, E.; Papież, M.; Gil, B. Slowing Down the “Magic Bullet”: Encapsulation of Imatinib in Fe-MOF for Cardiotoxicity Reduction and Improvement in Anticancer Activity. Molecules 2024, 29, 3818. https://doi.org/10.3390/molecules29163818
Strzempek W, Menaszek E, Papież M, Gil B. Slowing Down the “Magic Bullet”: Encapsulation of Imatinib in Fe-MOF for Cardiotoxicity Reduction and Improvement in Anticancer Activity. Molecules. 2024; 29(16):3818. https://doi.org/10.3390/molecules29163818
Chicago/Turabian StyleStrzempek, Weronika, Elżbieta Menaszek, Monika Papież, and Barbara Gil. 2024. "Slowing Down the “Magic Bullet”: Encapsulation of Imatinib in Fe-MOF for Cardiotoxicity Reduction and Improvement in Anticancer Activity" Molecules 29, no. 16: 3818. https://doi.org/10.3390/molecules29163818
APA StyleStrzempek, W., Menaszek, E., Papież, M., & Gil, B. (2024). Slowing Down the “Magic Bullet”: Encapsulation of Imatinib in Fe-MOF for Cardiotoxicity Reduction and Improvement in Anticancer Activity. Molecules, 29(16), 3818. https://doi.org/10.3390/molecules29163818