

Discovery of Antibacterial Compounds with Potential Multi-Pharmacology against Staphylococcus Mur ligase Family Members by In Silico Structure-Based Drug Screening

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
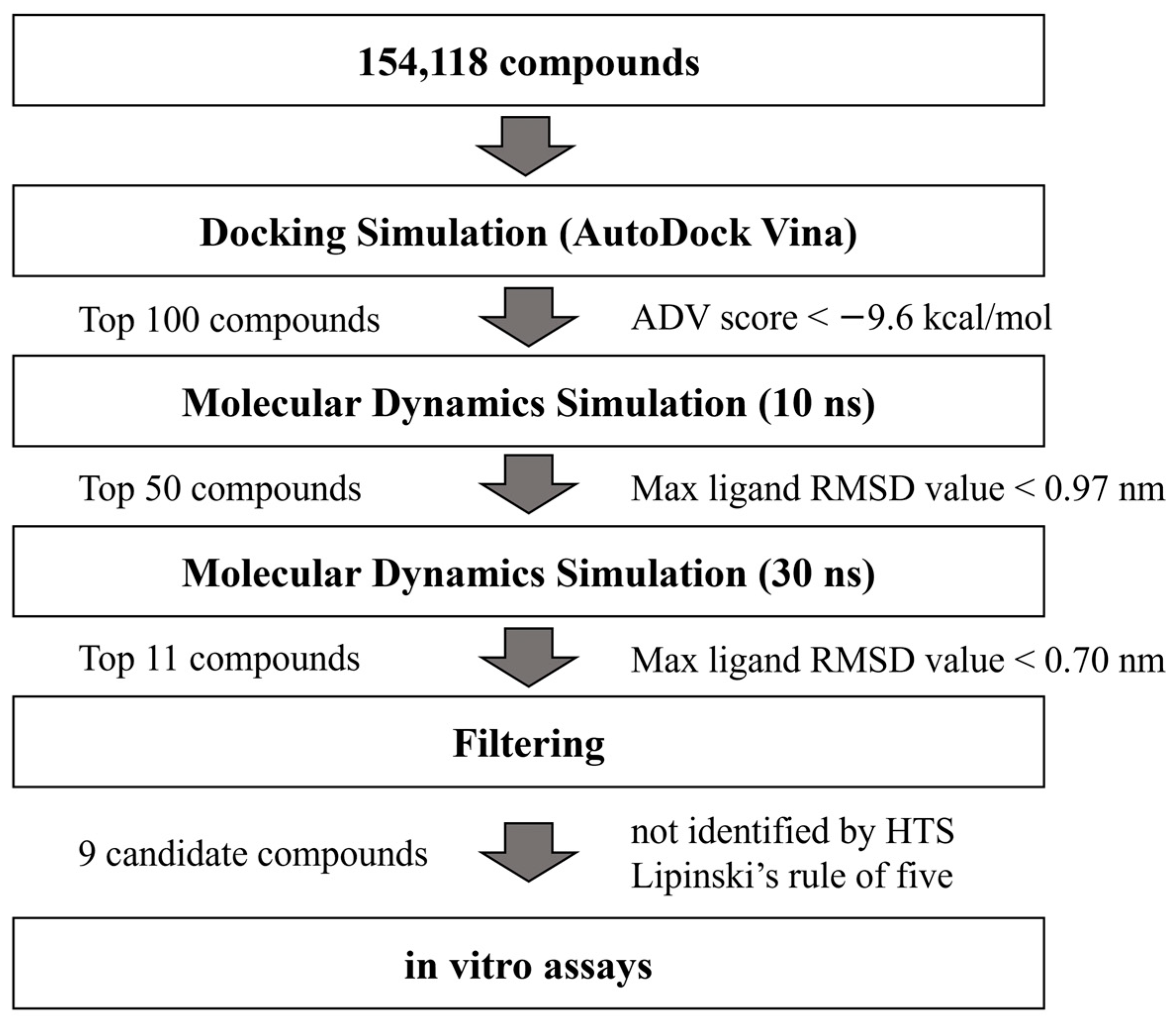
2.1. Hierarchical In Silico SBDS Targeting saMurE
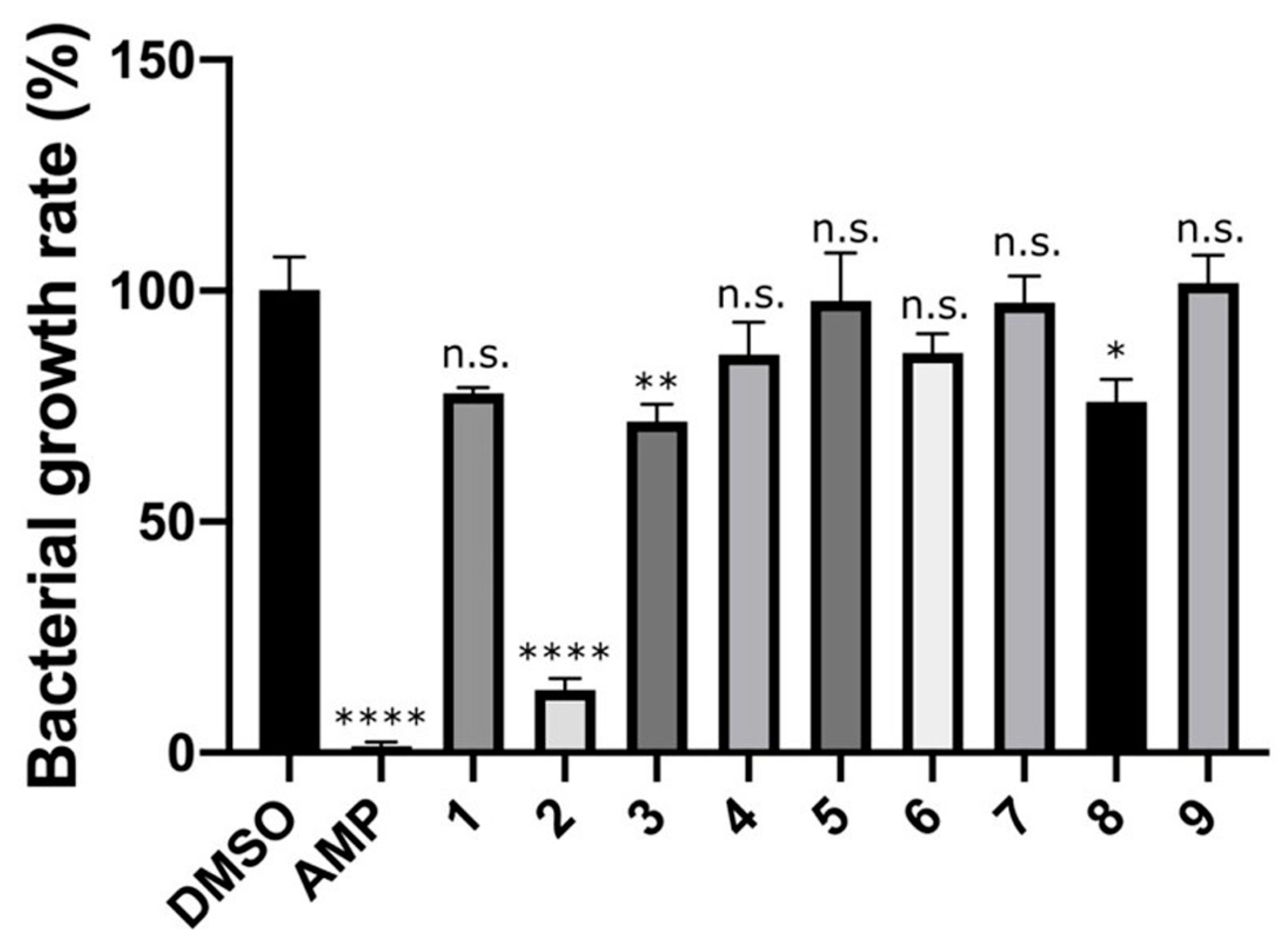
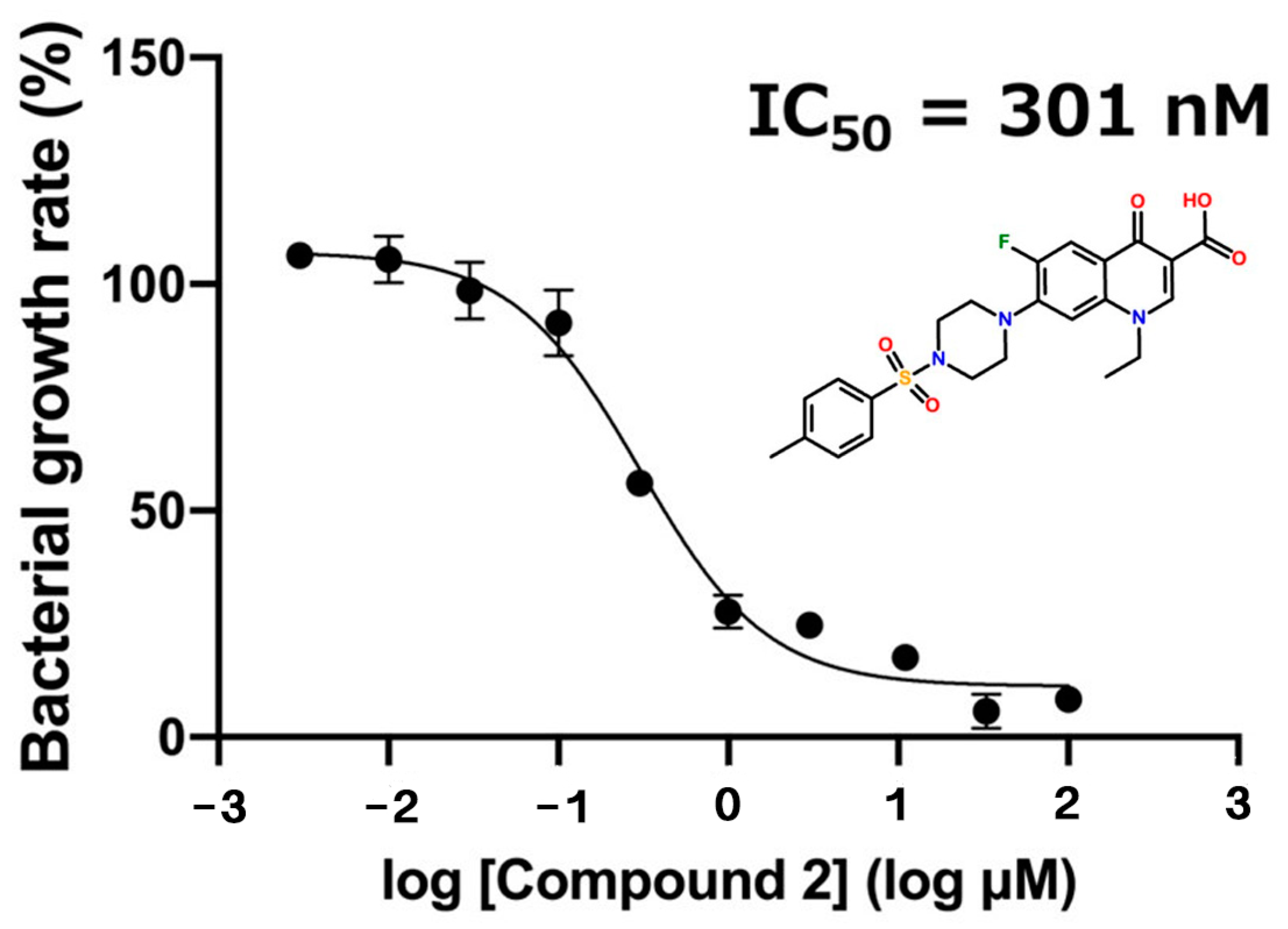
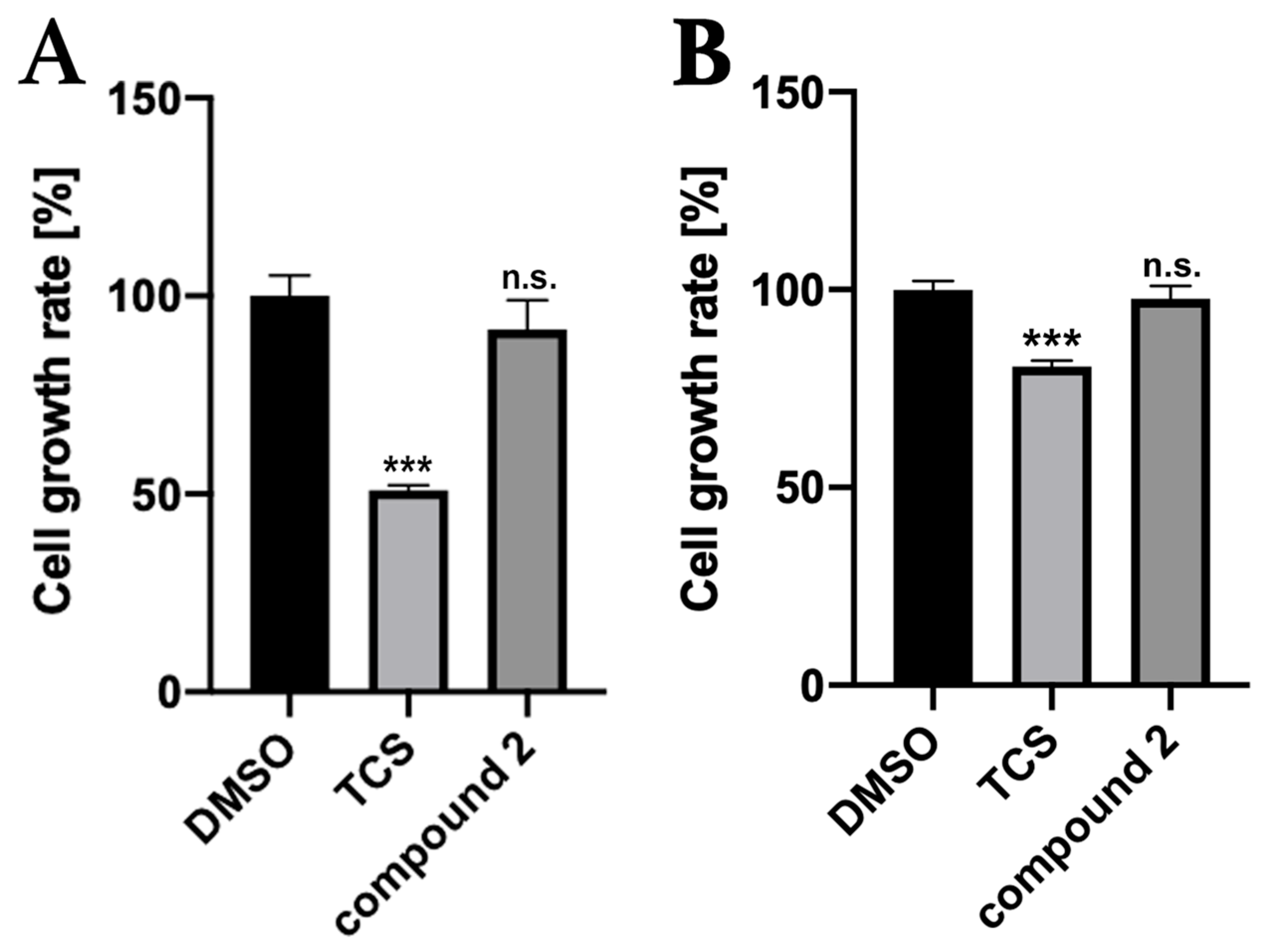
2.2. Growth Inhibition Assay against Staphylococcus epidermidis (S. epidermidis)
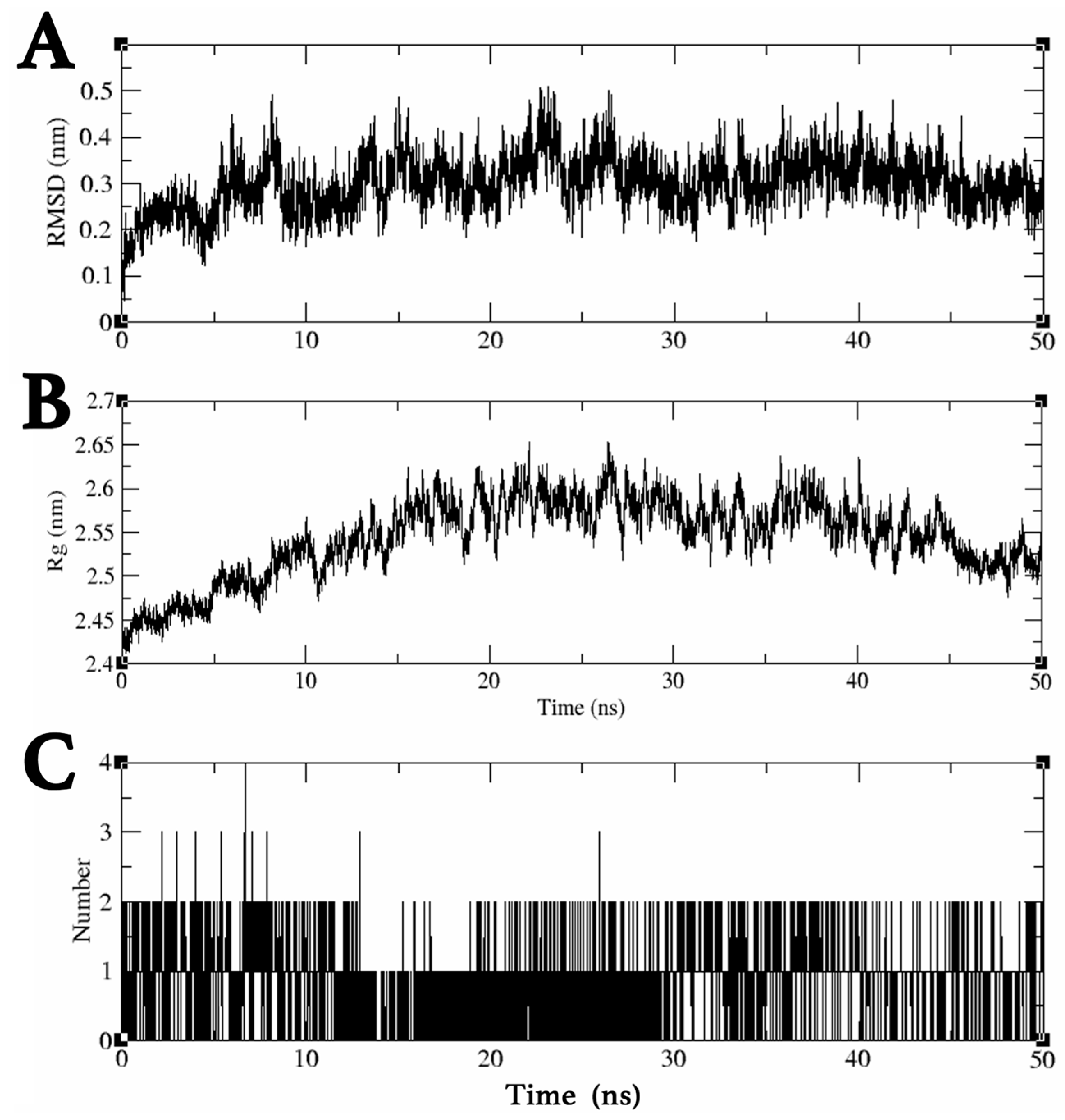
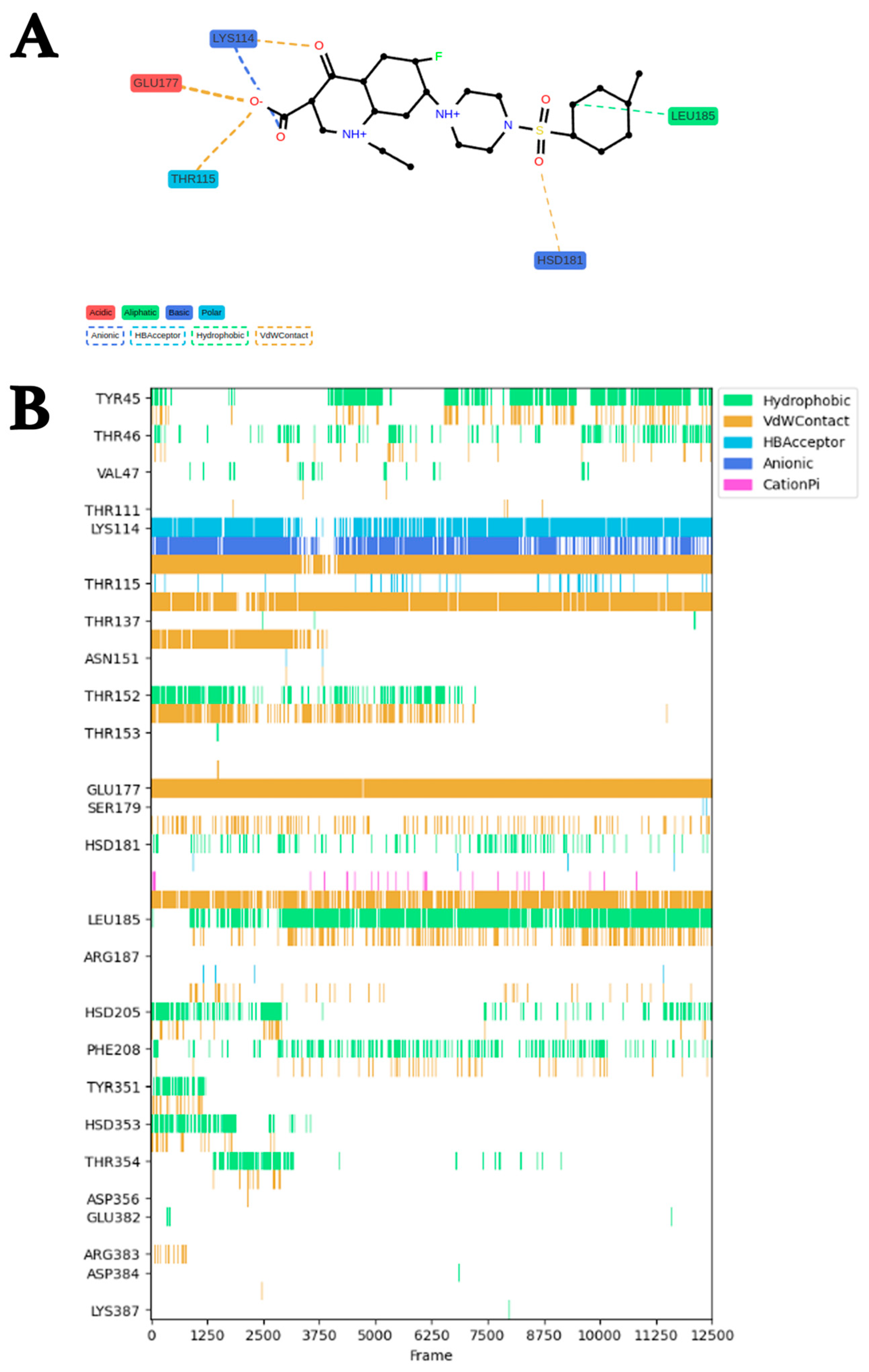
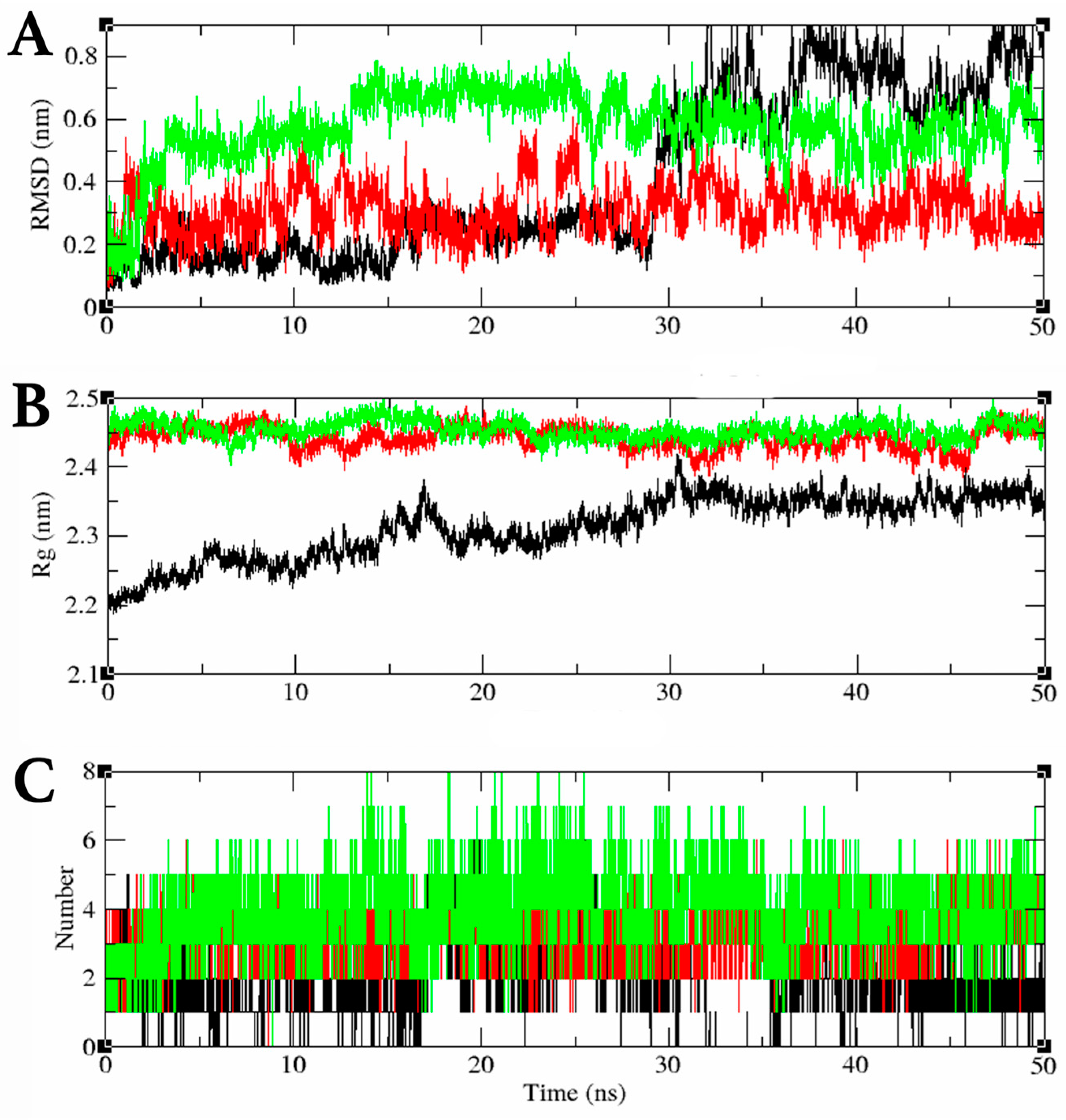
2.3. MDS Trajectory Data Analysis of the saMurE–Compound 2 Complex
2.4. Analysis of Binding Modes and Binding Free Energies of saMurE and Compound 2
2.5. Growth Inhibition Assay for Gram-Negative Bacterium
2.6. Prediction of Pharmacological Properties and Toxicity of Compound 2
2.7. Toxicity Assays for Mammalian-Derived Cells
2.8. Membrane Permeation Simulation of Compound 2 against Gram-Positive Bacteria
2.9. Interaction Analysis of Compound 2 and Other Members of the Mur ligase Family by MDSs
2.10. MDS Trajectory Analysis of Compound 2 and MurC, MurD, and MurF Complexes
3. Discussion
3.1. In Silico SBDS Constructed by Hierarchical Combination of DS and MS
3.2. Antimicrobial Spectrum of Compound 2
3.3. Potential Multiple Pharmacological Properties of Compound 2
3.4. In Silico Toxicity Prediction and In Vitro Cytotoxicity Assay Using Mammalian-Derived Cells
3.5. Major Group of Amino Acid Residues That Support Stable Binding of Compound 2
3.6. Diversity of Compound 2 Targets
4. Materials and Methods
4.1. Compound Structure Data Library
4.2. Pretreatment of Target Proteins
4.3. Docking Simulation
4.4. Molecular Dynamics Simulation
4.5. MDS Trajectory Data Analysis
4.6. Umbrella Sampling
4.7. Preparation of Compounds
4.8. Bacterial Growth Inhibition Assay
4.9. Toxicity Assays for Mammalian-Derived Cells
4.10. Ab Initio Fragment Molecular Orbital (FMO) Calculation
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus Aureus Host Interactions and Adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef]
- Lam, J.C.; Stokes, W. The Golden Grapes of Wrath—Staphylococcus Aureus Bacteremia: A Clinical Review. Am. J. Med. 2023, 136, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Tasneem, U.; Mehmood, K.; Majid, M.; Ullah, S.R.; Andleeb, S. Methicillin Resistant Staphylococcus Aureus: A Brief Review of Virulence and Resistance. J. Pak. Med. Assoc. 2022, 72, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Duval, R.E.; Grare, M.; Demoré, B. Fight Against Antimicrobial Resistance: We Always Need New Antibacterials but for Right Bacteria. Molecules 2019, 24, 3152. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.D.; Vollmer, W.; Foster, S.J. Different Walls for Rods and Balls: The Diversity of Peptidoglycan. Mol. Microbiol. 2014, 91, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The Architecture of the Gram-Positive Bacterial Cell Wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef]
- Gardete, S.; Ludovice, A.M.; Sobral, R.G.; Filipe, S.R.; de Lencastre, H.; Tomasz, A. Role of MurE in the Expression of β-Lactam Antibiotic Resistance in Staphylococcus Aureus. J. Bacteriol. 2004, 186, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Hervin, V.; Roy, V.; Agrofoglio, L.A. Antibiotics and Antibiotic Resistance—Mur Ligases as an Antibacterial Target. Molecules 2023, 28, 8076. [Google Scholar] [CrossRef] [PubMed]
- Budama-Kilinc, Y.; Gok, B.; Cetin Aluc, C.; Kecel-Gunduz, S. In Vitro and in Silico Evaluation of the Design of Nano-Phyto-Drug Candidate for Oral Use against Staphylococcus Aureus. PeerJ 2023, 11, e15523. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Šink, R.; Zidar, N.; Fic, A.; Contreras-Martel, C.; Dessen, A.; Patin, D.; Blanot, D.; Müller-Premru, M.; Gobec, S.; et al. Dual Inhibitor of MurD and MurE Ligases from Escherichia Coli and Staphylococcus Aureus. ACS Med. Chem. Lett. 2012, 3, 626–630. [Google Scholar] [CrossRef]
- Takeuchi, M.; Teshima, M.; Okubo, S.; Aoki, S. In Silico and in Vitro Identification of Compounds with Dual Pharmacological Activity against Metionyl-tRNA Synthetase and Isoleucyl-tRNA Synthetase of Staphylococcus Aureus. ChemistrySelect 2023, 8, e202300460. [Google Scholar] [CrossRef]
- Monobe, K.; Taniguchi, H.; Aoki, S. In Silico Identification of Potential Inhibitors against Staphylococcus Aureus Tyrosyl-TRNA Synthetase. Curr. Comput. Aided Drug Des. 2024, 20, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bouysset, C.; Fiorucci, S. ProLIF: A Library to Encode Molecular Interactions as Fingerprints. J. Cheminform. 2021, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment Molecular Orbital Method: An Approximate Computational Method for Large Molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Dehnbostel, F.O.; Preissner, R. Prediction Is a Balancing Act: Importance of Sampling Methods to Balance Sensitivity and Specificity of Predictive Models Based on Imbalanced Chemical Data Sets. Front. Chem. 2018, 6, 362. [Google Scholar] [CrossRef] [PubMed]
- Pogozheva, I.D.; Armstrong, G.A.; Kong, L.; Hartnagel, T.J.; Carpino, C.A.; Gee, S.E.; Picarello, D.M.; Rubin, A.S.; Lee, J.; Park, S.; et al. Comparative Molecular Dynamics Simulation Studies of Realistic Eukaryotic, Prokaryotic, and Archaeal Membranes. J. Chem. Inf. Model. 2022, 62, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Miyashita, N.; Im, W.; Feig, M.; Sugita, Y. Molecular Dynamics Simulations of Biological Membranes and Membrane Proteins Using Enhanced Conformational Sampling Algorithms. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 1635–1651. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Tang, Z.; Chang, C.A. Potential Mean Force from Umbrella Sampling Simulations: What Can We Learn and What Is Missed? J. Chem. Theory Comput. 2019, 15, 2433–2443. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N ⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Rossos, G.; Hadjikakou, S.K.; Kourkoumelis, N. Molecular Dynamics Simulation of 2-Benzimidazolyl-Urea with DPPC Lipid Membrane and Comparison with a Copper(II) Complex Derivative. Membranes 2021, 11, 743. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Choi, Y.K.; Kim, S.; Lee, J.; Im, W. CHARMM-GUI Membrane Builder for Lipid Nanoparticles with Ionizable Cationic Lipids and PEGylated Lipids. J. Chem. Inf. Model. 2021, 61, 5192–5202. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.S.; Berteotti, A.; Ronsisvalle, S.; Rocchia, W.; Cavalli, A. Steered Molecular Dynamics Simulations for Studying Protein–Ligand Interaction in Cyclin-Dependent Kinase 5. J. Chem. Inf. Model. 2014, 54, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L.; van der Spoel, D. G_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teshima, M.; Monobe, K.; Okubo, S.; Aoki, S. Discovery of Antibacterial Compounds with Potential Multi-Pharmacology against Staphylococcus Mur ligase Family Members by In Silico Structure-Based Drug Screening. Molecules 2024, 29, 3792. https://doi.org/10.3390/molecules29163792
Teshima M, Monobe K, Okubo S, Aoki S. Discovery of Antibacterial Compounds with Potential Multi-Pharmacology against Staphylococcus Mur ligase Family Members by In Silico Structure-Based Drug Screening. Molecules. 2024; 29(16):3792. https://doi.org/10.3390/molecules29163792
Chicago/Turabian StyleTeshima, Mio, Kohei Monobe, Saya Okubo, and Shunsuke Aoki. 2024. "Discovery of Antibacterial Compounds with Potential Multi-Pharmacology against Staphylococcus Mur ligase Family Members by In Silico Structure-Based Drug Screening" Molecules 29, no. 16: 3792. https://doi.org/10.3390/molecules29163792
APA StyleTeshima, M., Monobe, K., Okubo, S., & Aoki, S. (2024). Discovery of Antibacterial Compounds with Potential Multi-Pharmacology against Staphylococcus Mur ligase Family Members by In Silico Structure-Based Drug Screening. Molecules, 29(16), 3792. https://doi.org/10.3390/molecules29163792