
Synthesis of Benzofuran Derivatives via a DMAP-Mediated Tandem Cyclization Reaction Involving ortho-Hydroxy α-Aminosulfones


Abstract

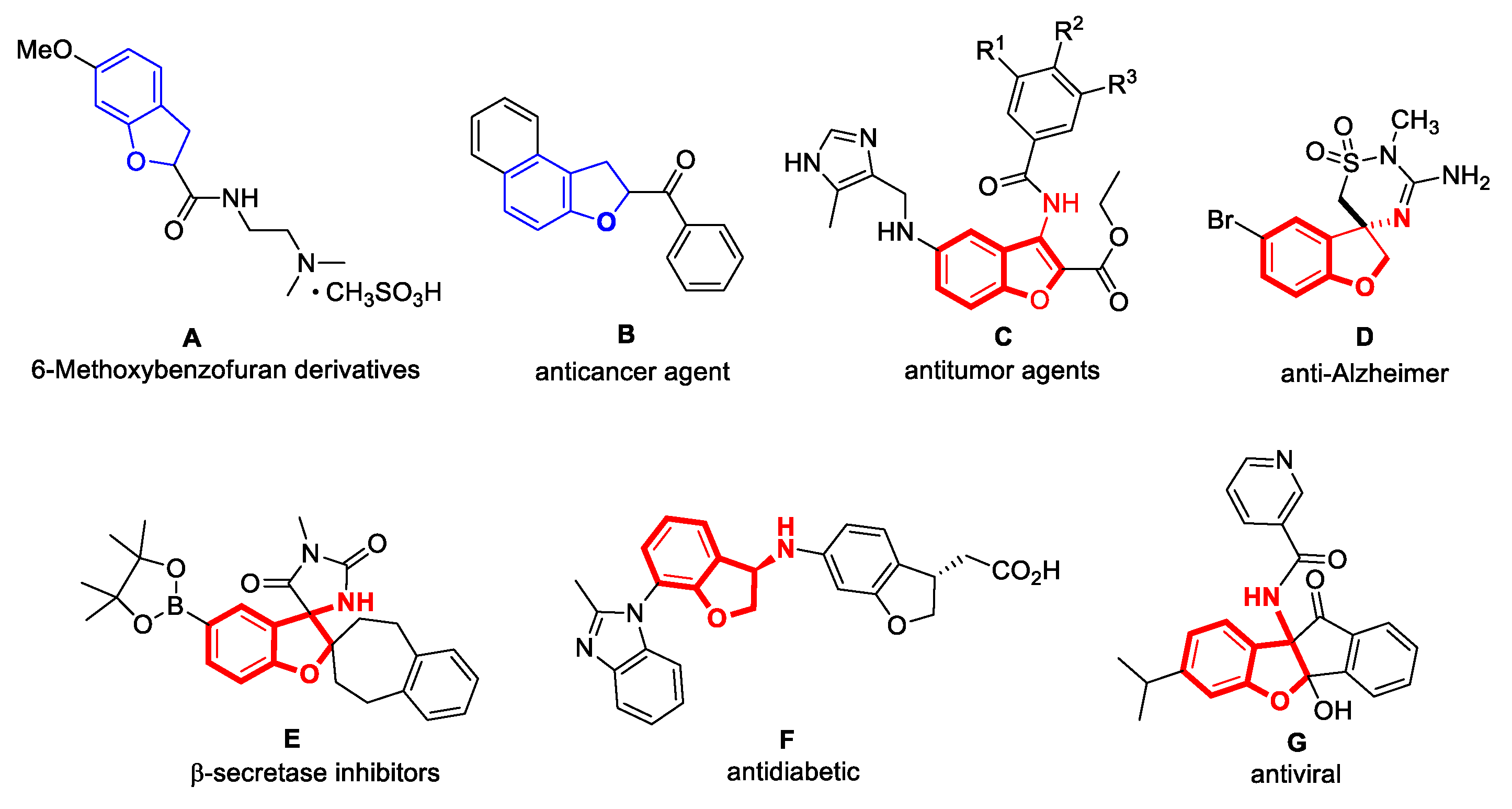
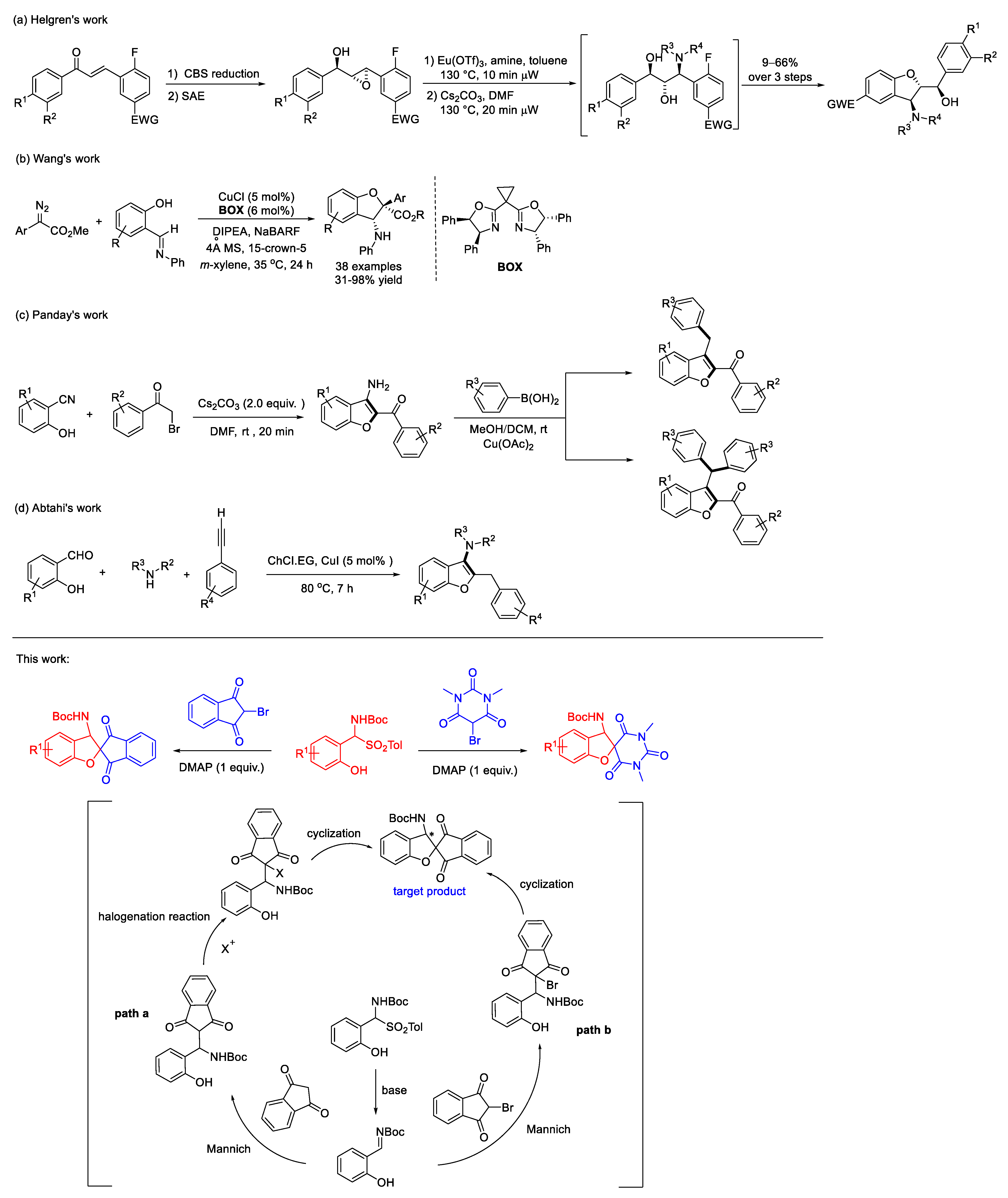
1. Introduction
2. Results and Discussion
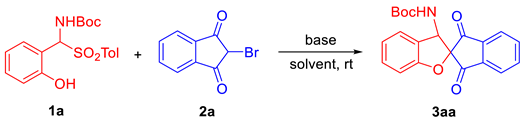
2.1. Optimization of Reaction Conditions
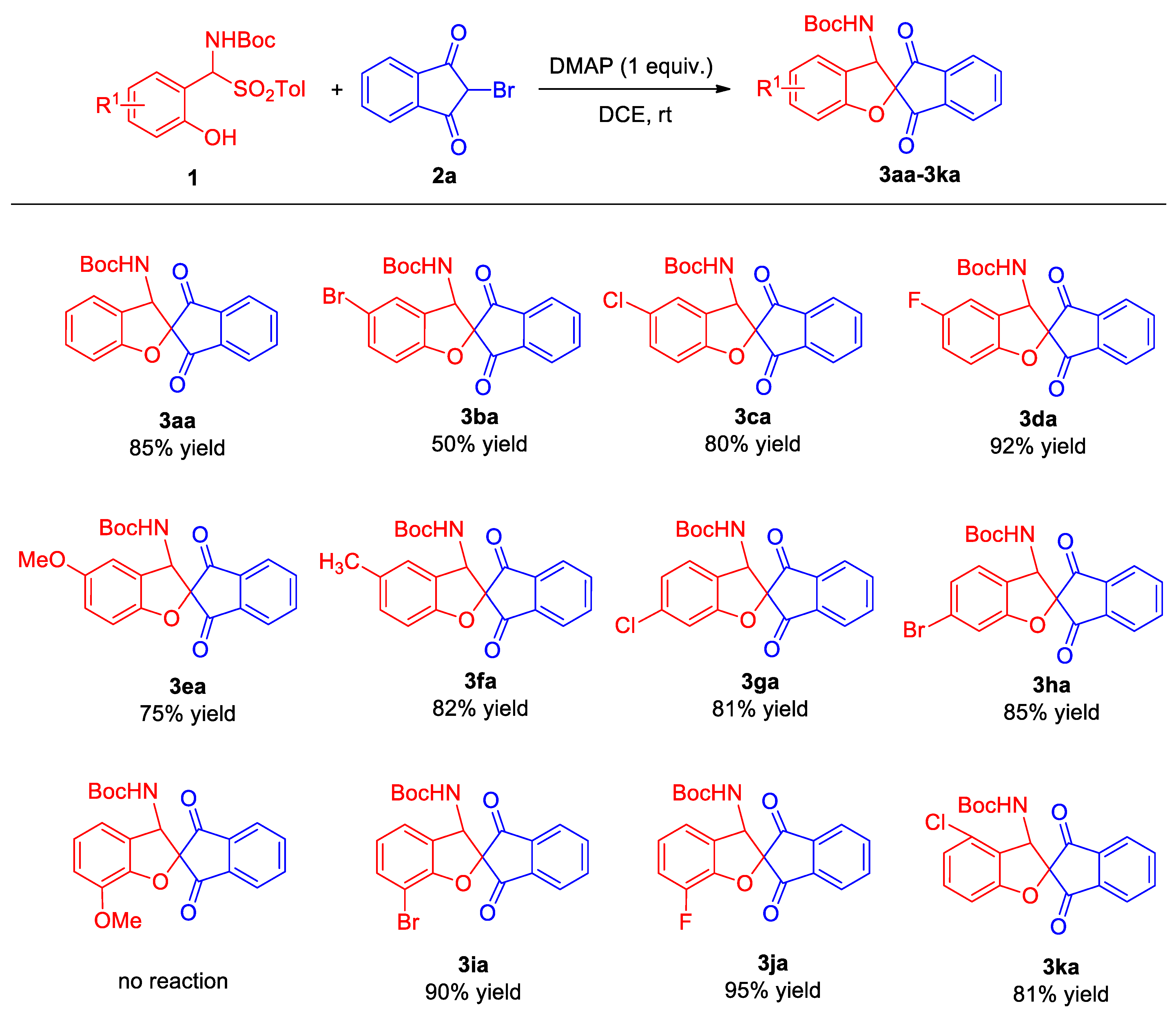
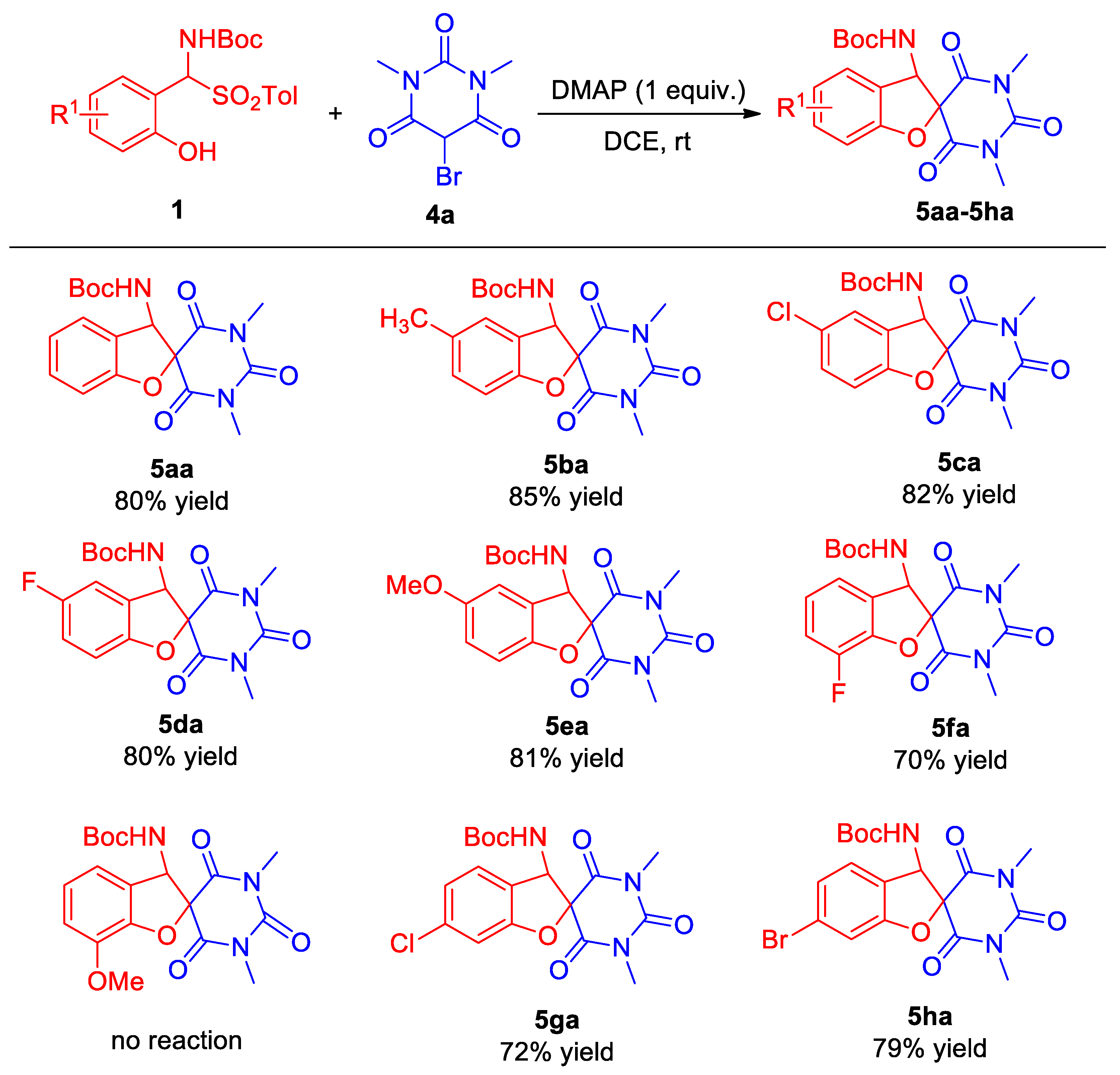
2.2. Substrate Scope
2.3. Scaled-Up Synthesis
2.4. Further Evaluation of Substrate Scope of Dicarbonyl α-Bromo-Substituted Compounds
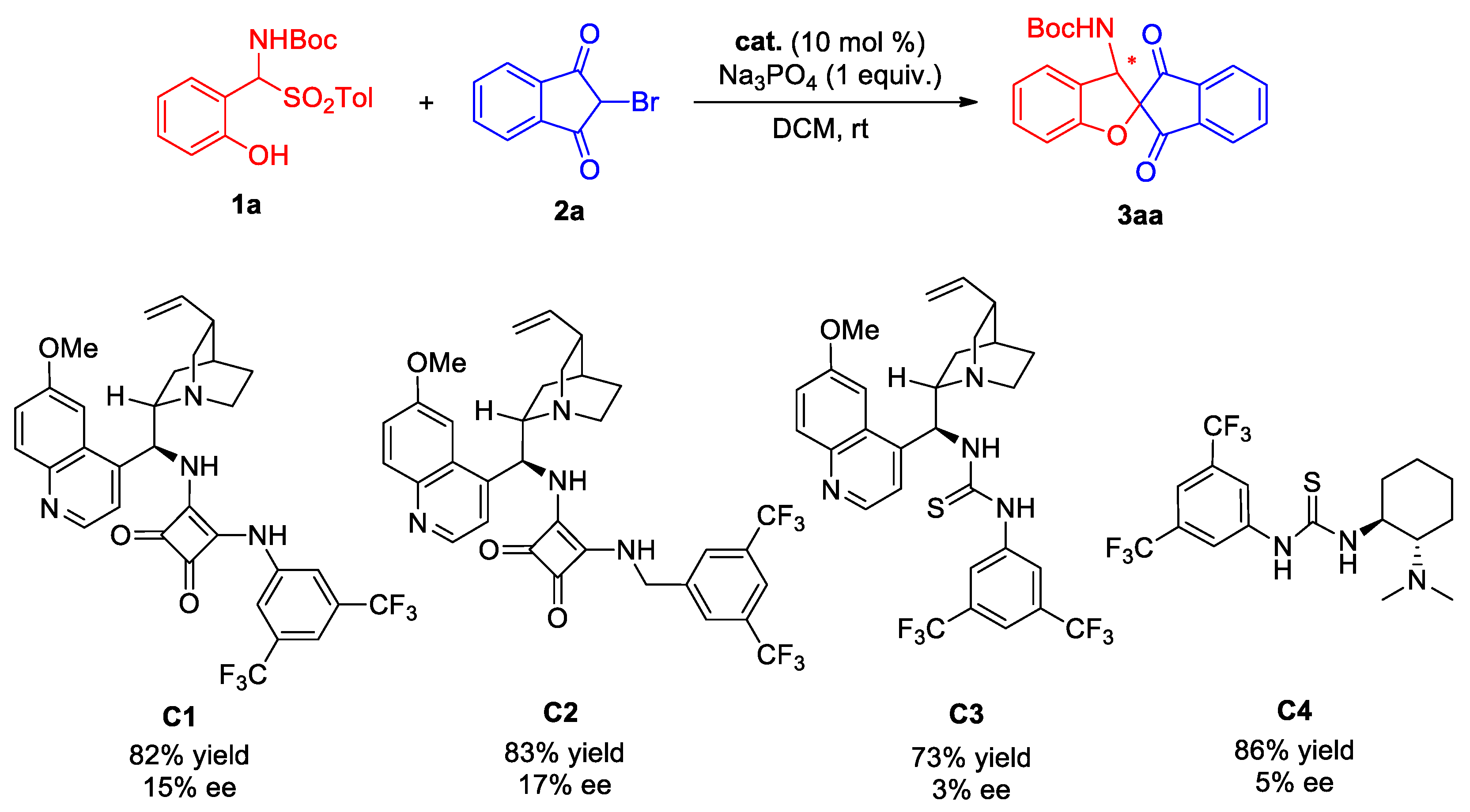
2.5. Asymmetric Catalytic Reaction Trials
3. Materials and Methods
3.1. General Information
3.2. Experimental Materials for Tandem Reactions
3.3. Procedure for the Synthesis of Aminobenzofuran Spiroindanone 3
- ortho-Hydroxy α-aminosulfone 1 (0.15 mmol), 2-bromo-1,3-indandione 2a (0.1 mmol), and 4-dimethylaminopyridine (DMAP) (1.0 equiv.) were combined in dry DCE (1.0 mL) in a 5 mL glass reaction vessel. The reaction system was stirred at room temperature for 20 h. After the completion of the reaction, which was monitored by thin-layer chromatography, the crude product was separated by means of a silica gel rapid column chromatography (ethyl acetate/petroleum ether 1:5) to obtain 3.
- tert-Butyl (1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3aa). Compound 3aa (31.0 mg, 85% yield) was isolated as a yellow solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 148–150 °C. 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 6.8 Hz, 1H, ArH), 7.98–7.88 (m, 3H, ArH), 7.32–7.26 (m, 2H, ArH), 7.02 (t, J = 7.6 Hz, 1H, ArH), 6.93 (d, J = 8.4 Hz, 1H, ArH), 5.70 (d, J = 8.8 Hz, 1H, NH), 5.11 (d, J = 8.4 Hz, 1H, CH), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ195.5, 194.1, 159.9, 154.9, 141.5, 141.4, 136.5, 136.3, 130.8, 124.8, 124.3, 123.7, 123.4, 122.3, 110.6, 88.2, 80.5, 59.7, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H19NNaO5 [M + Na]+ 388.1156, found 388.1151.
- tert-Butyl (5-bromo-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)-carbamate (3ba). Compound 3ba (22.2 mg, 50% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 140–142 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 6.8 Hz, 1H, ArH), 7.99–7.90 (m, 3H, ArH), 7.43–7.39 (m, 2H, ArH), 6.86 (d, J = 8.4 Hz, 1H, ArH), 5.69 (d, J = 8.8 Hz, 1H, NH), 5.07 (d, J = 8.8 Hz, 1H, CH), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 195.0, 193.6, 159.1, 154.7, 141.4, 136.8, 136.5, 133.7, 127.8, 126.1, 124.5, 123.5, 114.2, 112.3, 88.6, 80.9, 59.2, 27.8 ppm. HRMS (ESI): m/z calcd. for C21H1879BrNNaO5 [M + Na]+ 466.0261, found 466.0271; calcd. for C21H1881BrNNaO5 [M + Na]+ 468.0241, found 468.0252.
- tert-Butyl (5-chloro-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)-carbamate (3ca). Compound 3ca (31.9 mg, 80% yield) was isolated as a yellow solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 217–219 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 6.8 Hz, 1H, ArH), 7.99–7.88 (m, 3H, ArH), 7.28–7.25 (m, 2H, ArH), 6.90 (d, J = 8.4 Hz, 1H, ArH), 5.68 (d, J = 8.8 Hz, 1H, NH), 5.08 (d, J = 8.4 Hz, 1H, CH), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 195.0, 193.6, 158.6, 154.7, 141.4, 136.7, 136.5, 130.8, 127.2, 125.6, 124.9, 124.5, 123.5, 111.7, 88.6, 80.8, 59.3, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H18ClNNaO5 [M + Na]+ 422.0766, found 422.0764.
- tert-Butyl (5-fluoro-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3da). Compound 3da (35.3 mg, 92% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 213–215 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 6.8 Hz, 1H, ArH), 7.99–7.90 (m, 3H, ArH), 7.03–6.98 (m, 2H, ArH), 6.89 (dd, J1 = 3.8 Hz, J2 = 8.6 Hz, 1H, ArH), 5.69 (d, J = 8.8 Hz, 1H, NH), 5.09 (d, J = 8.4 Hz, 1H, CH), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 195.2, 193.9, 158.4 (d, 1JC–F = 238.9 Hz), 155.8, 154.7, 141.4, 136.7, 136.5, 125.0 (d, 3JC–F = 8.2 Hz), 124.4, 123.5, 117.4 (d, 2JC–F = 24.4 Hz), 111.8 (d, 2JC–F = 25.2 Hz), 111.2 (d, 3JC–F = 8.3 Hz), 88.7, 80.8, 59.5, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H18FNNaO5 [M + Na]+ 406.1062, found 406.1043.
- tert-Butyl (5-methoxy-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3ea). Compound 3ea (29.6 mg, 75% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 224–226 °C. 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 6.8 Hz, 1H, ArH), 7.98–7.86 (m, 3H, ArH), 6.89–6.81 (m, 3H, ArH), 5.67 (d, J = 8.8 Hz, 1H, NH), 5.10 (d, J = 8.8 Hz, 1H, CH), 3.78 (s, 3H, OMe), 1.23 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ155.5, 154.8, 153.9, 141.5, 141.4, 136.5, 136.3, 124.4, 124.3, 123.4, 116.7, 110.9, 109.9, 80.6, 59.9, 56.1, 28.2, 27.9 ppm. HRMS (ESI): m/z calcd. for C22H21NNaO6 [M + Na]+ 418.1261, found 418.1252.
- tert-Butyl (5-methyl-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3fa). Compound 3fa (31.1 mg, 82% yield) was isolated as a yellow solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 181–183 °C. 1H NMR (400 MHz, CDCl3): δ 8.08 (d, J = 6.8 Hz, 1H, ArH), 7.97–7.86 (m, 3H, ArH), 7.11–7.07 (m, 2H, ArH), 6.85 (d, J = 8.0 Hz, 1H, ArH), 5.65 (d, J = 8.8 Hz, 1H, NH), 5.06 (d, J = 8.4 Hz, 1H, CH), 2.31 (s, 3H, CH3), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 195.6, 194.3, 158.0, 154.8, 141.5, 141.4, 136.5, 136.3, 131.8, 131.3, 125.1, 124.3, 123.5, 123.4, 110.2, 88.4, 80.5, 59.8, 27.9, 20.7 ppm. HRMS (ESI): m/z calcd. for C22H21NNaO5 [M + Na]+ 402.1312, found 402.1310.
- tert-Butyl (6-chloro-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3ga). Compound 3ga (32.3 mg, 81% yield) was isolated as a yellow solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 225–227 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 6.8 Hz, 1H, ArH), 7.98–7.88 (m, 3H, ArH), 7.18 (d, J = 7.6 Hz, 1H, ArH), 7.02–6.98 (m, 2H, ArH), 5.65 (d, J = 8.8 Hz, 1H, NH), 5.05 (d, J = 8.8 Hz, 1H, CH), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 194.9, 193.5, 160.7, 154.8, 141.4, 136.7, 136.5, 136.4, 125.4, 124.5, 123.5, 122.7, 122.6, 111.5, 88.7, 80.8, 59.0, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H18ClNNaO5 [M + Na]+ 422.0766, found 422.0765.
- tert-Butyl (6-bromo-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3ha). Compound 3ha (37.7 mg, 85% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 164–166 °C. 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 6.8 Hz, 1H, ArH), 7.98–7.88 (m, 3H, ArH), 7.17–7.12 (m, 3H, ArH), 5.63 (d, J = 8.8 Hz, 1H, NH), 5.07 (d, J = 8.4 Hz, 1H, CH), 1.22 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 194.9, 193.5, 160.7, 154.8, 141.4, 136.7, 136.5, 125.8, 125.5, 124.4, 124.1, 123.5, 123.1, 114.4, 88.6, 80.8, 59.1, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H1879BrNNaO5 [M + Na]+ 466.0261, found 466.0271; calcd. for C21H1881BrNNaO5 [M + Na]+ 468.0241, found 468.0252.
- tert-Butyl (7-bromo-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3ia). Compound 3ia (39.9 mg, 90% yield) was isolated as a yellow solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 172–174 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 6.8 Hz, ArH), 7.99–7.88 (m, 3H, ArH), 7.46 (d, J = 8.0 Hz, 1H, ArH), 7.21 (d, J = 7.6 Hz, 1H, ArH), 6.92 (t, J = 7.6 Hz, 1H, ArH), 5.77 (d, J = 8.8 Hz, 1H, NH), 5.12 (d, J = 8.8 Hz, 1H, CH), 1.23 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 194.8, 193.3, 157.2, 154.8, 141.5 136.7, 136.5, 133.9, 125.2, 124.5, 123.8, 123.7, 123.5, 103.4, 87.6, 80.8, 60.0, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H1879BrNNaO5 [M + Na]+ 466.0261, found 466.0271, C21H1881BrNNaO5 [M + Na]+ 468.0241, found 468.0264.
- tert-Butyl (7-fluoro-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3ja). Compound 3ja (36.4 mg, 95% yield) was isolated as a yellow solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 175–177 °C. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 6.4 Hz, 1H, ArH), 7.99–7.90 (m, 3H, ArH), 7.12–7.05 (m, 2H, ArH), 7.00–6.95 (m, 1H, ArH), 5.74 (d, J = 8.8 Hz, 1H, NH), 5.12 (d, J = 8.8 Hz, 1H, CH), 1.23 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 194.7, 193.3, 154.8, 147.2 (d, 1J C–F = 247.0 Hz), 146.7 (2JC–F = 11.6 Hz), 141.44, 141.36, 136.7, 136.5, 127.2, 124.5, 123.5, 123.1 (d, 3J C–F = 5.5 Hz), 120.0 (d, 3JC–F = 3.3 Hz), 117.9 (d, 2JC–F = 16.5 Hz), 88.5, 80.8, 59.6, 27.8 ppm. HRMS (ESI): m/z calcd. for C21H18FNNaO5 [M + Na]+ 406.1062, found 406.1057.
- tert-Butyl (4-chloro-1′,3′-dioxo-1′,3′-dihydro-3H-spiro[benzofuran-2,2′-inden]-3-yl)carbamate (3ka). Compound 3ka (32.3 mg, 81% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 178–180 °C. 1H NMR (400 MHz, CDCl3): δ 8.10–8.08 (m, 1H, ArH), 7.99–7.89 (m, 3H, ArH), 7.26 (t, J = 8.2 Hz, 1H, ArH), 6.99 (d, J = 8.0 Hz, 1H, ArH), 6.88 (d, J = 8.4 Hz, 1H, ArH), 5.73 (d, J = 8.8 Hz, 1H, NH), 5.07 (d, J = 8.4 Hz, 1H, CH), 1.27 (s, 9H, CH3) ppm. 13C NMR (176 MHz, CDCl3): δ 194.7, 193.1, 161.1, 154.7, 141.8, 140.8, 136.7, 136.6, 132.0, 131.3, 124.5, 123.5, 122.9, 121.6, 109.2, 88.2, 80.6, 58.9, 27.9 ppm. HRMS (ESI): m/z calcd. for C21H18ClNNaO5 [M + Na]+ 422.0766, found 422.0748.
3.4. Procedure for the Synthesis of Aminobenzofuran Spirobarbituric 5
- tert-Butyl (1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5aa). Compound 5aa (30.0 mg, 80% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 198–200 °C. 1H NMR (400 MHz, CDCl3): δ 7.33 (t, J = 7.6 Hz, 1H, ArH), 7.22 (d, J = 7.6 Hz, 1H, ArH), 7.03–7.00 (m, 2H, ArH), 5.75 (d, J = 8.4 Hz, 1H, NH), 5.14 (d, J = 8.4 Hz, 1H, CH), 3.40 (s, 3H, CH3), 3.30 (s, 3H, CH3), 1.43 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 167.4, 165.2, 159.9, 155.0, 150.3, 131.2, 124.7, 122.5, 121.9, 110.8, 87.6, 81.3, 63.9, 29.2, 28.8, 28.1 ppm. HRMS (ESI): m/z calcd. for C18H21N3NaO6 [M + Na]+ 398.1322, found398.1307.
- tert-Butyl (1′,3′,5-trimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro[benzo-furan-2,5′-pyrimidin]-3-yl)carbamate (5ba). Compound 5ba (31.1 mg, 85% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 203–205 °C. 1H NMR (400 MHz, CDCl3): δ 7.11 (d, J = 8.4 Hz, 1H, ArH), 7.02 (s, 1H, ArH), 6.90 (d, J = 8.4 Hz, 1H, ArH), 5.70 (d, J = 8.8 Hz, 1H, NH), 5.12 (d, J = 8.4 Hz, 1H, CH), 3.38 (s, 3H, CH3), 3.28 (s, 3H, CH3), 2.30 (s, 3H, CH3), 1.42 (s, 9H, CH3) ppm. 13C NMR (176 MHz, CDCl3): δ 167.5, 165.3, 157.9, 154.9, 150.3, 132.0, 131.7, 124.9, 121.7, 110.3, 87.8, 81.3, 63.9, 29.2, 28.7, 28.1, 20.7 ppm. HRMS (ESI): m/z calcd. for C19H23N3NaO6 [M + Na]+ 412.1479, found 412.1471.
- tert-Butyl (5-chloro-1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro-[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5ca). Compound 5ca (33.5 mg, 82% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 218–220 °C. 1H NMR (400 MHz, CDCl3): δ 7.29 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H, ArH), 7.21 (s, 1H, ArH), 6.95 (d, J = 8.4 Hz, 1H, ArH), 5.73 (d, J = 8.8 Hz, 1H, NH), 5.12 (d, J = 8.4 Hz, 1H, CH), 3.39 (s, 3H, CH3), 3.29 (s, 3H, CH3), 1.42 (s, 9H, CH3) ppm. 13C NMR (176 MHz, CDCl3): δ 167.0, 164.8, 158.5, 154.8, 150.1, 131.2, 127.4, 124.8, 123.8, 111.8, 87.9, 81.6, 63.4, 29.3, 28.8, 28.1 ppm. HRMS (ESI): m/z calcd. for C18H20ClN3NaO6 [M + Na]+ 432.0933, found 432.0924.
- tert-Butyl (5-fluoro-1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro-[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5da). Compound 5da (31.4 mg, 80% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 205–207 °C. 1H NMR (400 MHz, CDCl3): δ 7.02 (td, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H, ArH), 6.96–6.93 (m, 2H, ArH), 5.74 (d, J = 8.8 Hz, 1H, NH), 5.19 (d, J = 8.4 Hz, 1H, CH), 3.39 (s, 3H, CH3), 3.29 (s, 3H, CH3), 1.42 (s, 9H, CH3) ppm. 13C NMR (176 MHz, CDCl3): δ 167.2, 165.0, 158.4 (1JC–F = 240.8 Hz), 155.7, 154.9, 150.1, 123.2 (3JC–F = 8.8 Hz), 117.8 (2JC–F = 24.6 Hz), 111.7 (2JC-F = 25.7 Hz), 111.3 (3JC–F = 8.1 Hz), 88.0, 81.6, 63.6, 29.2, 28.8, 28.1 ppm. HRMS (ESI): m/z calcd. for C18H20FN3NaO6 [M + Na]+ 416.1228, found 416.1209.
- tert-Butyl (5-methoxy-1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro-[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5ea). Compound 5ea (32.8 mg, 81% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 228–230 °C. 1H NMR (400 MHz, CDCl3): δ 6.93 (d, J = 8.8 Hz, 1H, ArH), 6.86 (dd, J1 = 2.6 Hz, J2 = 9.0 Hz, 1H, ArH), 6.74 (d, J = 2.0 Hz, 1H, ArH), 5.72 (d, J = 8.8 Hz, 1H, NH), 5.13 (d, J = 8.4 Hz, 1H, CH), 3.76 (s, 3H, OCH3), 3.39 (s, 3H, CH3), 3.29 (s, 3H, CH3), 1.43 (s, 9H, CH3) ppm. 13C NMR (176 MHz, CDCl3): δ 167.5, 165.3, 155.5, 154.9, 153.8, 150.3, 122.5, 117.0, 111.0, 109.8, 87.9, 81.4, 64.0, 56.1, 29.2, 28.7, 28.1 ppm. HRMS (ESI): m/z calcd. for C19H23N3NaO7 [M + Na]+ 428.1428, found 428.1417.
- tert-Butyl (7-fluoro-1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro-[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5fa). Compound 5fa (27.5 mg, 70% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 201–203 °C. 1H NMR (400 MHz, CDCl3): δ 7.13–7.09 (m, 1H, ArH), 7.01–6.94 (m, 2H, ArH), 5.79 (d, J = 8.4 Hz, 1H, NH), 5.17 (d, J = 8.4 Hz, 1H, CH), 3.40 (s, 3H, CH3), 3.30 (s, 3H, CH3), 1.42 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 166.8, 164.5, 155.0, 150.1, 147.1 (1JC–F = 247.6 Hz), 146.7 (2JC–F = 11.5 Hz), 125.5 (4JC–F = 2.5 Hz), 123.3 (3JC–F = 5.3 Hz), 119.8 (3JC–F = 3.4 Hz), 118.2 (2JC–F = 16.4 Hz), 87.9, 81.6, 63.9, 29.2, 28.8, 28.1 ppm. HRMS (ESI): m/z calcd. for C18H20FN3NaO6 [M + Na]+ 416.1228, found 416.1224.
- tert-Butyl (6-chloro-1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro-[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5ga). Compound 5ga (29.4 mg, 72% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 202–204 °C. 1H NMR (400 MHz, CDCl3): δ 7.14 (d, J = 8.0 Hz, 1H, ArH), 7.03–6.99 (m, 2H, ArH), 5.70 (d, J = 8.8 Hz, 1H, NH), 5.10 (d, J = 8.4 Hz, 1H, CH), 3.39 (s, 3H, CH3), 3.29 (s, 3H, CH3), 1.42 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 166.9, 164.8, 160.5, 154.9, 150.1, 136.8, 125.3, 122.8, 120.7, 111.6, 88.0, 81.6, 63.2, 29.2, 28.8, 28.1 ppm. HRMS (ESI): m/z calcd. for C18H20ClN3NaO6 [M + Na]+ 432.0933, found 432.0928.
- tert-Butyl (6-bromo-1′,3′-dimethyl-2′,4′,6′-trioxo-1′,3′,4′,6′-tetrahydro-2′H,3H-spiro-[benzofuran-2,5′-pyrimidin]-3-yl)carbamate (5ha). Compound 5ha (35.8 mg, 79% yield) was isolated as a white solid through a standard purification process involving column chromatography on silica gel (200–300 mesh), utilizing a mixture of petroleum ether and ethyl acetate (5:1) as the eluent. m.p. 198–200 °C. 1H NMR (400 MHz, CDCl3): δ 7.18–7.14 (m, 2H, ArH), 7.08 (d, J = 8.0 Hz, 1H, ArH), 5.68 (d, J = 8.8 Hz, 1H, NH), 5.20 (d, J = 8.8 Hz, 1H, CH), 3.38 (s, 3H, CH3), 3.27 (s, 3H, CH3), 1.42 (s, 9H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 166.9, 164.7, 160.5, 154.9, 150.1, 125.7, 125.6, 124.4, 121.3, 114.4, 87.9, 81.5, 63.3, 29.2, 28.7, 28.1 ppm. HRMS (ESI): m/z calcd. for C18H2179BrN3O6 [M + H]+ 454.0608, found 454.0305, calcd. for C18H2181BrN3O6 [M + H]+ 456.0588, found 456.0287.
3.5. Procedure for the Scaled-Up Synthesis of Compound 3aa and 5aa
3.6. Asymmetric Catalyzed Cyclization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khanam, H.; Uzzaman, S. Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Ojika, M.; Suzuki, S.; Murakami, M.; Sakagamia, Y. Iantherans A and B, unique dimeric polybrominated benzofurans as Na,K-ATPase inhibitors from a marine sponge, Ianthella sp. Bioorg. Med. Chem. 2001, 9, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Mitsui, C.; Illies, L.; Sato, Y.; Nakamura, E. Synthesis and properties of 2,3,6,7-tetraarylbenzo [1,2-b:4,5-b‘] difurans as hole-transporting material. J. Am. Chem. Soc. 2007, 129, 11902–11903. [Google Scholar] [CrossRef]
- Patel, P.; Shakya, R.; Vishakha, V.A.; Kurmi, B.D.; Verma, S.K.; Gupta, G.D.; Rajak, H. Furan and benzofuran derivatives as privileged scaffolds as anticancer agents: SAR and docking studies (2010 to till date). J. Mol. Struct. 2024, 1299, 137098–137118. [Google Scholar] [CrossRef]
- Soni, J.N.; Soman, S.S. Synthesis and antimicrobial evaluation of amide derivatives of benzodifuran-2-carboxylic acid. Eur. J. Med. Chem. 2014, 75, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Meng, Q.Y.; Daniliuc, C.G.; Studer, A. Aroyl fluorides as bifunctional reagents for dearomatizing fluoroaroylation of benzofurans. J. Am. Chem. Soc. 2022, 144, 7072–7079. [Google Scholar] [CrossRef]
- Abbas, A.A.; Dawood, K.M. Anticancer therapeutic potential of benzofuran scaffolds. RSC Adv. 2023, 13, 11096–11120. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V.; Hamidia, H.; Hajiabbas, P.; Amiria, T. Total synthesis of natural products containing benzofuran rings. RSC Adv. 2017, 7, 24470–24521. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Sun, L.Q.; Han, X.Y.; Wang, Y.J.; Xie, Z.S.; Xue, S.T.; Li, Z.R. Efficacy, mechanism, and structure–activity relationship of 6-methoxy benzofuran derivatives as a useful tool for senile osteoporosis. J. Med. Chem. 2023, 66, 1742–1760. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.J.; Lv, Z.S.; Feng, L.S.; Wang, Y.L.; Zhang, F.; Bai, L.Y.; Deng, J.L. Benzofuran derivatives and their anti-tubercular, anti-bacterial activities. Eur. J. Med. Chem. 2019, 162, 266–276. [Google Scholar] [CrossRef]
- Chen, Z.; Pitchakuntla, M.; Jia, Y.X. Synthetic approaches to natural products containing 2,3-dihydrobenzofuran skeleton. Nat. Prod. Rep. 2019, 36, 666. [Google Scholar] [CrossRef]
- Smith, D.T.; Vitaku, E.; Njardarson, J.T. Dearomatization approach to 2-trifluoromethylated benzofuran and dihydrobenzofuran products. Org. Lett. 2017, 19, 3508–3511. [Google Scholar] [CrossRef]
- Xiao, B.X.; Du, W.; Chen, Y.C. Asymmetric dearomatizative Diels–Alder reaction for the construction of hydrodibenzo[b,d]furan frameworks with tetrasubstituted stereogenic centers. Adv. Synth. Catal. 2017, 359, 1018–1027. [Google Scholar] [CrossRef]
- Danel, J.M.; Fleige, M.; Schluens, D.; Wollenburg, M.; Daniliuc, C.G.; Neugebauer, J.; Glorius, F. NHC-catalyzed enantioselective dearomatizing hydroacylation of benzofurans and benzothiophenes for the synthesis of spirocycles. ACS Catal. 2016, 6, 5735–5739. [Google Scholar]
- Dwarakanath, D.; Gaonkar, S.L. Advances in synthetic strategies and medicinal importance of benzofurans: A review. Asian J. Org. Chem. 2022, 11, e202200282. [Google Scholar] [CrossRef]
- Goyal, D.; Kaur, A.; Goyal, B. Benzofuran and indole: Promising scaffolds for drug development in alzheimer’s disease. Chem. Med. Chem. 2018, 13, 1275–1299. [Google Scholar] [CrossRef]
- Wang, Y.; Li, K.; Zou, Y.; Zhou, M.; Li, J.; Wu, C.; Tan, R.; Liao, Y.; Li, W.; Zheng, J. Metabolic activation of 3-aminodibenzofuran mediated by P450 enzymes and sulfotransferases. Toxicol. Lett. 2022, 360, 44–52. [Google Scholar] [CrossRef]
- Lauria, A.; Gentile, C.; Mingoia, F.; Piccionello, A.P.; Bartolotta, R.; Delisi, R.; Buscemi, S.; Martorana, A. Design synthesis, and biological evaluation of a new class of benzo[b]furan derivatives as antiproliferative agents, with in silico predicted antitubulin activity. Chem. Biol. Drug Des. 2018, 91, 39–49. [Google Scholar] [CrossRef]
- Cabrera-Pardo, J.R.; Fuentealba, J.; Gavilán, J.; Cajas, D.; Becerra, J.; Napiórkowska, M. Exploring the multi–target neuroprotective chemical space of benzofuran scaffolds: A new strategy in drug development for alzheimer’s disease. Front. Pharmacol. 2020, 10, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Chen, W.J.; Chen, F.Y.; Zhang, H.Y.; Xu, H.Y.; Zhou, Z.; Yi, W. Synthesis of 2-aminobenzofurans via base-mediated [3+2] annulation of N-phenoxy amides with gem-difluoroalkenes. Org. Chem. Front. 2021, 8, 4452–4458. [Google Scholar] [CrossRef]
- Li, L.S.; Li, C.Y.; Zhang, S.T.; Wang, X.R.; Fu, P.; Wang, Y. Catalytic asymmetric synthesis of 3,4′-piperidinoyl spirooxindoles via [3+3] annulation of 3-aminobenzofurans and isatin-derived enals. J. Org. Chem. 2024, 89, 5170–5180. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.X.; Zhang, Y.J.; Guo, Y.S.; Jin, Q.H.; Zhu, H.Y.; Xiu, H.S.; Liu, Z.H.; Wang, Y. The [4+1] cyclization reaction of 2-hydroxylimides and trimethylsulfoxonium iodide for the synthesis of 3-amino-2,3-dihydrobenzofurans. New J. Chem. 2022, 46, 18124–18127. [Google Scholar] [CrossRef]
- Ma, X.L.; Wang, Z.Q.; Liu, Z.R.; Li, Z. One-Pot Three-component synthesis of 2-methyl-3-aminobenzofurans using calcium carbide as a concise solid alkyne source. Chin. J. Chem. 2021, 39, 2990–2994. [Google Scholar] [CrossRef]
- Yang, W.L.; Sun, Z.T.; Zhang, J.; Li, Z.; Deng, W.P. Enantioselective synthesis of 3-amino-hydrobenzofuran-2,5-diones via Cu(i)-catalyzed intramolecular conjugate addition of imino esters. Org. Chem. Front. 2019, 6, 579–583. [Google Scholar] [CrossRef]
- Helgren, T.R.; Xu, L.L.; Sotelo, D.; Mehta, Y.R.; Korkmaz, M.A.; Pavlinov, I.; Aldrich, L.N. Microwave-assisted.; asymmetric synthesis of 3-amino-2,3-dihydrobenzofuran flavonoid derivatives from chalcones. Chem. Eur. J. 2018, 24, 4509–4514. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.S.; Li, R.D.; Wang, X.C. Copper-catalyzed asymmetric annulation reactions of carbenes with 2-iminyl- or 2-acyl-substituted phenols: Convenient access to enantioenriched 2,3-dihydrobenzofurans. Angew. Chem. Int. Ed. 2019, 58, 13885–13889. [Google Scholar] [CrossRef]
- Panday, A.K.; Ali, D.; Choudhury, L.H. Cs2CO3-mediated rapid room-temperature synthesis of 3-amino-2-aroyl benzofurans and their copper-catalyzed N-arylation reactions. ACS Omega 2020, 5, 3646–3660. [Google Scholar] [CrossRef] [PubMed]
- Abtahi, B.; Tavakol, H. CuI-catalyzed, one-pot synthesis of 3-aminobenzofurans in deep eutectic solvents. Appl. Organomet. Chem. 2021, 35, e6433. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Patil, S.A. Recent developments in biological activities of indanones. Eur. J. Med. Chem. 2017, 138, 182–198. [Google Scholar] [CrossRef]
- Turek, M.; Szczesna, D.; Koprowski, M.; Bałczewski, P. Synthesis of 1-indanones with a broad range of biological activity. Beilstein J. Org. Chem. 2017, 13, 451–494. [Google Scholar] [CrossRef]
- Yu, B.; Yu, D.Q.; Liu, H.M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Segovia, C.; Lebrêne, A.; Levacher, V.; Oudeyer, S.; Brière, J.F. Enantioselective catalytic transformations of barbituric acid derivatives. Catalysts 2019, 9, 131. [Google Scholar] [CrossRef]
- Bagherinejad, A.; Alizadeh, A. A review of the synthetic strategies toward spirobarbiturate-fused 3- to 7-membered rings. Org. Biomol. Chem. 2022, 20, 7188–7215. [Google Scholar] [CrossRef] [PubMed]
- Pigot, C.; Brunel, D.; Dumur, F. Indane-1,3-dione: From synthetic strategies to applications. Molecules. 2022, 27, 5976–6112. [Google Scholar] [CrossRef]
- Osman, E.E.A.; Hanafy, N.S.; George, R.F. Design and synthesis of some barbituric and 1,3-dimethylbarbituric acid derivatives: A non-classical scaffold for potential PARP1 inhibitors. Bioorg. Chem. 2020, 104, 104198. [Google Scholar]
- Sonoda, K.; Ujike, S.; Katayama, A.; Suzuki, N.; Kawaguchi, S.; Tsujita, T. Improving lipophilicity of 5-(1-acetyl-5-phenylpyrazolidin-3-ylidene)-1,3-dimethylbarbituric acid increases its efficacy to activate hypoxia-inducible factors. Bioorg. Med. Chem. 2022, 73, 117039. [Google Scholar] [CrossRef]
- Liu, M.M.; Yang, X.C.; Hua, Y.Z.; Chang, J.B.; Wang, M.C. Dinuclear zinc-catalyzed asymmetric tandem reaction of α-Hydroxy-1-indanone: Access to spiro [1-indanone-5,2′-γ-butyrolactones]. Org. Lett. 2019, 21, 7089–7093. [Google Scholar] [CrossRef]
- Schade, A.; Tchernook, I.; Bauer, M.; Oehlke, A.; Breugst, M.; Friedrich, J.; Spange, S. Kinetics of electrophilic alkylations of barbiturate and thiobarbiturate anions. J. Org. Chem. 2017, 82, 8476–8488. [Google Scholar] [CrossRef]
- Yan, X.B.; Shao, P.; Song, X.X.; Zhang, C.F.; Lu, C.; Liu, S.T.; Li, Y.L. Chemoselective syntheses of spirodihydrofuryl and spirocyclopropyl barbiturates via cascade reactions of barbiturate-based olefins and acetylacetone. Org. Biomol. Chem. 2019, 17, 2684–2690. [Google Scholar] [CrossRef]
- Alizadeh, A.; Bagherinejad, A.; Kayanian, J.; Vianello, R. An expedient metal-free cascade route to chromonyl diene scaffolds: Thermodynamic vs. kinetic control. RSC Adv. 2022, 12, 34946–34950. [Google Scholar] [CrossRef]
- Zhao, B.L.; Du, D.M. Organocatalytic cascade Michael/Michael reaction for the asymmetric synthesis of spirooxindoles containing five contiguous stereocenters. Chem. Commum. 2016, 52, 6162–6165. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Hou, X.Q.; Li, B.Y.; Du, D.M. Organocatalytic remote asymmetric inverse-electron-demand Oxa-Diels-Alder reaction of allyl ketones with isatin-derived unsaturated keto esters. Adv. Synth. Catal. 2020, 362, 5728. [Google Scholar] [CrossRef]
- Hou, X.Q.; Lin, Y.; Du, D.M. Organocatalytic domino annulation of in situ generated tert-butyl 2-hydroxybenzylidenecarbamates with 2-isothiocyanato-1-indanones for synthesis of bridged and fused ring heterocycles. Org. Chem. Front. 2021, 8, 4183–4187. [Google Scholar] [CrossRef]
- Ren, Q.; Siau, W.Y.; Du, Z.Y.; Zhang, K.; Wang, J. Expeditious assembly of a 2-Amino-4H-chromene skeleton by using an enantioselective mannich intramolecular ring cyclization–tautomerization cascade sequence. Chem. Eur. J. 2011, 17, 7781–7785. [Google Scholar] [CrossRef] [PubMed]
- Ahadi, S.; Khavasi, H.R.; Bazgir, A. Highly efficient construction of bisspirooxindoles containing vicinal spirocenters through an organocatalytic modified Feist–Bénary reaction. Chem. Eur. J. 2013, 19, 12553–12559. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, R.; Tajbakhsh, M.; Mohadjerani, M.; Lasemi, Z. Ethylenebis(N-methylimidazolium) ditribromide (EBMIDTB): An efficient reagent for the monobromination of 1,3-diketones and β-ketoesters. Monatsh. Chem. 2009, 140, 57–60. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Du, D.M. Chiral squaramide-catalyzed highly enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinones to nitroalkenes. Adv. Synth. Catal. 2011, 353, 1241–1246. [Google Scholar] [CrossRef]
- Anderson, J.C.; Koovits, P.J. An enantioselective tandem reduction/nitro-Mannich reaction of nitroalkenes using a simple thiourea organocatalyst. Chem. Sci. 2013, 4, 2897–2901. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yield b (%) |
| 1 | NaHCO3 | CH2Cl2 | 20 | 42 |
| 2 | Na2CO3 | CH2Cl2 | 20 | 50 |
| 3 | K2CO3 | CH2Cl2 | 20 | 52 |
| 4 | Cs2CO3 | CH2Cl2 | 20 | 41 |
| 5 | Et3N | CH2Cl2 | 20 | 45 |
| 6 | DABCO | CH2Cl2 | 20 | 31 |
| 7 | DBU | CH2Cl2 | 20 | 32 |
| 8 | TMG | CH2Cl2 | 20 | 36 |
| 9 | DMAP | CH2Cl2 | 20 | 70 |
| 10 | DMAP | DCE | 20 | 85 |
| 11 | DMAP | toluene | 20 | 54 |
| 12 | DMAP | CH3CN | 20 | 41 |
| 13 | DMAP | THF | 20 | 33 |
| 14 | DMAP | EtOH | 15 | 25 |
| 15 | DMAP | MTBE | 16 | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, R.-R.; Hou, X.-Q.; Du, D.-M. Synthesis of Benzofuran Derivatives via a DMAP-Mediated Tandem Cyclization Reaction Involving ortho-Hydroxy α-Aminosulfones. Molecules 2024, 29, 3725. https://doi.org/10.3390/molecules29163725
Zhu R-R, Hou X-Q, Du D-M. Synthesis of Benzofuran Derivatives via a DMAP-Mediated Tandem Cyclization Reaction Involving ortho-Hydroxy α-Aminosulfones. Molecules. 2024; 29(16):3725. https://doi.org/10.3390/molecules29163725
Chicago/Turabian StyleZhu, Rong-Rong, Xi-Qiang Hou, and Da-Ming Du. 2024. "Synthesis of Benzofuran Derivatives via a DMAP-Mediated Tandem Cyclization Reaction Involving ortho-Hydroxy α-Aminosulfones" Molecules 29, no. 16: 3725. https://doi.org/10.3390/molecules29163725
APA StyleZhu, R.-R., Hou, X.-Q., & Du, D.-M. (2024). Synthesis of Benzofuran Derivatives via a DMAP-Mediated Tandem Cyclization Reaction Involving ortho-Hydroxy α-Aminosulfones. Molecules, 29(16), 3725. https://doi.org/10.3390/molecules29163725