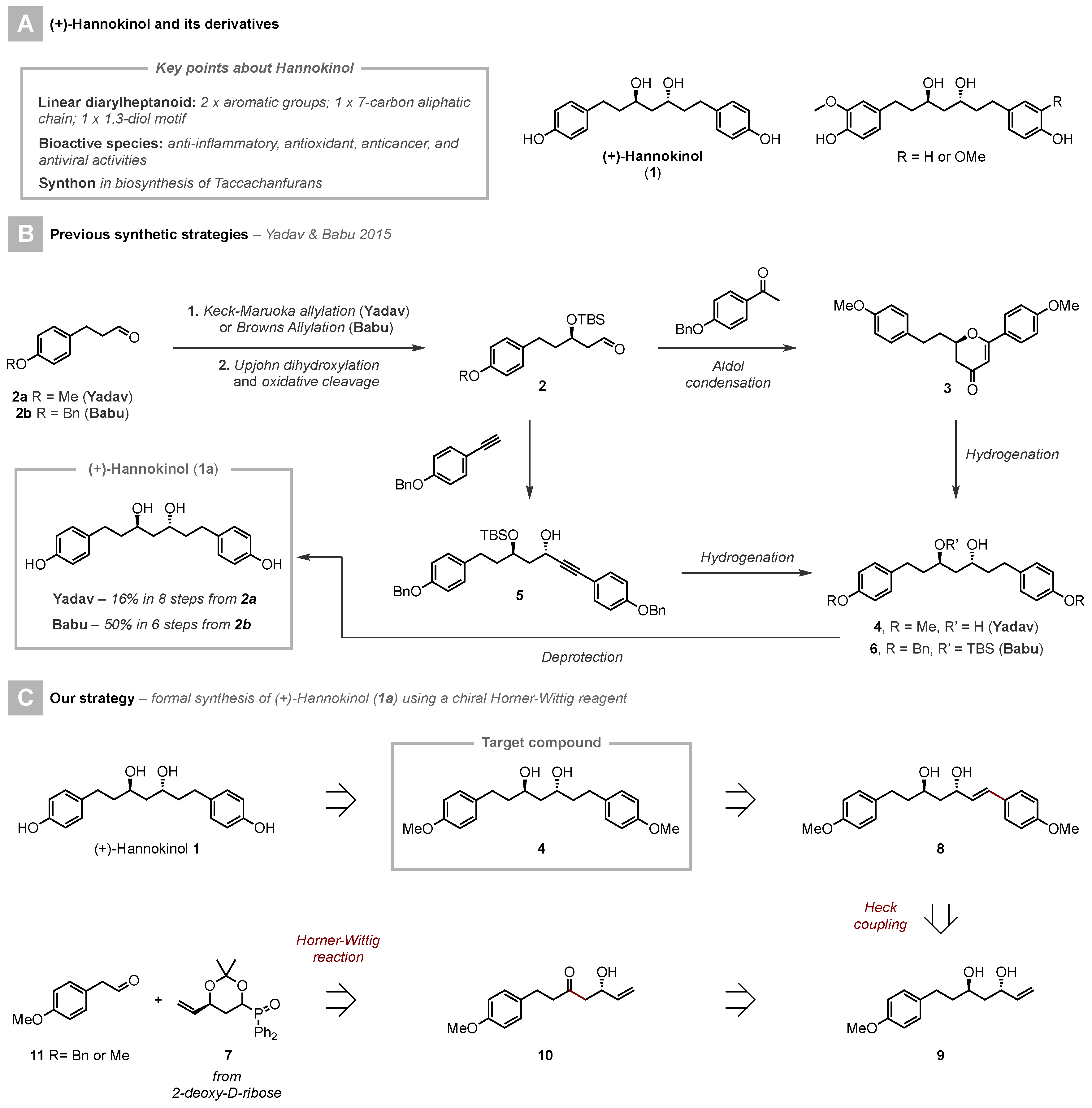
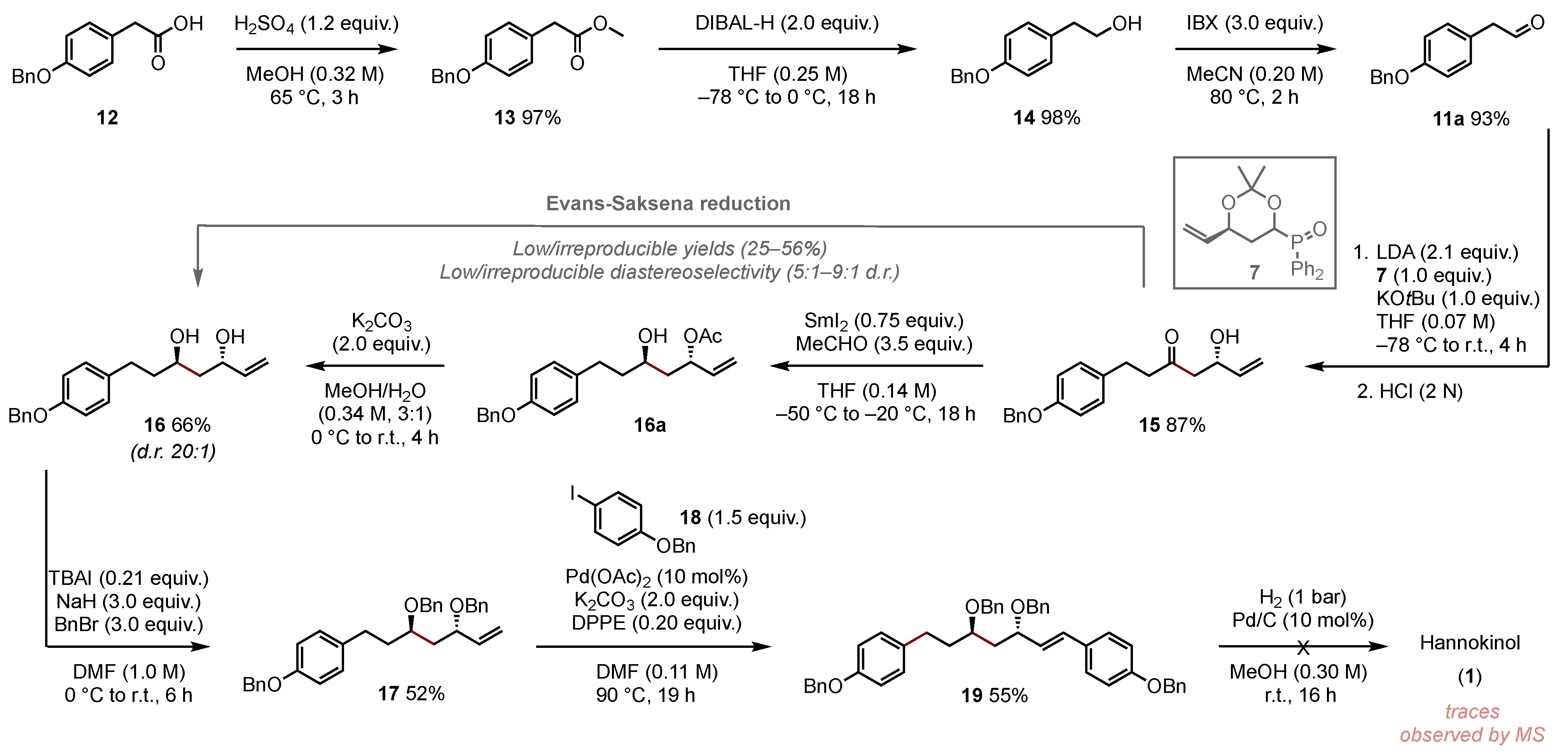
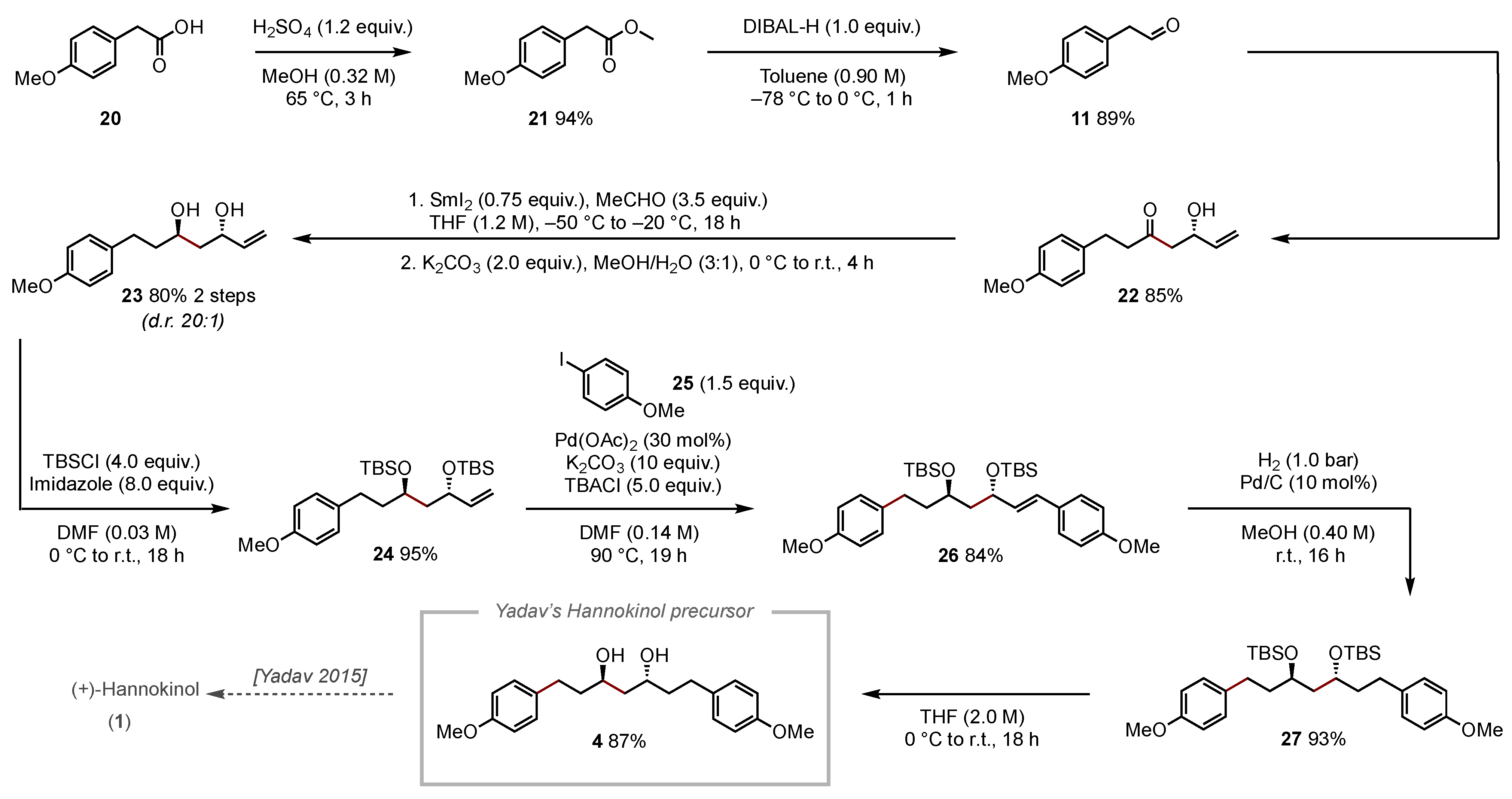
3.2. Synthesis and Charaecterization of Compounds
3.2.1. Methyl 2-(4-(Benzyloxy)phenyl)acetate (13)
To a solution of 2-(4-(benzyloxy)phenyl)acetic acid
12 (5.00 g, 20.6 mmol, 1.0 equiv.) in MeOH (65 mL) was added conc. H
2SO
4 (1.33 mL, 1.2 equiv.). The reaction mixture was heated under reflux (65 °C) for 3 h. After this time, the reaction mixture was allowed to cool down to room temperature, concentrated in vacuo, and dissolved in EA. The resulting solution was dried with sodium sulfate and filtered. The filtrate was concentrated in vacuo to afford a methyl ester
13 (5.13 g, yield 97%) as a yellow oil. TLC: R
f = 0.44 (EA/CyHex = 3:7) [KMnO
4].
1H NMR (600 MHz, CDCl
3) δ 7.43 (d,
J = 7.5 Hz, 2H), 7.39 (d,
J = 15.0 Hz, 2H), 7.33 (t,
J = 7.3 Hz, 1H), 7.20 (d,
J = 8.8 Hz, 2H), 6.94 (d,
J = 8.7 Hz, 2H), 5.06 (s, 2H), 3.69 (s, 3H), 3.57 (s, 2H).
13C NMR (151 MHz, CDCl
3) δ 172.5, 158.1, 137.2, 130.4, 128.7, 128.1, 127.6, 126.5, 115.1, 70.2, 52.1, 40.5. HRMS (ESI)
m/
z: [M + Na
+] calcd. for C
16H
16NaO
3: 279.0991; found: 279.0992. All the characterization data are consistent with the literature [
25].
3.2.2. 2-(4-(Benzyloxy)phenyl)ethan-1-ol (14)
To a solution of methyl 2-(4-(benzyloxy)phenyl)acetate
13 (3.70 g, 14.4 mmol, 1.0 equiv.) in anhydrous THF (58 mL) was added dropwise DIBAL-H (1.0 M solution in toluene, 28.9 mL, 28.9 mmol, 2.0 equiv.) at −78 °C. After the addition, the cold bath was removed, and the reaction mixture was stirred at room temperature for 7 h. Glycerol (8 mL) and K/Na-tartrate solution (20 mL) were then added at 0 °C. The mixture was stirred overnight at room temperature and then extracted three times with EA. The combined organic phase was washed with a sodium bicarbonate solution, dried with sodium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (EA/CyHex = 3:7) to give product
14 (3.26 g, yield 98%) as a colorless solid. TLC: R
f = 0.18 (EA/CyHex = 3:7) [KMnO
4].
1H NMR (600 MHz, CDCl
3) δ 7.43 (d,
J = 7.6 Hz, 2H), 7.39 (t,
J = 7.5 Hz, 2H), 7.32 (t,
J = 7.3 Hz, 1H), 7.15 (d,
J = 8.6 Hz, 2H), 6.94 (d,
J = 8.6 Hz, 2H), 5.06 (s, 2H), 3.83 (t,
J = 6.5 Hz, 2H), 2.82 (t,
J = 6.5 Hz, 2H), 1.43 (br, 1H).
13C NMR (151 MHz, CDCl
3) δ 157.7, 137.3, 130.9, 130.1, 128.7, 128.1, 127.6, 115.2, 70.2, 64.0, 38.5. All the characterization data are consistent with the literature [
25].
3.2.3. 2-(4-(Benzyloxy)phenyl)acetaldehyde (11a)
To a solution of 2-(4-(benzyloxy)phenyl)ethan-1-ol
14 (4.00 g, 17.5 mmol, 1.0 equiv.) in dry acetonitrile (88 mL) was added IBX (13.0 g, 52.6 mmol, 3.0 equiv.), and the suspension was stirred at 80 °C for 2 h and then cooled to room temperature. The suspension formed after cooling was filtered through a pad of celite and washed with EA. The filtrate was washed with a sodium bicarbonate solution, and the aqueous phase was extracted with EA (3 × 50 mL). The combined organic phases were dried over sodium sulfate and the solvent was removed under reduced pressure. The crude residue was purified by column chromatography (EA/CyHex = 2:8 → 3:7) to give product
11a (3.69 g, yield 93%) as a yellowish solid. TLC: R
f = 0.63 (EA/CyHex = 1:5) [KMnO
4].
1H NMR (400 MHz, CDCl
3) δ 9.73 (t,
J = 2.4 Hz, 1H), 7.47–7.38 (m, 4H), 7.36–7.30 (m, 1H), 7.14 (d,
J = 8.7 Hz, 2H), 6.98 (d,
J = 8.7 Hz, 2H), 5.07 (s, 2H), 3.63 (d,
J = 2.4 Hz, 2H).
13C NMR (101 MHz, CDCl
3) δ 199.8, 158.3, 137.0, 130.8, 128.8, 128.2, 124.1, 115.5, 70.2, 49.9. IR (ATR):
[cm
−1] = 3032, 2868, 1719, 1610, 1583, 1509, 1454, 1238, 1176, 1014, 927, 861, 612, 593. HRMS (ESI)
m/
z: [M + Na
+] calcd. for C
15H
14NaO
2: 249.0886; found: 249.0886. All the characterization data are consistent with the literature [
25].
3.2.4. (S)-1-(4-(Benzyloxy)phenyl)-5-hydroxyhept-6-en-3-one (15)
To a solution of diisopropylamine (2.15 mL, 15.3 mmol, 2.1 equiv.) in dry THF (73 mL) was added n-butyllithium (2.5 M in hexane, 6.13 mL, 15.3 mmol, 2.1 equiv.) at −78 °C. After the cooling was removed for 15 min, (S)-building block (2.50 g, 7.30 mmol, 1.0 equiv.) dissolved in dry THF (24 mL) was added slowly at −78 °C. The solution was allowed to stir for 1 h at this temperature. Following the addition of 2-(4-(benzyloxy)phenyl)acetaldehyde 11a (3.50 g, 15.5 mmol, 3.0 equiv.) dissolved in dry THF (9 mL), the reaction was allowed to warm to rt within 1 h. After the addition of potassium tert-butoxide (863 mg, 7.30 mmol, 1.0 equiv.), the reaction mixture was stirred for one more hour at room temperature and was quenched with a saturated aqueous ammonium chloride solution (30 mL), and the aqueous layer was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with hydrochloric acid (2 M, 2 × 50 mL), and the aqueous layer was again extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (100 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography (EA/CyHex = 3:7) to give the product 15 (1.97 g, yield 87%) as a light-yellow oil. TLC: Rf = 0.21 (EA/CyHex = 3:7) [KMnO4]. 1H NMR (400 MHz, CDCl3) δ 7.51–7.34 (m, 5H), 7.35–7.28 (m, 1H), 7.09 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 5.84 (ddd, J = 17.2, 10.5, 5.5 Hz, 1H), 5.28 (dt, J = 17.2, 1.4 Hz, 1H), 5.12 (dt, J = 10.5, 1.4 Hz, 1H), 5.04 (s, 2H), 4.56 (dtt, J = 7.0, 5.5, 1.5 Hz, 1H), 2.92–2.80 (m, 2H), 2.74 (ddd, J = 8.2, 6.8, 1.1 Hz, 2H), 2.62 (d, J = 5.5 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 210.4, 157.5, 139.2, 137.3, 133.2, 129.4, 128.7, 128.1, 127.6, 70.3, 68.8, 49.1, 45.5, 28.8. IR (ATR): [cm−1] = 3351, 3281, 3091, 3059, 3037, 3008, 2929, 2902, 2872, 1705, 1644, 1610, 1580, 1510, 1465, 1454, 1418, 1410, 1378, 1340, 1322, 1297, 1239, 1174, 1148, 1117, 1096, 1045, 1010, 994, 976, 917, 860, 845, 820, 807, 782, 747. [α]20D = −9.5 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C20H22NaO3: 333.1465; found: 333.1461.
3.2.5. (3S,5R)-7-(4-(Benzyloxy)phenyl)hept-1-ene-3,5-diol (16)
To a solution of (S)-1-(4-(benzyloxy)phenyl)-5-hydroxyhept-6-en-3-one 15 (530 mg, 1.71 mmol, 1.0 equiv.) in dry THF (12 mL) was added acetaldehyde (337 µL, 5.98 mmol, 3.5 equiv.). At −50 °C, SmI2 (0.1 M in THF, 12.8 mL, 1.28 mmol, 0.75 equiv.) was added slowly under the exclusion of light. The reaction mixture was allowed to stir at −20 °C for 18 h. After the addition of a saturated aqueous sodium bicarbonate solution (15 mL), the layers were separated, and the aqueous layer was extracted with EA (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by column chromatography (EA/CyHex = 2:8) to give the product 16 (563 mg, yield 93%) as a colorless oil.
To a solution of (3S,5R)-7-(4-(benzyloxy)phenyl)-5-hydroxyhept-1-en-3-yl acetate (500 mg, 1.41 mmol, 1.0 equiv.) in MeOH/H2O (4,2 mL, 3:1) was added potassium carbonate (389 mg, 2.82 mmol, 2.0 equiv.) at 0 °C. The reaction mixture was stirred for 4 h at 0 °C and then allowed to warm up to room temperature. Upon completion of the reaction indicated by TLC analysis, H2O (10 mL) and ethyl acetate (10 mL) were added, the layers were separated, and the aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (15 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by column chromatography (EA/CyHex = 3:7) to give the product 16 (313 mg, yield 71%) as a colorless oil. TLC: Rf = 0.29 (EA/CyHex = 1:1) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.43 (d, J = 7.5 Hz, 1H), 7.38 (t, J = 7.6 Hz, 5H), 7.32 (t, J = 7.3 Hz, 2H), 7.11 (d, J = 8.5 Hz, 5H), 6.90 (d, J = 8.6 Hz, 5H), 5.93 (ddd, J = 17.3, 10.5, 5.5 Hz, 3H), 5.29 (d, J = 17.2 Hz, 2H), 5.15 (s, 1H), 5.04 (s, 6H), 4.48 (dt, J = 5.4, 1.7 Hz, 2H), 3.97 (dd, J = 4.7, 3.2 Hz, 2H), 2.73 (ddd, J = 13.8, 9.8, 5.7 Hz, 3H), 2.62 (ddd, J = 13.9, 9.7, 6.4 Hz, 3H), 2.02 (s, 9H), 1.90–1.56 (m, 12H). 13C NMR (151 MHz, CDCl3) δ 157.1, 140.7, 137.2, 134.2, 129.3, 128.6, 127.9, 127.5, 114.9, 114.5, 70.8, 70.1, 68.7, 42.3, 39.4, 31.2. [α]20D = 16.8 (c = 1.0, DCM). IR (ATR): [cm−1] = 3286, 3090, 3066, 3035, 3015, 2928, 2906, 2852, 1646, 1611, 1582, 1511, 1422, 1402, 1382, 1349, 1312, 1296, 1278, 1240, 1173, 1134, 1110, 1069, 1038, 1026, 1014, 1001, 991, 975, 918, 903, 863, 837, 825, 808, 781, 734, 696. HRMS (ESI) m/z: [M + Na+] calcd. for C20H24NaO3: 335.1620; found: 335.1618.
3.2.6. ((((3S,5R)-7-(4-(Benzyloxy)phenyl)hept-1-ene-3,5-diyl)bis(oxy))bis(methylene))dibenzene (17)
To a solution of (3S,5R)-7-(4-(benzyloxy)phenyl)hept-1-ene-3,5-diol 16 (34.9 mg, 109 µmol, 1.0 equiv.) in 1.0 mL of dry DMF was added tetra-n-butylammonium iodide (8.40 mg, 22.9 µmol, 0.20 equiv.) and NaH (13.1 mg, 60% in mineral oil, 327 µmol, 3.0 equiv.). The reaction mixture was stirred for 30 min at 0 °C. Then, benzyl bromide (39.0 µL, 56.2 mg, 328 µmol, 3.0 equiv.) was added to the mixture and stirred for 5 h at room temperature under argon. After reaction completion, the crude mixture was directly purified by column chromatography (EA/CyHex = 1:9 → 2:8). A further separation was carried out using preparative HPLC (MeCN:H2O = 9:1) to give product 17 (27.7 mg, yield 52%) as a colorless solid. TLC: Rf = 0.54 (EA/CyHex = 2:8) [KMnO4]. 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.1 Hz, 2H), 7.40 (t, J = 7.3 Hz, 2H), 7.37–7.26 (m, 11H), 7.08 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 5.79 (ddd, J = 17.5, 10.3, 7.5 Hz, 1H), 5.31–5.19 (m, 2H), 5.06 (s, 2H), 4.58 (d, J = 11.6 Hz, 1H), 4.50 (d, J = 11.4 Hz, 1H), 4.35 (d, J = 11.4 Hz, 1H), 4.24 (d, J = 11.6 Hz, 1H), 4.01 (td, J = 8.0, 4.5 Hz, 1H), 3.79–3.68 (m, 1H), 2.64 (t, J = 8.0 Hz, 2H), 1.94–1.73 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 157.0, 139.1, 138.9, 138.7, 134.8, 129.3, 128.6, 128.3, 127.9 (d, J = 1.7 Hz), 127.9, 127.5 (d, J = 4.1 Hz), 116.7, 114.8, 75.0, 71.2, 70.3, 70.1, 41.2, 36.3, 30.4. IR (ATR): [cm−1] = 3087, 3063, 3030, 3007, 2860, 1610, 1583, 1509, 1496, 1453, 1420, 1380, 1355, 1310, 1298, 1237, 1175, 1089, 1067, 1026, 993, 924, 860, 820, 732, 694, 644, 613, 560, 532, 514, 458. [α]20D = 16.6 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C34H36NaO3: 515.2564; found: 515.2567.
3.2.7. 4,4′-((3S,5R,E)-3,5-Bis(benzyloxy)hept-1-ene-1,7-diyl)bis((benzyloxy)benzene) (19)
In a 4 mL vial was added palladium (II) acetate (1.23 mg, 5.48 µmol, 10 mol%), DPPE (4.37 mg, 11.0 µmol, 20 mol%), potassium carbonate (15.1 mg, 110 µmol, 2.0 equiv.), and 100 µL of dry DMF. A solution of alkene 17 (27.0 mg, 54.8 mmol, 1.0 equiv.) and 1-benzyloxy-4-iodobenzene 18 (25.5 mg, 82.2 µmol, 1.5 equiv.) in 700 µL of dry DMF was added dropwise. The reaction mixture was stirred for 18 h at 90 °C under argon. The solution was then cooled to room temperature and purified directly by column chromatography (EA:CyHex = 1:9). A small amount of the solid was dissolved in acetonitrile and separated via preparative HPLC (MeCN:H2O = 9:1). It was again purified by silica gel column chromatography (EA/CyHex = 1:9) to give 19 in the form of a slightly brownish solid (20.3 mg, yield 55%). TLC: Rf = 0.49 (EA/CyHex = 2:8) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.46–7.27 (m, 20H), 7.04 (dd, J = 15.7, 8.7 Hz, 4H), 6.94 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 6.48 (d, J = 15.9 Hz, 1H), 5.99 (dd, J = 15.9, 8.0 Hz, 1H), 5.12 (s, 1H), 5.08 (s, 1H), 5.03 (s, 2H), 4.59 (d, J = 11.7 Hz, 1H), 4.49 (d, J = 11.4 Hz, 1H), 4.35 (d, J = 11.3 Hz, 1H), 4.27 (d, J = 11.7 Hz, 1H), 4.12 (td, J = 8.5, 4.1 Hz, 1H), 3.81–3.67 (m, 1H), 2.63 (t, J = 8.0 Hz, 2H), 1.87 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 158.7, 157.1, 139.0, 138.9, 137.4, 137.1, 134.9, 131.7, 129.5, 128.7 (d, J = 6.5 Hz), 128.5 (d, J = 2.6 Hz), 128.1 (d, J = 6.3 Hz), 128.0, 127.8, 127.7–127.5 (m), 115.2, 114.9, 75.1, 71.3, 70.3, 70.2 (d, J = 2.0 Hz), 41.6, 36.5, 30.5. IR (ATR): [cm−1] = 3088, 3063, 3031, 2924, 2858, 1729, 1606, 1583, 1509, 1454, 1381, 1297, 1240, 1174, 1070, 1026, 969, 913, 860, 819, 735, 696, 613, 512, 458. [α]20D = −20.3 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C47H46NaO4: 697.3281; found: 697.3288.
3.2.8. Methyl 2-(4-Methoxyphenyl)acetate (21)
To a solution of 4-methoxyphenylacetic acid
20 (6.00 g, 36.0 mmol, 1.0 equiv.) in MeOH (112 mL) was added conc. H
2SO
4 (2.32 mL, 43.3 mmol, 1.2 equiv.). The reaction mixture was heated under reflux (65 °C) for 3 h. The resulting mixture was cooled to room temperature, and the solvent was removed under vacuum. The crude residue was redissolved in EA, dried with sodium sulfate, filtered, concentrated, and purified by silica gel column chromatography (EA/CyHex = 1:1) to give the methyl ester (6.10 g, yield 94%) as a colorless oil. TLC: R
f = 0.82 (EA/CyHex = 1:1) [KMnO
4].
1H NMR (600 MHz, CDCl
3) δ 7.20 (d,
J = 8.6 Hz, 2H), 6.86 (d,
J = 8.6 Hz, 2H), 3.79 (s, 3H), 3.69 (s, 3H), 3.57 (s, 2H).
13C NMR (151 MHz, CDCl
3) δ 172.4, 158.9, 130.4, 126.2, 114.1, 55.4, 52.1, 40.4. IR (ATR):
[cm
−1] = 2952, 1732, 1612, 1511, 1299, 1242, 1151, 1012, 788. HRMS (ESI)
m/
z: [M + Na
+] calcd. for C
10H
12NaO
3: 203.0678; found: 203.0679. All the characterization data are consistent with the literature [
32].
3.2.9. 2-(4-Methoxyphenyl)acetaldehyde (11)
To a solution of methyl 2-(4-methoxyphenyl)acetate
21 (5.00 g, 27.8 mmol, 1.0 equiv.) in anhydrous toluene (70 mL) at −78 °C was added dropwise diisobutylaluminium hydride (1.2 M solution in toluene, 25.4 mL, 30.5 mmol, 1.1 equiv.). The reaction was stirred for 1 h. The reaction was quenched with MeOH (15 mL) and then poured into DCM, then washed sequentially with 1 M HCl (25 mL) and brine (25 mL). The organic phase was concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (DE/PE = 1:10) to give product
11 (3.70 g, yield 89%) as a colorless oil. TLC: R
f = 0.81 (DE/PE = 1:10) [KMnO
4].
1H NMR (400 MHz, CDCl
3) δ 9.72 (t,
J = 2.4 Hz, 1H), 7.13 (d,
J = 8.8 Hz, 2H), 6.91 (d,
J = 8.7 Hz, 2H), 3.81 (s, 3H), 3.63 (d,
J = 2.4 Hz, 2H).
13C NMR (101 MHz, CDCl
3) δ 200.1, 159.4, 131.1, 124.2, 114.9, 55.7, 50.2. IR (ATR):
[cm
−1] = 1721, 1611, 1511, 1245, 1177, 1032, 906, 825, 726, 648, 524. HRMS (ESI)
m/
z: [M + Na
+] calcd. for C
9H
10NaO
2: 173.0681; found: 173.0568. All the characterization data are consistent with the literature [
35].
3.2.10. 5-Hydroxy-1-(4-methoxyphenyl)hept-6-en-3-one (22)
To a solution of diisopropylamine (1.70 mL, 12.3 mmol, 2.1 equiv.) in dry THF (58 mL) was added n-butyllithium (2.5 M in hexane, 6.40 mL, 12.3 mmol, 2.1 equiv.) at −78 °C. After the cooling was removed for 15 min, (S)-building block 7 (2.00 g, 5.84 mmol, 1.0 equiv.) dissolved in dry THF (19 mL) was added slowly at −78 °C. The solution was allowed to stir for 1 h at this temperature. Following the addition of 2-(4-methoxyphenyl)acetaldehyde 11 (2.63 g, 17.5 mmol, 3.0 equiv.) dissolved in dry THF (9 mL), the reaction was allowed to warm to rt within 1 h. After the addition of potassium tert-butoxide (690 mg, 5.80 mmol, 1.0 equiv.), the reaction mixture was stirred for an additional hour at rt. The reaction was quenched with a saturated aqueous ammonium chloride solution (30 mL), and the aqueous layer was extracted with DCM (3 × 50 mL). The combined organic layers were washed with hydrochloric acid (2 M, 2 × 50 mL), and the aqueous layer was again extracted with DCM (3 × 50 mL). The combined organic layers were washed with brine (100 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography (EA/PE = 1:4) to give the product 22 (1.16 g, yield 85%) as a light-yellow oil. TLC: Rf = 0.26 (EA/PE = 1:4) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.08 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 5.83 (ddd, J = 16.6, 10.6, 5.6 Hz, 1H), 5.27 (d, J = 17.1 Hz, 1H), 5.14–5.08 (m, 1H), 4.61–4.52 (m, 1H), 3.77 (s, 3H), 2.88 (s, 1H), 2.84 (t, J = 7.6 Hz, 2H), 2.73 (t, J = 7.6 Hz, 2H), 2.60 (d, J = 6.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 210.2, 158.1, 139.2, 132.8, 129.3, 115.0, 114.0, 68.7, 55.3, 49.1, 45.5, 28.7. IR (ATR): [cm−1] = 2932, 1707, 1611, 1583, 1510, 1463, 1441, 1404, 1365, 1299, 1242, 1177, 1108, 1084, 1032, 976, 927, 823. [α]20D = +11.3 (c = 1.21, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C14H18NaO3: 257.1148; found: 257.1148.
3.2.11. (3S,5R)-5-Hydroxy-7-(4-methoxyphenyl)hept-1-en-3-yl acetate (23a)
To a solution of 5-hydroxy-1-(4-methoxyphenyl)hept-6-en-3-one 23a (412 mg, 1.40 mmol, 1.0 equiv.) in dry THF (3 mL) was added acetaldehyde (347 µL, 6.15 mmol, 3.5 equiv.). At −50 °C, SmI2 (0.1 M in THF, 13.2 mL, 1.32 mmol, 0.80 equiv.) was added slowly under the exclusion of light. The reaction mixture was allowed to stir at −20 °C for 18 h. After the addition of a saturated aqueous sodium bicarbonate solution (15 mL), the layers were separated, and the aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by column chromatography (EA/CyHex = 1:4) to give the product 23a (426 mg, yield 87%) as a colorless oil. TLC: Rf = 0.165 (EA/CyHex = 1:4) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.11 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 5.84 (ddd, J = 16.9, 10.5, 6.0 Hz, 1H), 5.52 (ddt, J = 9.5, 5.6, 3.6 Hz, 1H), 5.27 (dt, J = 17.3, 1.3 Hz, 1H), 5.16 (dt, J = 10.5, 1.3 Hz, 1H), 3.78 (s, 3H), 3.56 (tt, J = 7.8, 3.5 Hz, 1H), 2.75 (ddd, J = 14.8, 9.8, 5.5 Hz, 1H), 2.62 (ddd, J = 13.8, 9.7, 6.6 Hz, 2H), 2.08 (s, 3H), 1.84–1.63 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 171.7, 158.1, 136.8, 129.6, 116.6, 72.4, 67.0, 55.6, 43.0, 39.4, 31.5, 21.4. IR (ATR): [cm−1] = 2928, 1608, 1510, 1463, 1247, 1174, 1037, 834, 774. [α]20D = −3.1 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C16H22NaO4: 301.1410; found: 301.1410.
3.2.12. (3S,5R)-7-(4-Methoxyphenyl)hept-1-ene-3,5-diol (23)
To a solution of (3S,5R)-5-hydroxy-7-(4-methoxyphenyl)hept-1-en-3-yl acetate 23a (300 mg, 1.08 mmol, 1.0 equiv.) in MeOH/H2O (4 mL, 3:1) was added potassium carbonate (297 mg, 2.15 mmol, 2.0 equiv.) at 0 °C. The reaction mixture was stirred for 4 h at 0 °C and then allowed to warm up to room temperature. Upon completion of the reaction indicated by TLC analysis, H2O (10 mL) and ethyl acetate (10 mL) were added, the layers were separated, and the aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (15 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and the crude residue was purified by column chromatography (EA/CyHex = 3:7) to give the product 23 (234 mg, yield 92%) as a colorless oil. TLC: Rf = 0.195 (EA/CyHex = 3:7) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.11 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 5.92 (ddd, J = 17.1, 10.5, 5.5 Hz, 1H), 5.28 (d, J = 17.3 Hz, 1H), 5.14 (d, J = 10.5 Hz, 1H), 4.51–4.44 (m, 1H), 4.00–3.93 (m, 1H), 3.78 (s, 3H), 2.73 (ddd, J = 13.8, 9.9, 5.7 Hz, 1H), 2.62 (ddd, J = 13.9, 9.7, 6.4 Hz, 1H), 2.25 (s, 2H), 1.87–1.61 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 158.0, 140.8, 134.1, 129.4, 114.6, 114.0, 70.9, 68.9, 55.4, 42.5, 39.5, 31.3. IR (ATR): [cm−1] = 3354, 2935, 1611, 1510, 1242, 1177, 1034, 922, 822, 526. [α]20D = +15.4 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C14H20NaO3: 259.1304; found: 259.1305.
3.2.13. (5R,7S)-5-(4-Methoxyphenethyl)-2,2,3,3,9,9,10,10-octamethyl-7-vinyl-4,8-dioxa-3,9-disilaundecane (24)
To a dry round-bottom flask was added diol 23 (200 mg, 846 µmol, 1.0 equiv.) in DMF (5 mL), and the solution was cooled to 0 °C for 20 min. Imidazole (460 mg, 6.77 mmol, 8.0 equiv.) and tert-butyldimethylsilyl chloride (511 mg, 3.39 mmol, 4.0 equiv.) were added, and the solution was allowed to stir at rt for 18 h. Following the addition of distilled water (15 mL) and diethyl ether (15 mL), the layers were separated, and the aqueous layer was extracted with diethyl ether (3 × 15 mL). The combined organic layers were washed with brine (15 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and flash chromatography (EA/CyHex = 1:99) on silica gel afforded product 24 in 95% yield (374 mg, dr > 99:1) as a colorless oil. TLC: Rf = 0.195 (EA/CyHex = 3:7) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.10 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.4 Hz, 2H), 5.81 (ddd, J = 17.2, 10.3, 7.0 Hz, 1H), 5.12 (d, J = 17.2 Hz, 1H), 5.03 (d, J = 10.3 Hz, 1H), 4.19 (d, J = 6.7 Hz, 1H), 3.87–3.80 (m, 1H), 3.79 (s, 3H), 2.60 (dddd, J = 38.9, 13.7, 10.6, 5.7 Hz, 1H), 1.82–1.69 (m, 3H), 1.65 (dt, J = 13.5, 6.0 Hz, 1H), 0.92 (s, 9H), 0.89 (s, 9H), 0.19–−0.17 (m, 12H). 13C NMR (151 MHz, CDCl3) δ 157.9, 142.2, 134.9, 129.4, 114.2, 72.1, 69.4, 55.4, 46.4, 40.0, 30.6, 26.1 (d, J = 5.0 Hz), 18.3, −3.7, −3.8, −4.0, −4.4. IR (ATR): [cm−1] = 2928, 1511, 1463, 1246, 1073, 833, 773. [α]20D = +1.0 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C26H48O3NaSi2: 487.3034; found: 487.3034.
3.2.14. (5R,7S)-5-(4-Methoxyphenethyl)-7-((E)-4-methoxystyryl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecane (26)
An oven-dried round-bottom flask was charged with (3S,5R)-7-(4-methoxyphenyl)hept-1-ene-3,5-diol 24 (320 mg, 688 µmol, 1.0 equiv.), palladium (II) acetate (46.4 mg, 210 µmol, 30 mol%), potassium carbonate (951 mg, 6.88 mmol, 10 equiv.), and tetra-n-butylammonium chloride (957 mg, 3.44 mmol, 5.0 equiv.). The flask was evacuated and backfilled with argon three times. 4-iodoanisole 25 (242 mg, 1.03 mmol, 1.5 equiv.) dissolved in DMF (5 mL; degassed by freeze–pump–thaw method) was added to the mixture. The mixture was allowed to stir at 90 °C for 19 h. After this time, the reaction mixture was allowed to cool down to room temperature and diluted with ethyl acetate (10 mL) and water (10 mL). The combined aqueous layers were extracted with ethyl acetate (3 × 10 mL), and the combined organic layers were washed with brine (8 mL), dried with sodium sulfate, and filtered. The filtrate was concentrated in vacuo, and flash chromatography (DE/PE = 1:50) on silica gel afforded the product 26 (300 mg, yield 84%, dr > 20:1) as a colorless oil. TLC: Rf = 0.195 (DE/PE = 1:50) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.29 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 8.5 Hz, 2H), 6.37 (d, J = 15.9 Hz, 1H), 6.00 (dd, J = 15.8, 7.5 Hz, 1H), 4.34 (q, J = 6.9 Hz, 1H), 3.87 (p, J = 5.8 Hz, 1H), 3.82 (s, 3H), 3.78 (s, 3H), 2.60 (tq, J = 19.6, 6.8 Hz, 2H), 1.89–1.68 (m, 4H), 0.92 (s, 9H), 0.90 (s, 9H), 0.09–0.02 (m, 12H). 13C NMR (151 MHz, CDCl3) δ 159.3, 157.8, 134.8, 131.5, 130.0, 129.4, 129.1, 127.7, 114.2, 113.9, 71.8, 69.3, 55.4 (d, J = 7.6 Hz), 46.7, 40.1, 30.6, 18.3, −3.5, −3.9, −4.0, −4.3.z IR (ATR): [cm−1] = 2929, 1607, 1502, 1275, 1250, 1182, 1040, 906, 823, 809, 731. [α]20D −23.0 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C33H54O4NaSi2: 593.3561; found: 593.3452.
3.2.15. (5R,7R)-5,7-bis(4-Methoxyphenethyl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecane (27)
In an oven-dried round-bottom flask, (5R,7S)-5-(4-methoxyphenethyl)-7-((E)-4-methoxystyryl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecane 26 (200 mg, 350 µmol, 1.0 equiv.) was dissolved in methanol (8 mL), and Pd/C (10% on activated charcoal, 112 mg, 105 µmol, 30 mol%) was added. The reaction mixture was stirred for 8 h at room temperature under a hydrogen atmosphere (1 atm). The suspension was then filtered through a pad of celite and washed (3 × 10 mL) with ethyl acetate. The filtrate was concentrated in vacuo, and flash chromatography (EA/CyHex = 1:9) on silica gel afforded product 27 (193 mg, yield 93%) as a colorless oil. TLC: Rf = 0.21 (EA/CyHex = 1:17) [KMnO4]. 1H NMR (600 MHz, CDCl3) δ 7.09 (d, J = 8.1 Hz, 4), 6.83 (d, J = 8.6 Hz, 4H), 3.80 (d, J = 6.2 Hz, 8H), 2.67–2.53 (m, 4H), 1.72 (dt, J = 12.1, 5.8 Hz, 6H), 0.91 (s, 18H), 0.07 (d, J = 6.8 Hz, 12H). 13C NMR (151 MHz, CDCl3) δ 157.9, 134.8, 129.4, 114.0, 69.9, 55.4, 45.4, 40.1, 30.6, 26.1, 18.3, −3.9. IR (ATR): [cm−1] = 2928, 1511, 1462, 1244, 1176, 1037, 832, 771. [α]20D = +2.1 (c = 1.0, DCM). HRMS (ESI) m/z: [M + Na+] calcd. for C33H56O4NaSi2: 595.3717; found: 595.3608.
3.2.16. (3R,5R)-1,7-bis(4-Methoxyphenyl)heptane-3,5-diol (4)
First, HF-pyridine (70% HF, 250 µL, 1.75 mmol, 20 equiv.) was dissolved in a pyridine/MeOH mixture (890 µL, 6:1) at 0 °C in a Teflon vial. In another Teflon vial, (5
R,7
R)-5,7-bis(4-methoxyphenethyl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecane
27 (50.0 mg, 871 μmol, 1.0 equiv.) was dissolved in THF (0.8 mL), and the solution was cooled to 0 °C. The prepared HF-pyridine solution was then transferred to the THF solution, and the reaction mixture was stirred at rt for 18 h. Following the addition of an excess of methoxytrimethylsilane (7 mL) at 0 °C and dilution with toluene (10 mL), the volatile compounds were removed in vacuo. The residue was diluted with toluene (5 mL) again, and the procedure was repeated three times to remove all of the pyridine. The crude residue was purified by silica gel column chromatography (EA/CyHex = 2:5) to give 1,3 diol
4 (26.2 mg, yield 87%) as a white solid.
1H NMR (600 MHz, CDCl
3) δ 7.11 (d,
J = 8.4 Hz, 4H), 6.83 (d,
J = 8.6 Hz, 4H), 3.97 (dd,
J = 8.4, 5.1 Hz, 2H), 3.78 (s, 6H), 2.71 (ddd,
J = 15.0, 9.7, 5.7 Hz, 2H), 2.61 (ddd,
J = 13.8, 9.5, 6.5 Hz, 2H), 2.12 (s, 2H), 1.83 (ddd,
J = 14.3, 8.7, 5.6 Hz, 2H), 1.80 (d,
J = 5.7 Hz, 2H), 1.73 (dddd,
J = 13.9, 10.4, 6.5, 4.6 Hz, 2H), 1.66 (t,
J = 5.6 Hz, 2H).
13C NMR (151 MHz, CDCl
3) δ 158.0, 134.0, 129.4, 114.1, 69.1, 55.4, 42.7, 39.4, 31.4. IR (ATR):
[cm
−1] = 3374, 2939, 1612, 1511, 1300, 1244, 1176, 1033, 814, 514. [α]
20D = +6.5 (c = 1.0, DCM). HRMS (ESI)
m/
z: [M + Na
+] calcd. for C
21H
28NaO
4: 367.1988; found: 367.1879. All the characterization data are consistent with the literature [
11].


 ,
, {kind=link}
{kind=link}
{kind=link}