
A Series of Novel 1-H-isoindole-1,3(2H)-dione Derivatives as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: In Silico, Synthesis and In Vitro Studies

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
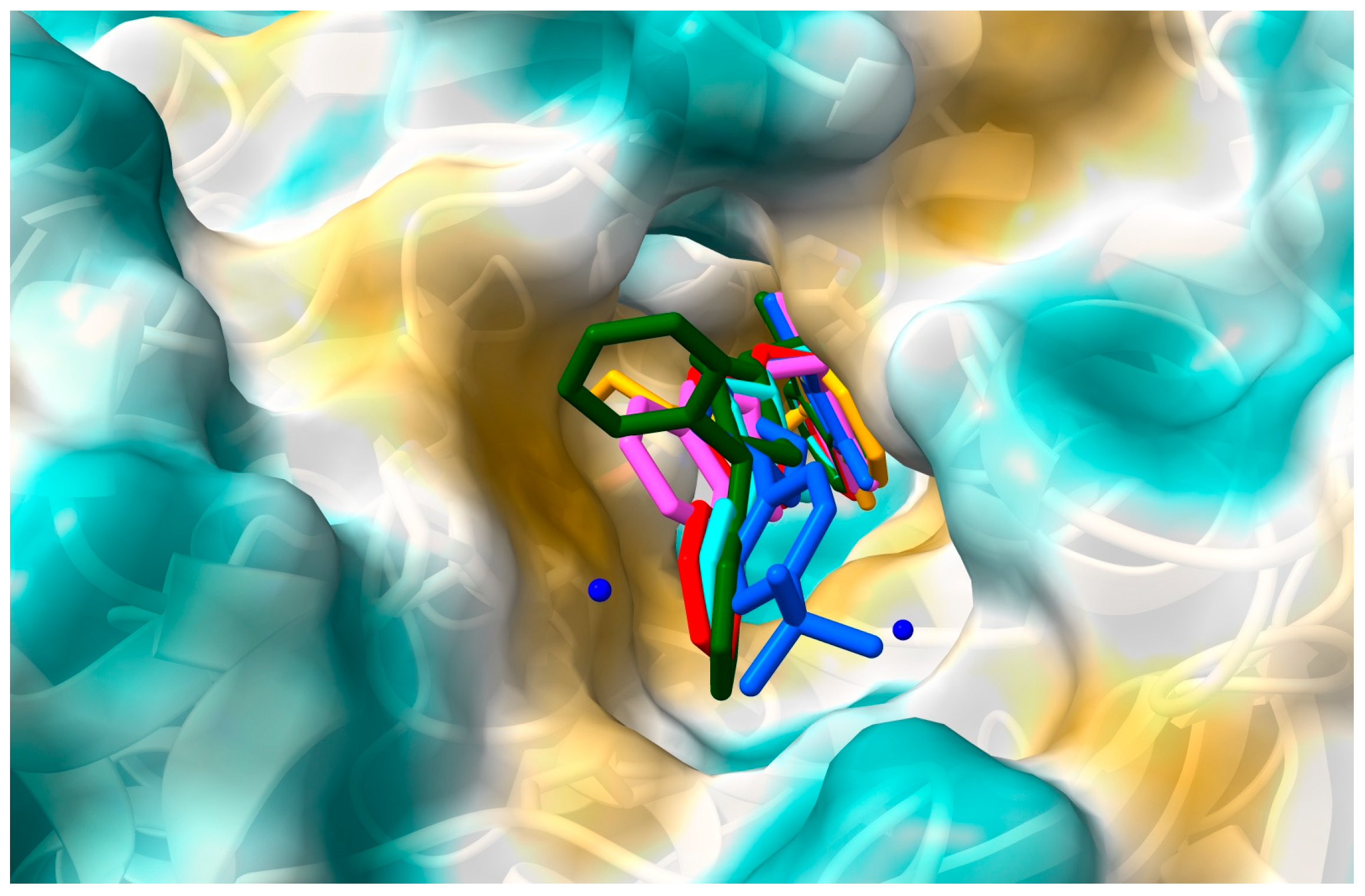
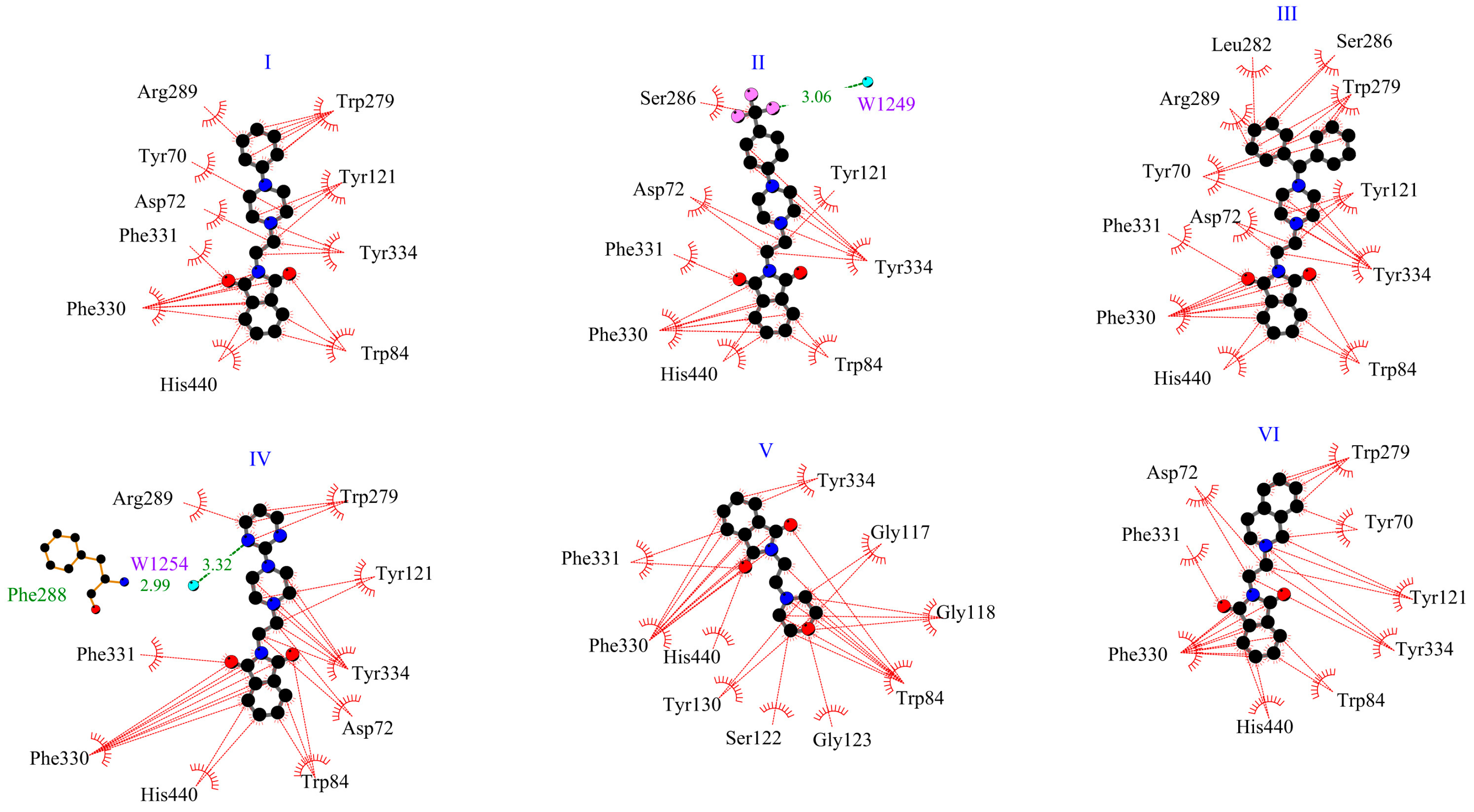
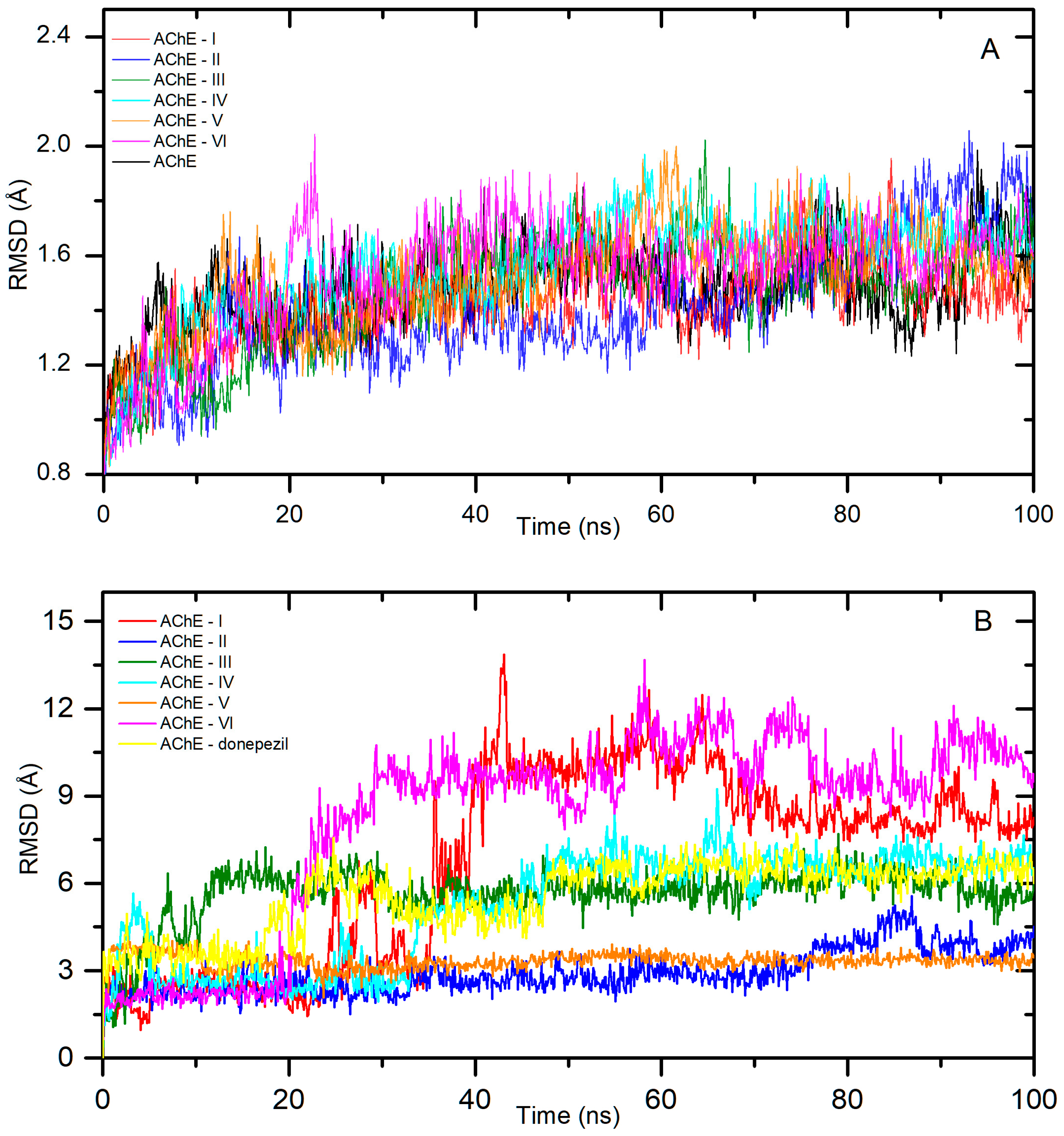
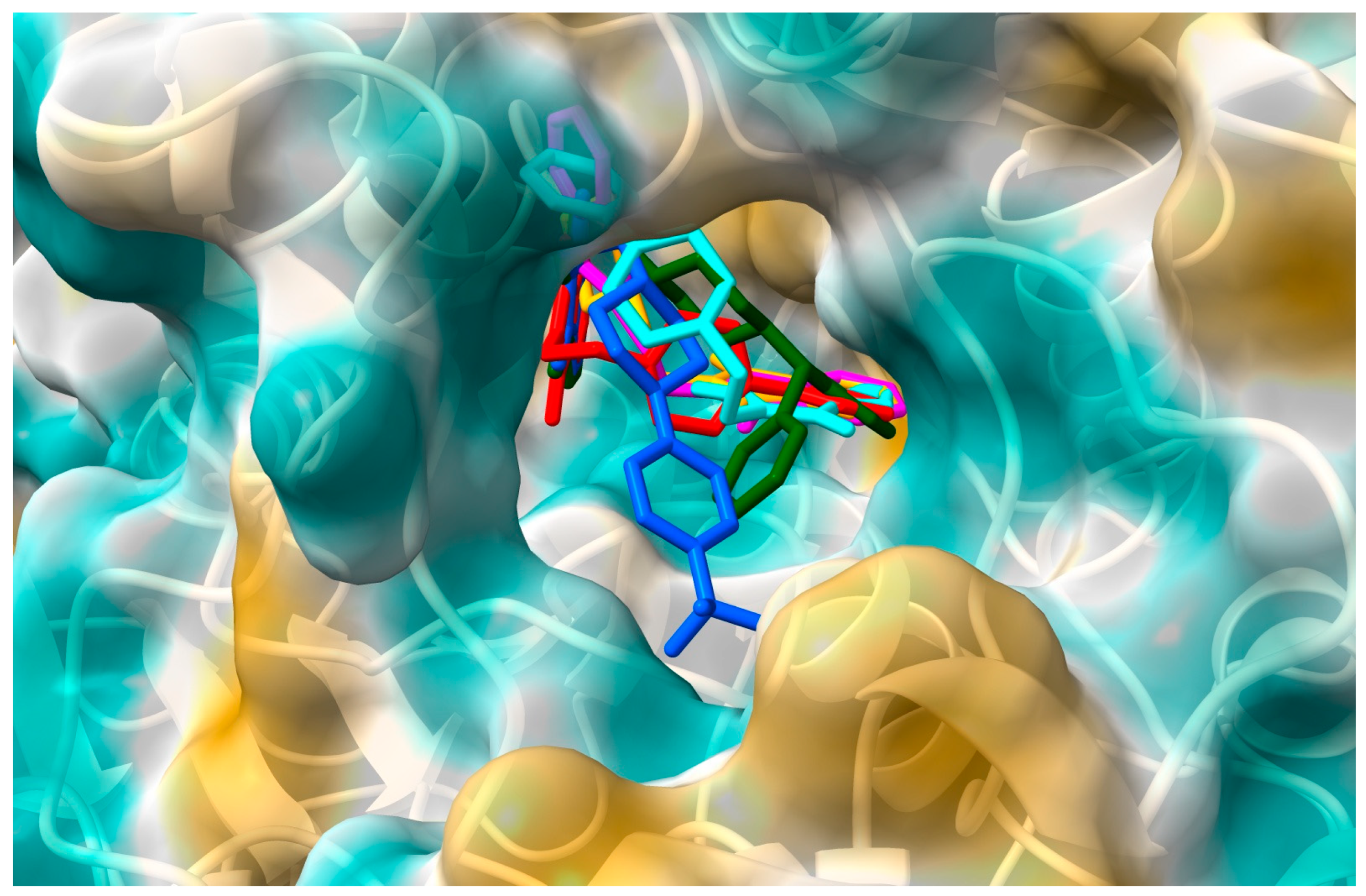
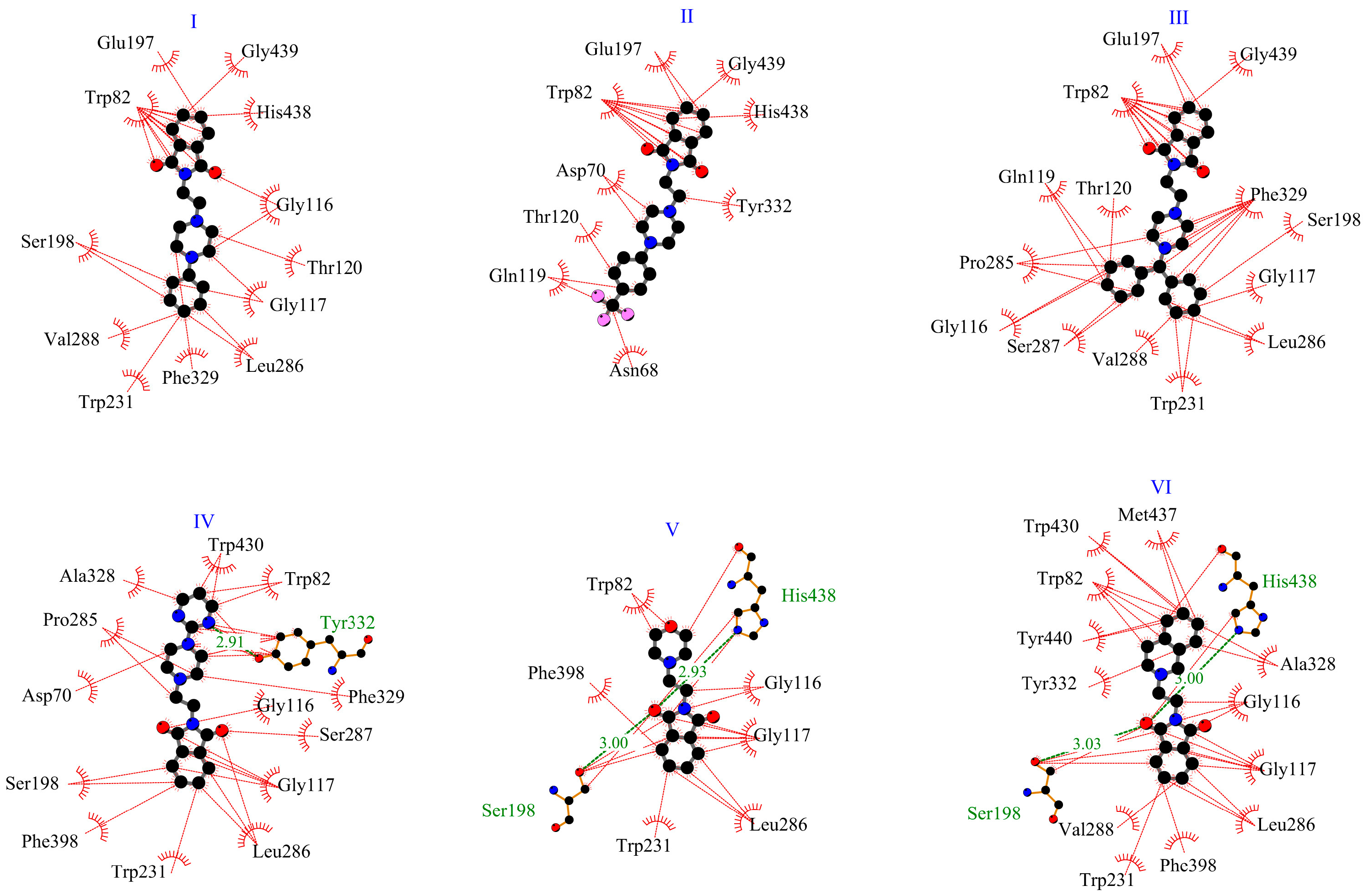
2.1. In Silico Interactions Studies
2.1.1. Interactions with Acetylcholinesterase Enzyme (AChE)
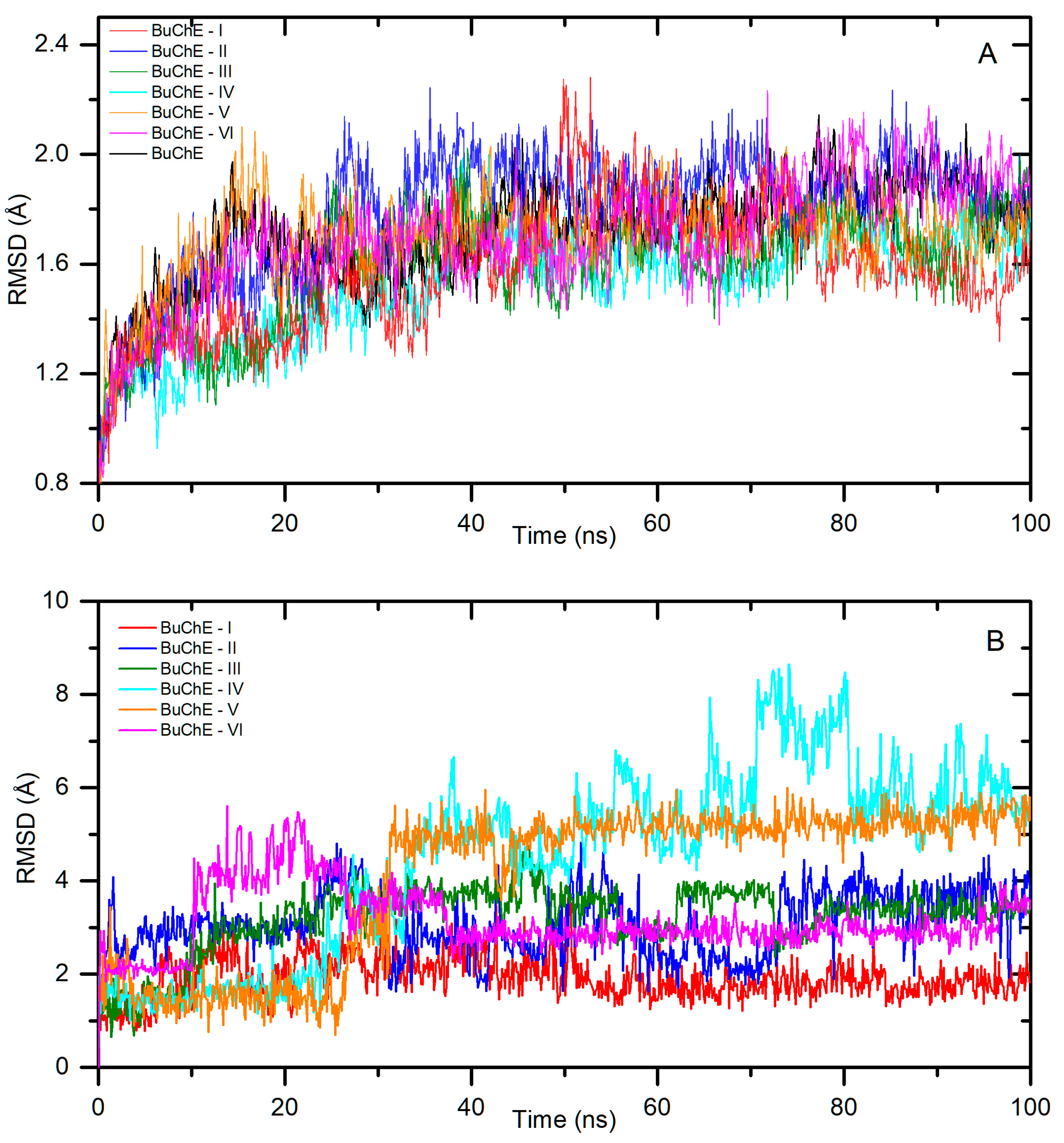
2.1.2. Interactions with Butyrylcholinesterase Enzyme (BuChE)
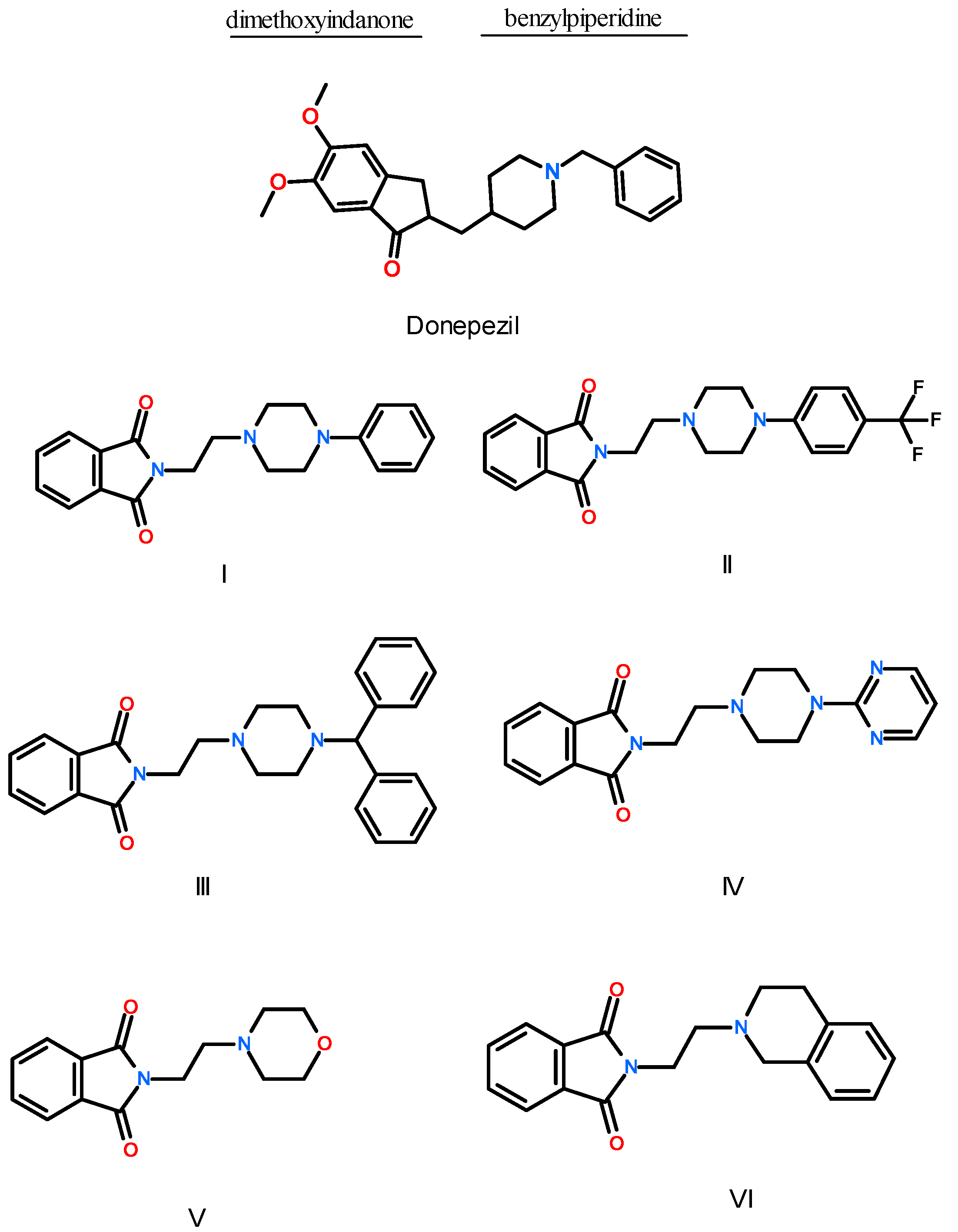
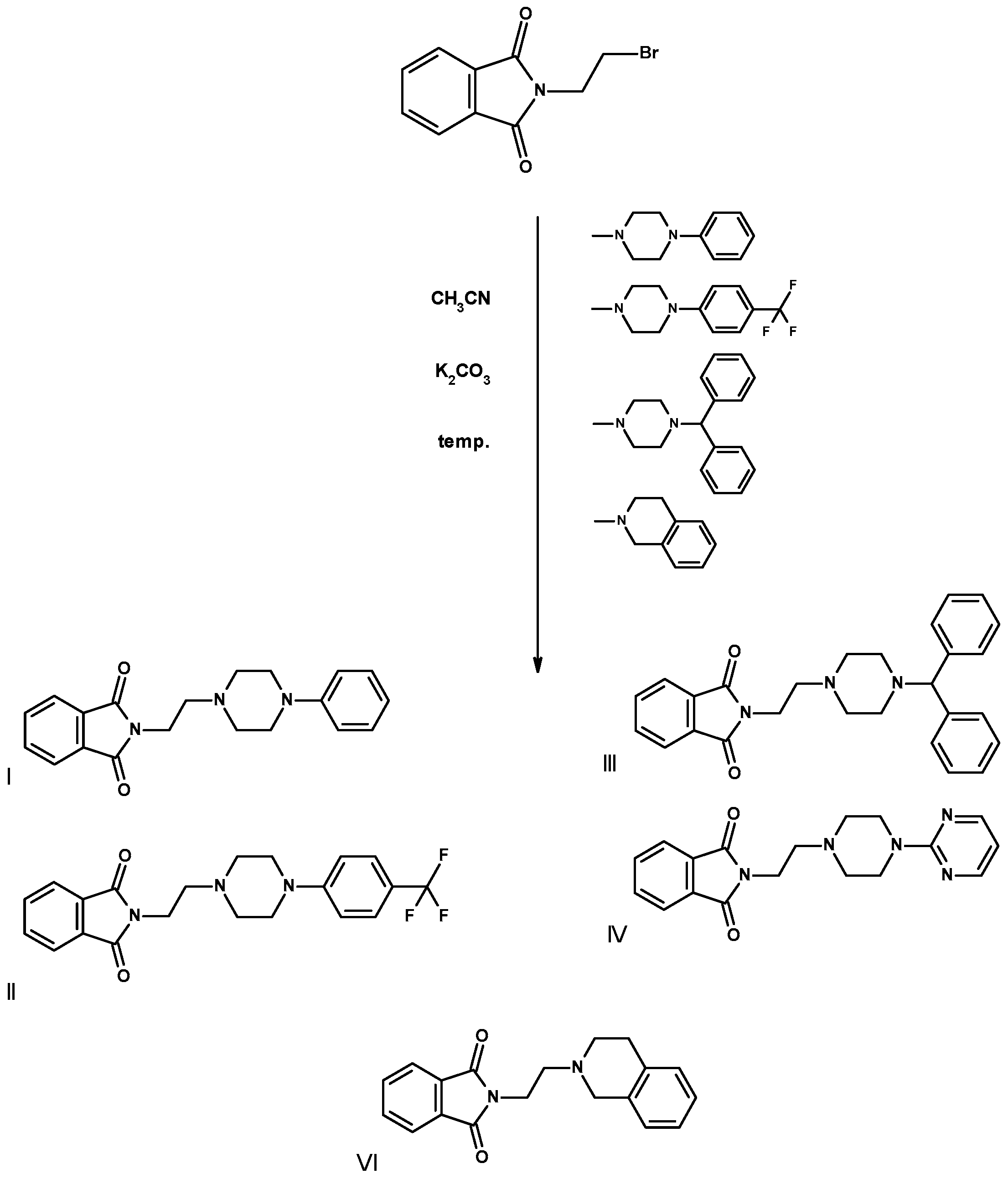
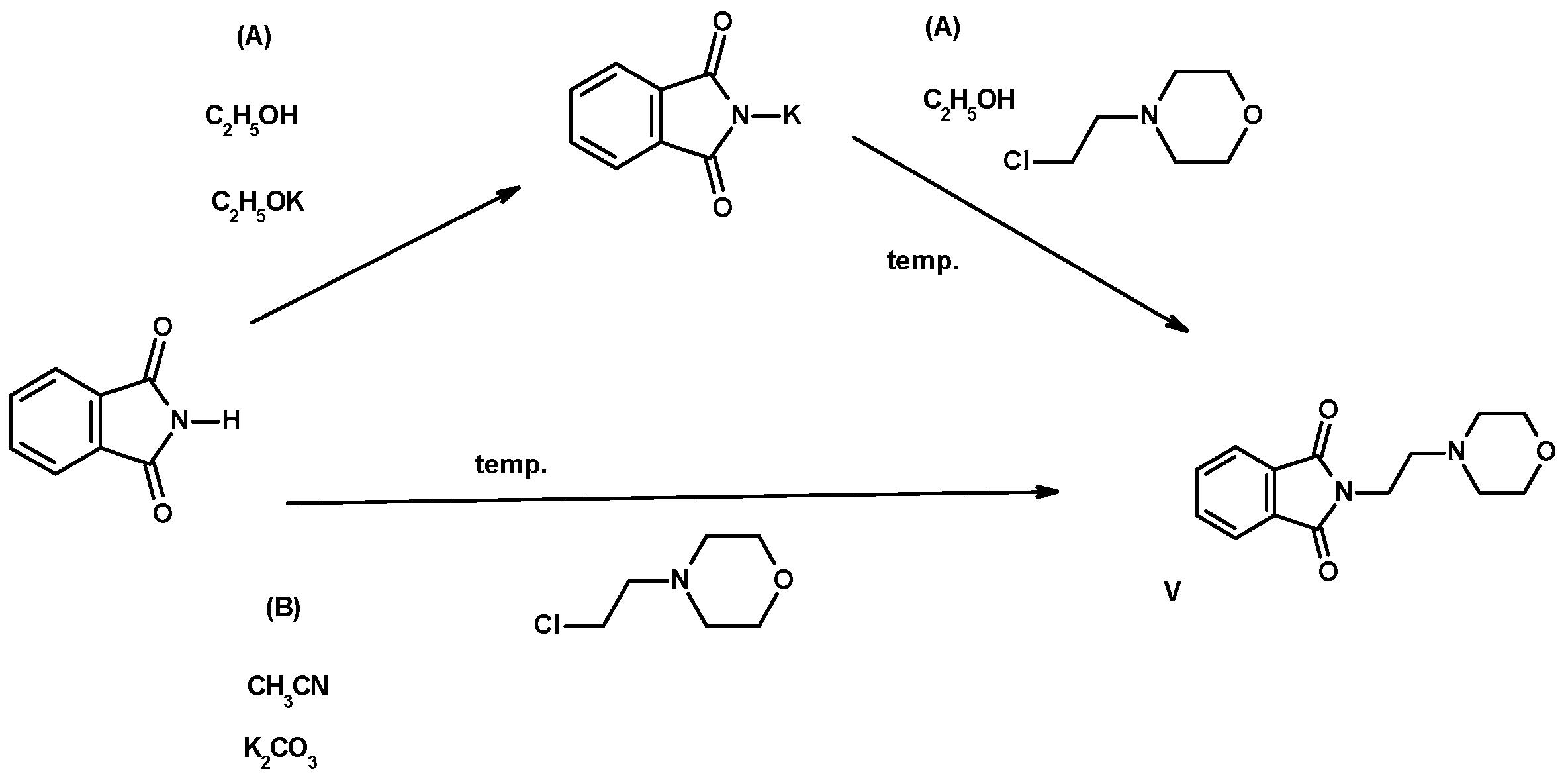
2.2. Synthesis of Compounds I–VI
2.3. In Vitro Studies
2.4. Blood–Brain Barrier and Toxicity
3. Materials and Methods
3.1. Chemistry
3.1.1. Instrumentation and Chemicals
3.1.2. Synthesis of Compounds I–VI
3.2. In Silico Studies
3.3. In Vitro Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anand, R.; Kaushal, A.; Wani, W.Y.; Gill, K.D. Road to Alzheimer’s disease: The Pathomechanism underlying. Pathobiology 2012, 79, 55–71. [Google Scholar] [CrossRef]
- Peng, Y.; Jin, H.; Xue, Y.H.; Chen, Q.; Yao, S.Y.; Du, M.Q.; Liu, S. Current and future therapeutic strategies for Alzheimer’s disease: An overview of drug development bottlenecks. Front. Aging Neurosci. 2023, 15, 1206572. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and β-amyloid protein in Alzheimer’s disease. NeuroMol. Med. 2011, 13, 223–250. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J.F. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef] [PubMed]
- Leuzinger, W. Structure and Function of Acetylcholinesterase. Prog. Brain Res. 1969, 31, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Yu, Q.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Bhardwaj, B.; Waiker, D.K.; Tripathi, A.; Shrivastava, S.K. Discovery of novel dual acetylcholinesterase and butyrylcholinesterase inhibitors using machine learning and structure-based drug design. J. Mol. Struct. 2023, 1286, 135517. [Google Scholar] [CrossRef]
- Alzahrani, A.Y.A.; Ullah, H.; Rahim, F.; Khan, F.; Wadood, A.; Taha, M.; Al-Bagawi, A.; Fareid, M.; Othman, M.S. Synthesis, in vitro biological evaluation and in silico molecular docking study of hydroxy-quinoline based sulfonohydrazide derivatives as potential acetylcholinesterase and butyrylcholinesterase inhibitors. J. Mol. Struct. 2024, 1306, 137884. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Dong, J.; Tang, L. Alzheimer’s disease: Updated multi-targets therapeutics are in clinical and in progress. Eur. J. Med. Chem. 2022, 238, 114464. [Google Scholar] [CrossRef]
- Gok, M.; Cicek, C.; Bodur, E. Butyrylcholinesterase in lipid metabolism: A new outlook. J. Neurochem. 2024, 168, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Kareem, R.T.; Abedinifar, F.; Mahmood, E.A.; Ebadi, A.G.; Rajabi, F.; Vessally, E. The recent development of donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s agents: Highlights from 2010 to 2020. RSC Adv. 2021, 11, 30781–30797. [Google Scholar] [CrossRef]
- Mohsin, N.U.A.; Ahmad, M. Donepezil: A review of the recent structural modifications and their impact on anti-alzheimer activity. Brazilian J. Pharm. Sci. 2020, 56, e18325. [Google Scholar] [CrossRef]
- Mohammadi-Farani, A.; Ahmadi, A.; Nadri, H.; Aliabadi, A. Synthesis, docking and acetylcholinesterase inhibitory assessment of 2-(2-(4-Benzylpiperazin-1-yl)ethyl)isoindoline-1,3-dione derivatives with potential anti-Alzheimer effects. DARU J. Pharm. Sci. 2013, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Farani, A.; Abdi, N.; Moradi, A.; Aliabadi, A. 2-(2-(4-Benzoylpiperazin-1-yl)ethyl)isoindoline-1,3-dione derivatives: Synthesis, docking and acetylcholinesterase inhibitory evaluation as anti-alzheimer agents. Iran. J. Basic Med. Sci. 2017, 20, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Farani, A.; Darbandi, S.S.; Aliabadi, A. Synthesis and acetylcholinesterase inhibitory evaluation of 4-(1,3-dioxoisoindolin-2-yl)-N-phenyl benzamide derivatives as potential anti-alzheimer agents. Iran. J. Pharm. Res. 2016, 15, 313–320. [Google Scholar] [PubMed]
- Ignasik, M.; Bajda, M.; Guzior, N.; Prinz, M.; Holzgrabe, U.; Malawska, B. Design, synthesis and evaluation of novel 2-(Aminoalkyl)-isoindoline-1,3- dione derivatives as dual-binding site acetylcholinesterase inhibitors. Arch. Pharm. 2012, 345, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Wiȩckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.A.; Malawska, B. Structure-based search for new inhibitors of cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608–5632. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Bajda, M.; Skrok, M.; Kurpiewska, K.; LewiÅski, K.; Brus, B.; Pišlar, A.; Kos, J.; Gobec, S.; Malawska, B. Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and β-amyloid aggregation inhibitors with neuroprotective properties. Eur. J. Med. Chem. 2015, 92, 738–749. [Google Scholar] [CrossRef]
- Guzior, N.; Bajda, M.; Rakoczy, J.; Brus, B.; Gobec, S.; Malawska, B. Isoindoline-1,3-dione derivatives targeting cholinesterases: Design, synthesis and biological evaluation of potential anti-Alzheimer’s agents. Bioorg. Med. Chem. 2015, 23, 1629–1637. [Google Scholar] [CrossRef]
- Hassanzadeh, M.; Hassanzadeh, F.; Khodarahmi, G.A.; Rostami, M.; Azimi, F.; Nadri, H.; Homayouni Moghadam, F. Design, synthesis, and bio-evaluation of new isoindoline-1,3-dione derivatives as possible inhibitors of acetylcholinesterase. Res. Pharm. Sci. 2021, 16, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Karim, N.; Khan, I.; Khan, I.; Halim, S.A.; Khalid, A.; Abdalla, A.N.; Rehman, N.U.; Khan, A.; Al-Harrasi, A. Antiamnesic Effects of Novel Phthalimide Derivatives in Scopolamine-Induced Memory Impairment in Mice: A Useful Therapy for Alzheimer’s Disease. ACS Omega 2023, 8, 8052–8065. [Google Scholar] [CrossRef]
- Marciniak, A.; Kotynia, A.; Szkatuła, D.; Krzyżak, E. The 2-hydroxy-3-(4-aryl-1-piperazinyl)propyl Phthalimide Derivatives as Prodrugs—Spectroscopic and Theoretical Binding Studies with Plasma Proteins. Int. J. Mol. Sci. 2022, 23, 7003. [Google Scholar] [CrossRef]
- Szkatuła, D.; Krzyżak, E.; Stanowska, P.; Duda, M.; Wiatrak, B. A new n-substituted 1h-isoindole-1,3(2h)-dione derivative—Synthesis, structure and affinity for cyclooxygenase based on in vitro studies and molecular docking. Int. J. Mol. Sci. 2021, 22, 7678. [Google Scholar] [CrossRef]
- Kułaga, D.; Drabczyk, A.K.; Satała, G.; Latacz, G.; Rózga, K.; Plażuk, D.; Jaśkowska, J. Design and synthesis of new potent 5-HT7 receptor ligands as a candidate for the treatment of central nervous system diseases. Eur. J. Med. Chem. 2022, 227, 113931. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Ariceptρ): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal Structure of Human Butyrylcholinesterase and of Its Complexes with Substrate and Products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef]
- Brown, N. Bioisosteres in Medicinal Chemistry. Bioisosteres Med. Chem. 2012, 54, 1–237. [Google Scholar] [CrossRef]
- Mokrosz, J.L.; Dereń-Wesołek, A.; Tatarczyńska, E.; Duszyńska, B.; Bojarski, A.J.; Mokrosz, M.J.; Chojnacka-Wójcik, E. 8-[4-[2-(1,2,3,4-tetrahydroisoquinolinyl)]butyl]-8-azaspiro[4.5]decane-7,9- dione: A new 5-HT1A receptor ligand with the same activity profile as buspirone. J. Med. Chem. 1996, 39, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Dileep, K.V.; Ihara, K.; Mishima-Tsumagari, C.; Kukimoto-Niino, M.; Yonemochi, M.; Hanada, K.; Shirouzu, M.; Zhang, K.Y.J. Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery. Int. J. Biol. Macromol. 2022, 210, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 {R}evision {C}.01 2016. Available online: https://gaussian.com/citation/ (accessed on 4 July 2024).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple LigandÀProtein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model 2011, 51, 26. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024, 2024, W513–W520. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Affinity (Docking) (kcal/mol) | Binding Free Energy (MD) (kcal/mol) | ||
|---|---|---|---|---|
| ACHE | BuCHE | ACHE | BuCHE | |
| I | −9.7 | −9.9 | −12.87 ± 1.66 | −13.78 ± 1.42 |
| II | −9.3 | −9.9 | −15.15 ± 1.91 | −13.94 ± 1.71 |
| III | −10.1 | −11.0 | −15.71 ± 2.37 | −15.41 ± 1.81 |
| IV | −9.2 | −9.4 | −7.08 ± 1.43 | −7.18 ± 1.37 |
| V | −8.2 | −8.3 | −7.35 ± 1.74 | −1.91 ± 0.51 |
| VI | −10.2 | −10.2 | −14.34 ± 2.02 | −7.03 ± 0.94 |
| Donepezil | −10.9 | −10.0 | - | - |
| Comp. | Formula | m.p. (°C) Ethanol | Yield (%) | TLC Rf Ethyl Acetate (EA) | FT-IR (cm−1) C=O Arene -CH2- |
|---|---|---|---|---|---|
| I | C20H21N3O2 335.4 | 156–158 | 78.85 | 0.87 | 1715, 1775 695, 710 |
| II | C21H20F3N3O2 403.40 | 137–139 | 56.6 | 0.74 | 1720, 1770 700, 715 |
| III | C27H27N3O2 425.52 | 150–152 | 75.7 | 0.84 | 1710, 1770 710 |
| IV | C18H19N5O2 337.40 | 113–115 | 49.79 | 0.80 | 1710, 1780 710 |
| V | C14H16N2O3 260.29 | 134–136 | 47.20 (A) 86.54 (B) | 0.78 | 1700, 1780 3030, 2850 |
| VI | C19H18N2O2 306.40 | 128–130 | 55.48 | 0.85 | 1710, 1780 735 3030, 2850 |
| Compound | IC50 [μM] | Selectivity | ||
|---|---|---|---|---|
| AChE | BuChE | AChE | BuChE | |
| I | 1.12 ± 0.30 | 24.67 ± 1.74 | 22.02 | 0.05 |
| II | 2.31 ± 0.36 | 24.71 ± 3.78 | 10.70 | 0.09 |
| III | 1.94 ± 0.43 | 21.24 ± 4.84 | 10.95 | 0.09 |
| IV | 3.90 ± 0.43 | 68.88 ± 7.82 | 17.66 | 0.06 |
| V | 16.20 ± 2.37 | >1000 | ||
| VI | 1.14 ± 0.17 | 52.11 ± 9.73 | 45.71 | 0.02 |
| Donepezil | 0.04 ± 0.01 | 3.29 ± 0.85 | 82.55 | 0.01 |
| Compound | ||||||
|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | |
| Carcinogenicity | inactive | inactive | inactive | inactive | inactive | inactive |
| Immunotoxicity | inactive | inactive | inactive | inactive | inactive | inactive |
| Mutagenicity | inactive | inactive | inactive | inactive | inactive | inactive |
| Cytotoxicity | inactive | inactive | inactive | inactive | inactive | inactive |
| LD50 (mg/kg) | 4000 | 728 | 4000 | 4000 | 4000 | 315 |
| class | 5 | 4 | 5 | 5 | 5 | 4 |
| BBB (probability) | 0.98 | 0.98 | 0.98 | 0.99 | 0.92 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzyżak, E.; Marciniak, A.; Szkatuła, D.; Jankowska, K.A.; Dobies, N.; Kotynia, A. A Series of Novel 1-H-isoindole-1,3(2H)-dione Derivatives as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: In Silico, Synthesis and In Vitro Studies. Molecules 2024, 29, 3528. https://doi.org/10.3390/molecules29153528
Krzyżak E, Marciniak A, Szkatuła D, Jankowska KA, Dobies N, Kotynia A. A Series of Novel 1-H-isoindole-1,3(2H)-dione Derivatives as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: In Silico, Synthesis and In Vitro Studies. Molecules. 2024; 29(15):3528. https://doi.org/10.3390/molecules29153528
Chicago/Turabian StyleKrzyżak, Edward, Aleksandra Marciniak, Dominika Szkatuła, Klaudia A. Jankowska, Natalia Dobies, and Aleksandra Kotynia. 2024. "A Series of Novel 1-H-isoindole-1,3(2H)-dione Derivatives as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: In Silico, Synthesis and In Vitro Studies" Molecules 29, no. 15: 3528. https://doi.org/10.3390/molecules29153528
APA StyleKrzyżak, E., Marciniak, A., Szkatuła, D., Jankowska, K. A., Dobies, N., & Kotynia, A. (2024). A Series of Novel 1-H-isoindole-1,3(2H)-dione Derivatives as Acetylcholinesterase and Butyrylcholinesterase Inhibitors: In Silico, Synthesis and In Vitro Studies. Molecules, 29(15), 3528. https://doi.org/10.3390/molecules29153528