
AI-Assisted Rational Design and Activity Prediction of Biological Elements for Optimizing Transcription-Factor-Based Biosensors


Abstract
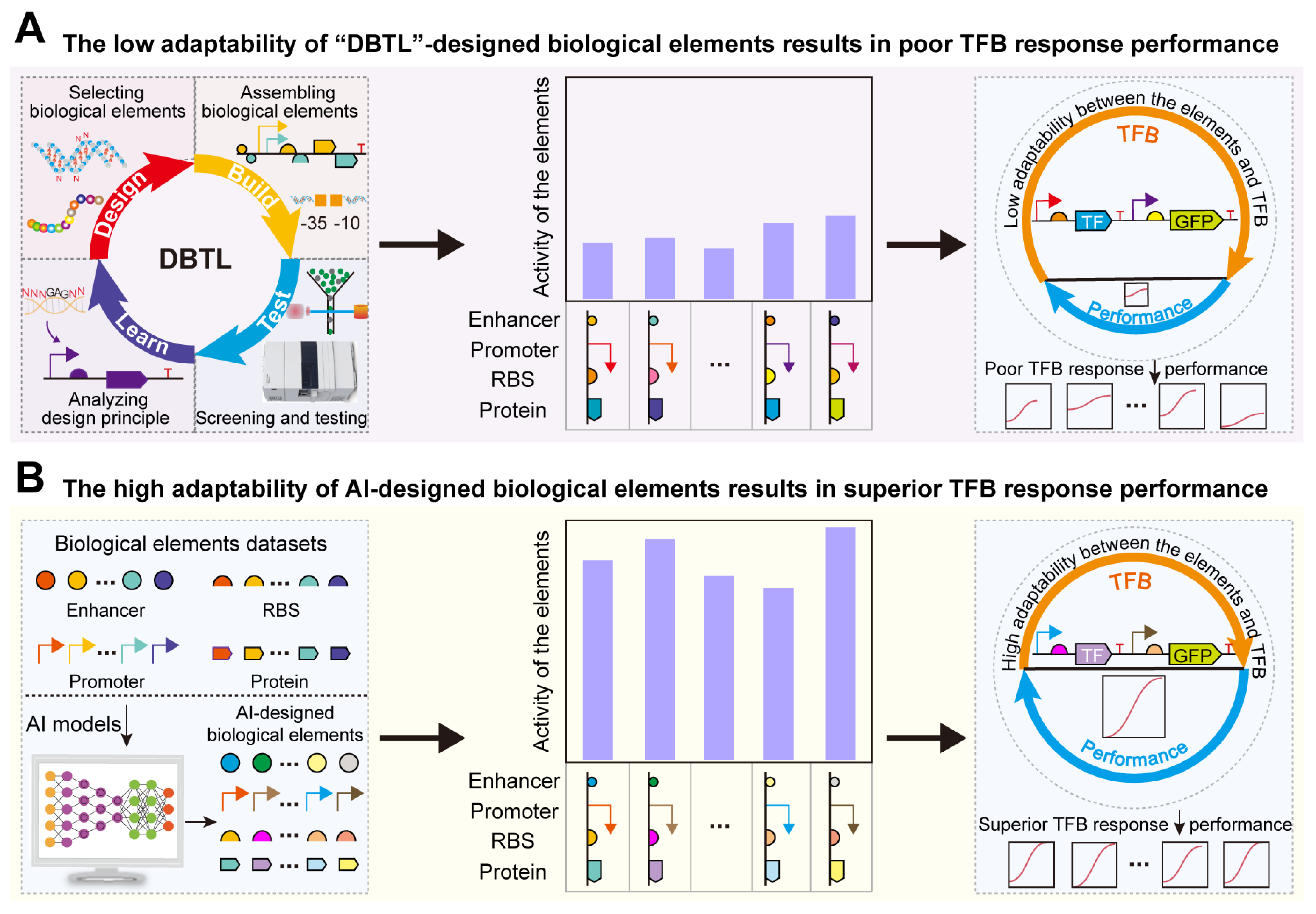
1. Introduction
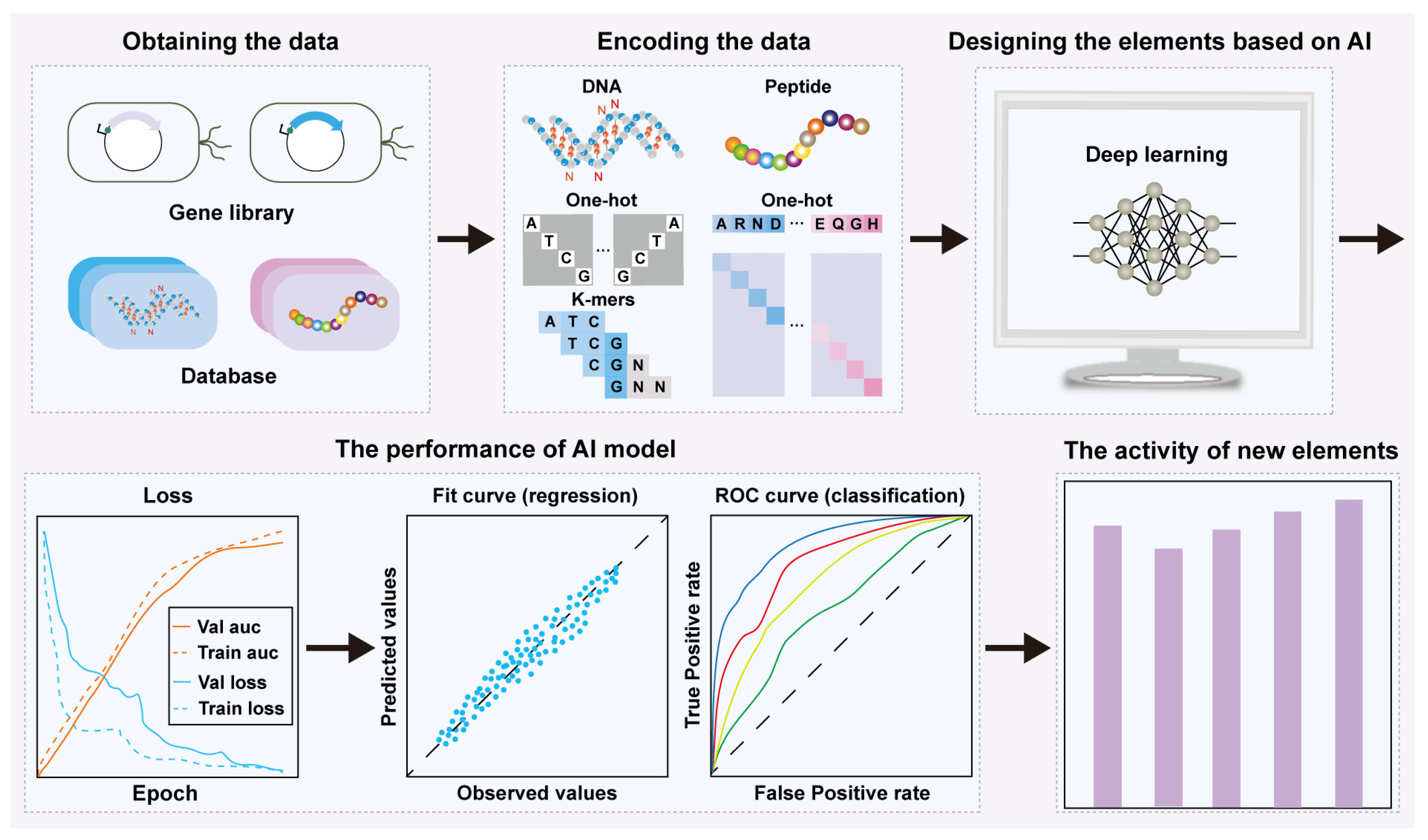
2. AI-Based Rational Design and Activity Prediction of Bio-Elements
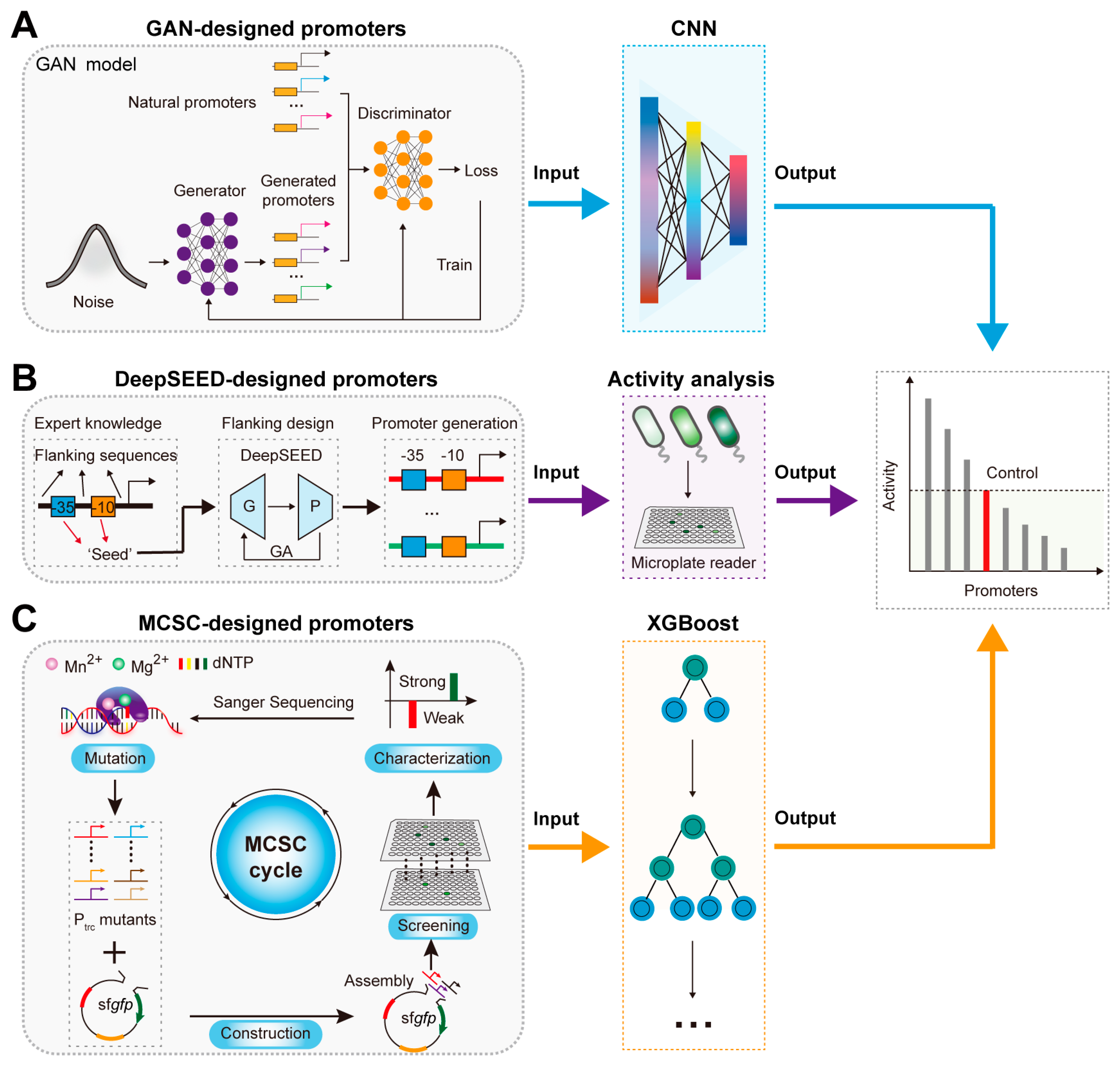
2.1. AI-Assisted Rational Design and Activity Prediction of Promoters
2.2. AI-Assisted Rational Design and Activity Prediction of Enhancers
2.3. AI-Assisted Rational Design and Activity Prediction of RBS
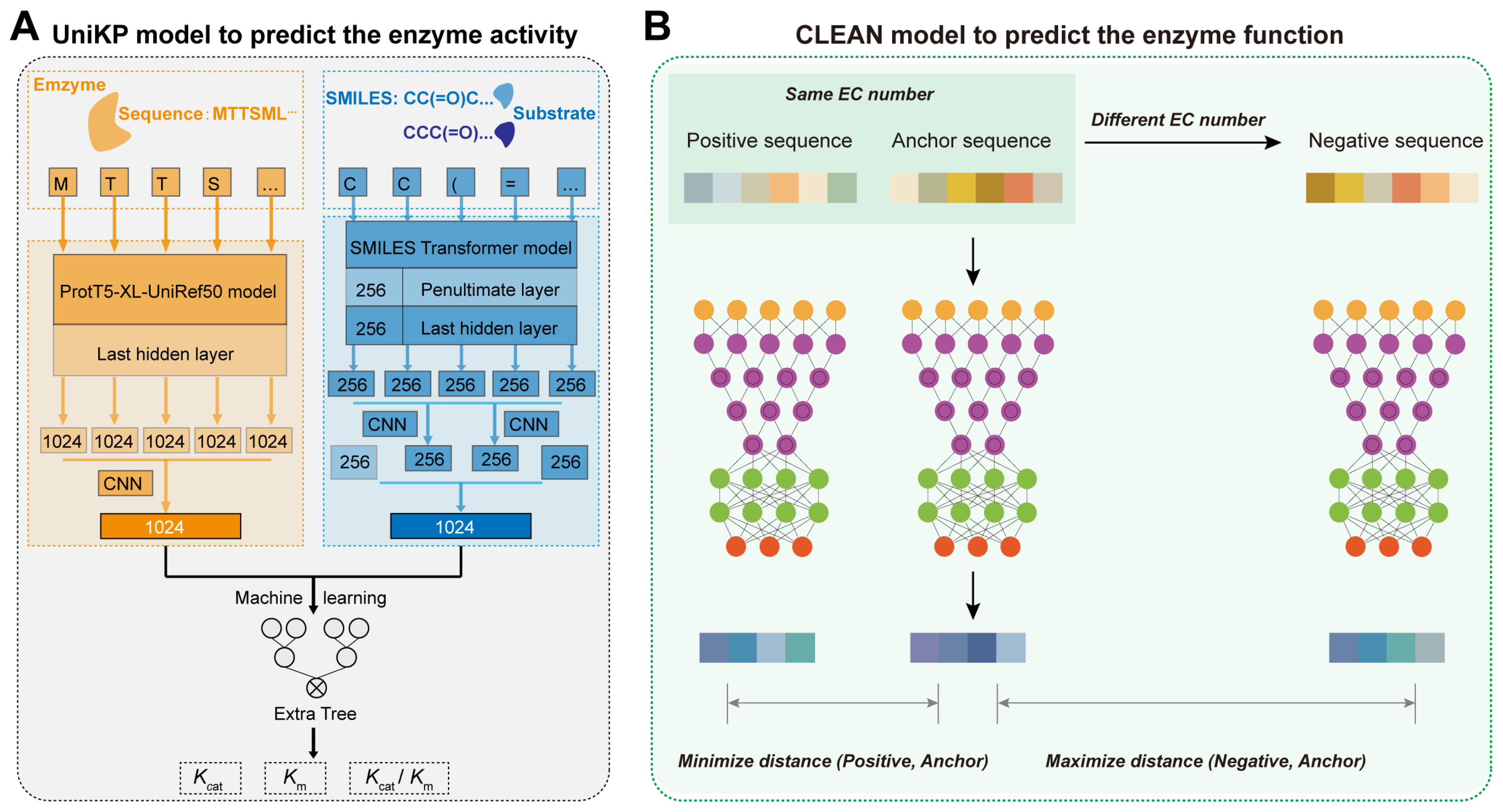
2.4. AI-Assisted Design of Protein Sequences and Structures and Prediction of Functional Activity
3. Optimizing the TFB Response Performance Based on AI-Designed Biological Elements
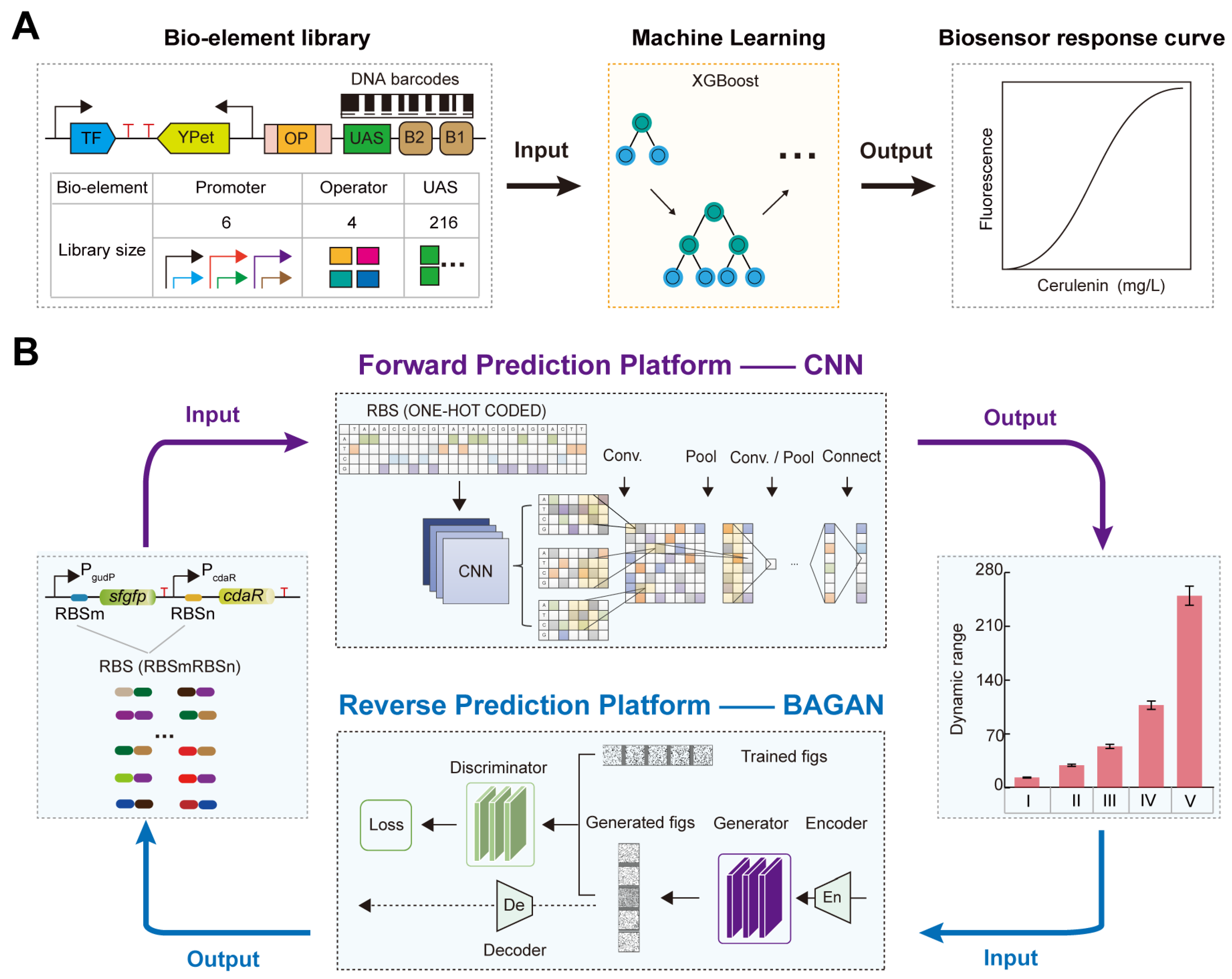
3.1. AI-Designed Promoters for Regulating TFB Response Performance
3.2. AI-Designed RBS for Regulating TFB Response Performance
3.3. AI-Optimized Transcription Factor Regulating the Dynamic Range of TFB
4. Applications of Optimized TFB
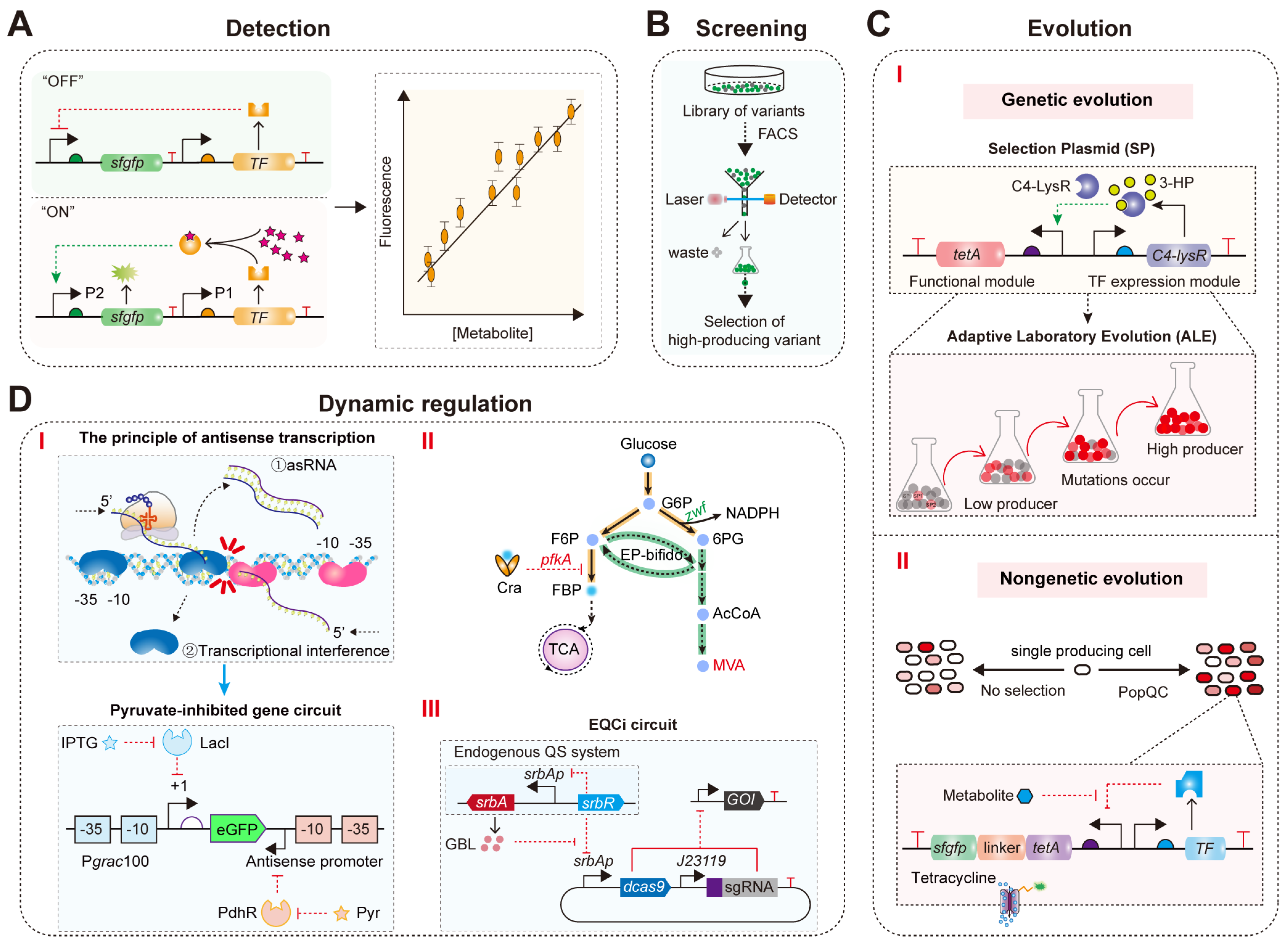
4.1. Real-Time Detection of Target Metabolite Concentrations
4.2. High-Throughput Screening of High-Titer Strains for Target Metabolites
4.3. Directed Evolution
4.4. Dynamic Regulation of Microbial Intracellular Metabolism
5. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Portela, R.M.C.; Vogl, T.; Kniely, C.; Fischer, J.E.; Oliveira, R.; Glieder, A. Synthetic core promoters as universal parts for fine-tuning expression in different yeast species. ACS Synth. Biol. 2016, 6, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Weingarten Gabbay, S.; Nir, R.; Lubliner, S.; Sharon, E.; Kalma, Y.; Weinberger, A.; Segal, E. Systematic interrogation of human promoters. Genome Res. 2019, 29, 171–183. [Google Scholar] [CrossRef]
- Reeve, B.; Hargest, T.; Gilbert, C.; Ellis, T. Predicting translation initiation rates for designing synthetic biology. Front. Bioeng. Biotechnol. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Avsec, Ž.; Agarwal, V.; Visentin, D.; Ledsam, J.R.; Grabska-Barwinska, A.; Taylor, K.R.; Assael, Y.; Jumper, J.; Kohli, P.; Kelley, D.R. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 2021, 18, 1196–1203. [Google Scholar] [CrossRef]
- Lynch, S.A.; Gill, R.T. Synthetic biology: New strategies for directing design. Metab. Eng. 2012, 14, 205–211. [Google Scholar] [CrossRef]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-learning methods for ligand–protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef]
- Handelman, G.S.; Kok, H.K.; Chandra, R.V.; Razavi, A.H.; Lee, M.J.; Asadi, H. eDoctor: Machine learning and the future of medicine. J. Intern. Med. 2018, 284, 603–619. [Google Scholar] [CrossRef]
- Guest, D.; Cranmer, K.; Whiteson, D. Deep larning and its application to LHC physics. Annu. Rev. Nucl. Part. Sci. 2018, 68, 161–181. [Google Scholar] [CrossRef]
- Roscher, R.; Bohn, B.; Duarte, M.F.; Garcke, J. Explainable machine learning for scientific insights and discoveries. IEEE Access 2020, 8, 42200–42216. [Google Scholar] [CrossRef]
- Wang, H.; Fu, T.; Du, Y.; Gao, W.; Huang, K.; Liu, Z.; Chandak, P.; Liu, S.; Van Katwyk, P.; Deac, A.; et al. Scientific discovery in the age of artificial intelligence. Nature 2023, 620, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2021, 23, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, P.; Radivojevic, T.; García Martín, H. Opportunities at the intersection of synthetic biology, machine learning, and automation. ACS Synth. Biol. 2019, 8, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Merchant, A.; Batzner, S.; Schoenholz, S.S.; Aykol, M.; Cheon, G.; Cubuk, E.D. Scaling deep learning for materials discovery. Nature 2023, 624, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.Y.; et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 2021, 5, 613. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Yang, W.J.; Peng, S.H.; Liu, F. A survey of convolutional neural networks: Analysis, applications, and prospects. IEEE Trans. Neural Netw. Learn. Syst. 2022, 33, 6999–7019. [Google Scholar] [CrossRef]
- Greg, V.H.; Carlos, M.; Gonzalo, N. A review on the long short-term memory model. Artif. Intell. Rev. 2020, 53, 5929–5955. [Google Scholar]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.T.; Chen, K.Y.; Yu, Y.M.; Le, N.Q.K.; Chua, M.C.H. Prediction of anticancer peptides based on an ensemble model of deep learning and machine learning using ordinal positional encoding. Brief. Bioinform. 2023, 24, bbac630. [Google Scholar] [CrossRef]
- Wang, Y.B.; Wu, H.X.; Zhang, J.J.; Gao, Z.F.; Wang, J.M.; Yu, P.S.; Long, M.S. PredRNN: A recurrent neural network for spatiotemporal predictive learning. IEEE Trans. Pattern Anal. Mach. Intell. 2023, 45, 2208–2225. [Google Scholar] [CrossRef]
- Ian, G.; Jean, P.A.; Mehdi, M.; Xu, B.; David, W.F.; Sherjil, O.; Aaron, C.; Yoshua, B. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar]
- Jin, X.B.; Gong, W.T.; Kong, J.L.; Bai, Y.T.; Su, T.L. PFVAE: A planar flow-based variational auto-encoder prediction model for time series data. Mathematics 2022, 10, 610. [Google Scholar] [CrossRef]
- Wang, J.M.; Chu, Y.Y.; Mao, J.S.; Jeon, H.N.; Jin, H.Y.; Zeb, A.; Jang, Y.; Cho, K.H.; Song, T.; No, K.T. De novo molecular design with deep molecular generative models for PPI inhibitors. Brief. Bioinform. 2022, 23, bbac285. [Google Scholar] [CrossRef] [PubMed]
- Zrimec, J.; Fu, X.Z.; Muhammad, A.S.; Skrekas, C.; Jauniskis, V.; Speicher, N.K.; Börlin, C.S.; Verendel, V.; Chehreghani, M.H.; Dubhashi, D.; et al. Controlling gene expression with deep generative design of regulatory DNA. Nat. Commun. 2022, 13, 5099. [Google Scholar] [CrossRef]
- He, X.; Samee, M.A.H.; Blatti, C.; Sinha, S.; Ohler, U. Thermodynamics-based models of transcriptional regulation by enhancers: The roles of synergistic activation, cooperative binding and short-range repression. PLoS Comput. Biol. 2010, 6, 1000935. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Liu, P.; Nielsen, A.A.K.; Brophy, J.A.N.; Clancy, K.; Peterson, T.; Voigt, C.A. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 2013, 10, 659–664. [Google Scholar] [CrossRef]
- Yasmeen, E.; Wang, J.; Riaz, M.; Zhang, L.D.; Zuo, K.J. Designing artificial synthetic promoters for accurate, smart, and versatile gene expression in plants. Plant Commun. 2023, 4, e1000935. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.J.; Yu, S.Y.; Wang, K.; Tan, Y.M.; Zhao, X.Y.; Liu, S.; Zhou, J.W.; Wang, X.L. AI and knowledge-based method for rational design of Escherichia coli sigma70 promoters. Acs Synth. Biol. 2024, 13, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Yan, J.W.; Zhang, J.; Liu, L.L.; Chen, Y.; Buskirk, A.R. Riboformer: A deep learning framework for predicting context-dependent translation dynamics. Nat. Commun. 2024, 15, 2011. [Google Scholar] [CrossRef]
- Krenn, M.; Pollice, R.; Guo, S.Y.; Aldeghi, M.; Cervera-Lierta, A.; Friederich, P.; Gomes, G.D.; Häse, F.; Jinich, A.; Nigam, A.; et al. On scientific understanding with artificial intelligence. Nat. Rev. Phys. 2022, 4, 761–769. [Google Scholar] [CrossRef]
- Wang, X.; Liu, L.; Li, S.; Wei, L.; Wang, H.; Wang, Y. Synthetic promoter design in Escherichia coli based on a deep generative network. Nucleic Acids Res. 2020, 48, 6403–6412. [Google Scholar] [CrossRef] [PubMed]
- Jores, T.; Tonnies, J.; Wrightsman, T.; Buckler, E.S.; Cuperus, J.T.; Fields, S.; Queitsch, C. Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters. Nat. Plants 2021, 7, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Sajid, S.; Zveushe, O.K.; de Dios, V.R.; Nabi, F.; Lee, Y.K.; Kaleri, A.R.; Ma, L.; Zhou, L.; Zhang, W.; Dong, F. Pretreatment of rice straw by newly isolated fungal consortium enhanced lignocellulose degradation and humification during composting. Bioresour. Technol. 2022, 354, 127150. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.; Stogios, P.J.; Savchenko, A.; Mahadevan, R. Advances in engineering and optimization of transcription factor-based biosensors for plug-and-play small molecule detection. Curr. Opin. Biotechnol. 2022, 76, 102753. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.J.; Zhang, F. Applications and tuning strategies for transcription factor-based metabolite biosensors. Biosensors 2023, 13, 428. [Google Scholar] [CrossRef]
- Ding, N.; Zhou, S.; Deng, Y. Transcription-factor-based biosensor engineering for applications in synthetic biology. ACS Synth. Biol. 2021, 10, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Miyake, R.; Ling, H.; Foo, J.L.; Fugono, N.; Chang, M.W. Transporter-driven engineering of a genetic biosensor for the detection and production of short-branched chain fatty acids in Saccharomyces cerevisiae. Front. Bioeng. Biotechnol. 2022, 10, 838732. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sha, Y.; Kumar, V.; Xu, Z.; Zhai, R.; Jin, M. Transcription factor-based biosensor: A molecular-guided approach for advanced biofuel synthesis. Biotechnol. Adv. 2024, 72, 108339. [Google Scholar] [CrossRef]
- Mao, Y.; Huang, C.; Zhou, X.; Han, R.; Deng, Y.; Zhou, S. Genetically encoded biosensor engineering for application in directed evolution. J. Microbiol. Biotechnol. 2023, 33, 1257–1267. [Google Scholar] [CrossRef]
- Teng, Y.X.; Gong, X.Y.; Zhang, J.L.; Obideen, Z.; Yan, Y.J. Investigating and engineering an 1,2-propanediol-responsive transcription factor-based biosensor. ACS Synth. Biol. 2024, 13, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Zhou, Y.Y.; Zou, Y.S.; Jiang, T.; Gong, X.Y.; Yan, Y.J. Identifying, characterizing, and engineering a phenolic acid-responsive transcriptional factor from Bacillus amyloliquefaciens. ACS Synth. Biol. 2023, 12, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Tang, X.L.; Kardashliev, T. Transcription factor-based biosensors in high-throughput screening: Advances and applications. Biotechnol. J. 2018, 13, 1700648. [Google Scholar] [CrossRef] [PubMed]
- d’Oelsnitz, S.; Nguyen, V.; Alper, H.S.; Ellington, A.D. Evolving a generalist biosensor for bicyclic monoterpenes. ACS Synth. Biol. 2022, 11, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Zhang, R.; Wang, J.; Yan, Y. Engineering of a TrpR-based biosensor for altered dynamic range and ligand preference. ACS Synth. Biol. 2022, 11, 2175–2183. [Google Scholar] [CrossRef]
- Ding, N.; Yuan, Z.; Zhang, X.; Chen, J.; Zhou, S.; Deng, Y. Programmable cross-ribosome-binding sites to fine-tune the dynamic range of transcription factor-based biosensor. Nucleic Acids Res. 2020, 48, 10602–10613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, H.; Xu, H.; Wei, L.; Liu, L.; Hu, Z.; Wang, X. Deep flanking sequence engineering for efficient promoter design using DeepSEED. Nat. Commun. 2023, 14, 6309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yuan, Z.; Wu, L.; Zhou, S.; Deng, Y. Precise prediction of promoter strength based on a de novo synthetic promoter library coupled with machine learning. ACS Synth. Biol. 2021, 11, 92–102. [Google Scholar] [CrossRef]
- Qiao, H.; Zhang, S.; Xue, T.; Wang, J.; Wang, B. iPro-GAN: A novel model based on generative adversarial learning for identifying promoters and their strength. Comput. Methods Programs Biomed. 2022, 215, 106625. [Google Scholar] [CrossRef]
- de Almeida, B.P.; Reiter, F.; Pagani, M.; Stark, A. DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 2022, 54, 613–624. [Google Scholar] [CrossRef]
- Liao, M.; Zhao, J.P.; Tian, J.; Zheng, C.H. iEnhancer-DCLA: Using the original sequence to identify enhancers and their strength based on a deep learning framework. BMC Bioinform. 2022, 23, 480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Holowko, M.B.; Hayman Zumpe, H.; Ong, C.S. Machine learning guided batched design of a bacterial ribosome binding site. ACS Synth. Biol. 2022, 11, 2314–2326. [Google Scholar] [CrossRef] [PubMed]
- Höllerer, S.; Papaxanthos, L.; Gumpinger, A.C.; Fischer, K.; Beisel, C.; Borgwardt, K.; Benenson, Y.; Jeschek, M. Large-scale DNA-based phenotypic recording and deep learning enable highly accurate sequence-function mapping. Nat. Commun. 2020, 11, 3551. [Google Scholar] [CrossRef] [PubMed]
- Donatas, R.; Vykintas, J.; Laurynas, K.; Elzbieta, R.; Jan, Z.; Simona, P.; Irmantas, R.; Audrius, L.; Wissam, A.; Otto, S.; et al. Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 2021, 3, 324–333. [Google Scholar]
- Karimi, M.; Zhu, S.; Cao, Y.; Shen, Y. De novo protein design for novel folds using guided conditional wasserstein generative adversarial networks (gcWGAN). J. Chem. Inf. Model. 2019, 60, 5667–5681. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yuan, L.; Lu, H.; Li, G.; Chen, Y.; Engqvist, M.K.M.; Kerkhoven, E.J.; Nielsen, J. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 2022, 5, 662–672. [Google Scholar] [CrossRef]
- Yu, H.; Deng, H.; He, J.; Keasling, J.D.; Luo, X. UniKP: A unified framework for the prediction of enzyme kinetic parameters. Nat. Commun. 2023, 14, 8211. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Cui, H.; Li, J.C.; Luo, Y.; Jiang, G.; Zhao, H. Enzyme function prediction using contrastive learning. Science 2023, 379, 1358. [Google Scholar] [CrossRef]
- Busby, S.; Ebright, R.H. Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 1994, 79, 743–746. [Google Scholar] [CrossRef]
- Curran, K.A.; Crook, N.C.; Karim, A.S.; Gupta, A.; Wagman, A.M.; Alper, H.S. Design of synthetic yeast promoters via tuning of nucleosome architecture. Nat. Commun. 2014, 5, 4002. [Google Scholar] [CrossRef]
- Huang, Y.K.; Yu, C.H.; Ng, I.S. Precise strength prediction of endogenous promoters from Escherichia coli and J-series promoters by artificial intelligence. J. Taiwan Inst. Chem. Eng. 2024, 160, 105211. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2011, 44, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Taskiran, I.I.; Spanier, K.I.; Dickmänken, H.; Kempynck, N.; Pančíková, A.; Ekşi, E.C.; Hulselmans, G.; Ismail, J.N.; Theunis, K.; Vandepoel, R.; et al. Cell-type-directed design of synthetic enhancers. Nature 2023, 626, 212–220. [Google Scholar] [CrossRef]
- Wolfe, J.C.; Mikheeva, L.A.; Hagras, H.; Zabet, N.R. An explainable artificial intelligence approach for decoding the enhancer histone modifications code and identification of novel enhancers in Drosophila. Genome Biol. 2021, 22, 308. [Google Scholar] [CrossRef]
- Hamamoto, R.; Takasawa, K.; Shinkai, N.; Machino, H.; Kouno, N.; Asada, K.; Komatsu, M.; Kaneko, S. Analysis of super-enhancer using machine learning and its application to medical biology. Brief. Bioinform. 2023, 24, bbad107. [Google Scholar] [CrossRef] [PubMed]
- Peterman, N.; Levine, E. Sort-seq under the hood: Implications of design choices on large-scale characterization of sequence-function relations. BMC Genom. 2016, 17, 206. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef]
- Ding, W.; Nakai, K.; Gong, H. Protein design via deep learning. Brief. Bioinform. 2022, 23, 102. [Google Scholar] [CrossRef] [PubMed]
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R.; Milles, L.; Wicky, B.; Courbet, A.; de Haas, R.; Bethel, N.; et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 2022, 378, 49–55. [Google Scholar] [CrossRef]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Lin, C.H.; Lane, H.Y. De novo peptide and protein design using generative adversarial networks: An update. J. Chem. Inf. Model. 2022, 62, 761–774. [Google Scholar] [CrossRef]
- Scalvini, B.; Sheikhhassani, V.; Mashaghi, A. Topological principles of protein folding. Phys. Chem. Chem. Phys. 2021, 23, 21316–21328. [Google Scholar] [CrossRef] [PubMed]
- Baker, D. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef]
- Goverde, C.A.; Wolf, B.; Khakzad, H.; Rosset, S.; Correia, B.E. De novo protein design by inversion of the AlphaFold structure prediction network. Protein Sci. 2023, 32, 4653. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Design of cyclic peptides targeting protein–protein interactions using AlphaFold. Int. J. Mol. Sci. 2023, 24, 13257. [Google Scholar] [CrossRef]
- Bryant, P.; Elofsson, A. Peptide binder design with inverse folding and protein structure prediction. Commun. Chem. 2023, 6, 229. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Topf, M. Critical assessment of techniques for protein structure prediction, fourteenth round. In CASP 14 Abstract Book; CASP, 2020; pp. 1–344. Available online: https://www.predictioncenter.org/casp14/doc/CASP14_Abstracts.pdf (accessed on 15 June 2024).
- Tellechea-Luzardo, J.; Stiebritz, M.T.; Carbonell, P. Transcription factor-based biosensors for screening and dynamic regulation. Front. Bioeng. Biotechnol. 2023, 11, 1118702. [Google Scholar] [CrossRef] [PubMed]
- Hartline, C.J.; Zhang, F. The growth dependent design constraints of transcription-factor-based metabolite biosensors. ACS Synth. Biol. 2022, 11, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Mannan, A.A.; Liu, D.; Zhang, F.; Oyarzún, D.A. Fundamental design principles for transcription-factor-based metabolite biosensors. ACS Synth. Biol. 2017, 6, 1851–1859. [Google Scholar] [CrossRef]
- Chen, Y.; Ho, J.M.L.; Shis, D.L.; Gupta, C.; Long, J.; Wagner, D.S.; Ott, W.; Josić, K.; Bennett, M.R. Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat. Commun. 2018, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Song, J.; Zhang, H.; Lin, Y.; Han, S.; Huang, Y.; Zheng, S. Development of a transcription factor-based diamine biosensor in Corynebacterium glutamicum. ACS Synth. Biol. 2021, 10, 3074–3083. [Google Scholar] [CrossRef]
- Peters, G.; De Paepe, B.; De Wannemaeker, L.; Duchi, D.; Maertens, J.; Lammertyn, J.; De Mey, M. Development of N-acetylneuraminic acid responsive biosensors based on the transcriptional regulator NanR. Biotechnol. Bioeng. 2018, 115, 1855–1865. [Google Scholar] [CrossRef]
- Zhou, Y.; Yuan, Y.; Wu, Y.; Li, L.; Jameel, A.; Xing, X.; Zhang, C. Encoding genetic circuits with DNA barcodes paves the way for machine learning-assisted metabolite biosensor response curve profiling in yeast. ACS Synth. Biol. 2022, 11, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Kasey, C.M.; Zerrad, M.; Li, Y.; Cropp, T.A.; Williams, G.J. Development of transcription factor-based designer macrolide biosensors for metabolic engineering and synthetic biology. ACS Synth. Biol. 2017, 7, 227–239. [Google Scholar] [CrossRef]
- Greco, F.V.; Pandi, A.; Erb, T.J.; Grierson, C.S.; Gorochowski, T.E. Harnessing the central dogma for stringent multi-level control of gene expression. Nat. Commun. 2021, 12, 1738. [Google Scholar] [CrossRef]
- Ding, N.; Sun, L.; Zhou, X.; Zhang, L.; Deng, Y.; Yin, L. Enhancing glucaric acid production from myo-inositol in Escherichia coli by eliminating cell-to-cell variation. Appl. Environ. Microbiol. 2024, 90, e00149-24. [Google Scholar] [CrossRef]
- Ding, N.; Zhang, G.; Zhang, L.; Shen, Z.; Yin, L.; Zhou, S.; Deng, Y. Engineering an AI-based forward-reverse platform for the design of cross-ribosome binding sites of a transcription factor biosensor. Comput. Struct. Biotechnol. J. 2023, 21, 2929–2939. [Google Scholar] [CrossRef]
- Xiao, Y.; Jiang, W.; Zhang, F. Developing a genetically encoded, cross-species biosensor for detecting ammonium and regulating biosynthesis of cyanophycin. ACS Synth. Biol. 2017, 6, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Trabelsi, H.; Koch, M.; Faulon, J.L. Building a minimal and generalizable model of transcription factor–based biosensors: Showcasing flavonoids. Biotechnol. Bioeng. 2018, 115, 2292–2304. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Fu, Y.; Bao, Y.; Wang, Z.; Lei, B.; Zheng, W.; Wang, C.; Liu, Y. DeepSATA: A deep learning-based sequence analyzer incorporating the transcription factor binding affinity to dissect the effects of non-coding genetic variants. Int. J. Mol. Sci. 2023, 24, 12023. [Google Scholar] [CrossRef]
- Han, K.; Shen, L.C.; Zhu, Y.H.; Xu, J.; Song, J.N.; Yu, D.J. MAResNet: Predicting transcription factor binding sites by combining multi-scale bottom-up and top-down attention and residual network. Brief. Bioinform. 2022, 23, 445. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.J.; Sun, X.Y.; Wu, J.; Mei, J.; Huang, L.Q.; He, R.J.; Nie, L.P.; Chen, Y.; Lyu, Q. Learning useful representations of DNA sequences from ChIP-Seq datasets for exploring transcription factor binding specificities. IEEE-ACM Trans. Comput. Biol. Bioinform. 2022, 19, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Baumann, L.; Rajkumar, A.S.; Morrissey, J.P.; Boles, E.; Oreb, M. A yeast-based biosensor for screening of short- and medium-chain fatty acid production. ACS Synth. Biol. 2018, 7, 2640–2646. [Google Scholar] [CrossRef]
- Yu, W.; Xu, X.; Jin, K.; Liu, Y.; Li, J.; Du, G.; Lv, X.; Liu, L. Genetically encoded biosensors for microbial synthetic biology: From conceptual frameworks to practical applications. Biotechnol. Adv. 2023, 62, 108077. [Google Scholar] [CrossRef]
- Seok, J.Y.; Han, Y.H.; Yang, J.S.; Yang, J.; Lim, H.G.; Kim, S.G.; Seo, S.W.; Jung, G.Y. Synthetic biosensor accelerates evolution by rewiring carbon metabolism toward a specific metabolite. Cell Rep. 2021, 36, 109589. [Google Scholar] [CrossRef]
- Xu, X.; Li, X.; Liu, Y.; Zhu, Y.; Li, J.; Du, G.; Chen, J.; Ledesma-Amaro, R.; Liu, L. Pyruvate-responsive genetic circuits for dynamic control of central metabolism. Nat. Chem. Biol. 2020, 16, 1261–1268. [Google Scholar] [CrossRef]
- Su, B.; Lai, P.; Deng, M.R.; Zhu, H. Design of a dual-responding genetic circuit for high-throughput identification of L-threonine-overproducing Escherichia coli. Bioresour. Technol. 2024, 395, 130407. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, Y.; Feng, T.; Shen, J.; Lu, H.; Zhang, Y.; Chou, H.H.; Luo, X.; Keasling, J.D. Dynamic upregulation of the rate-limiting enzyme for valerolactam biosynthesis in Corynebacterium glutamicum. Metab. Eng. 2023, 77, 89–99. [Google Scholar] [CrossRef]
- Rogers, J.K.; Church, G.M. Genetically encoded sensors enable real-time observation of metabolite production. Proc. Natl. Acad. Sci. USA 2016, 113, 2388–2393. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.K.; Guzman, C.D.; Taylor, N.D.; Raman, S.; Anderson, K.; Church, G.M. Synthetic biosensors for precise gene control and real-time monitoring of metabolites. Nucleic Acids Res. 2015, 43, 7648–7660. [Google Scholar] [CrossRef]
- Mitchler, M.M.; Garcia, J.M.; Montero, N.E.; Williams, G.J. Transcription factor-based biosensors: A molecular-guided approach for natural product engineering. Curr. Opin. Biotechnol. 2021, 69, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Kortmann, M.; Mack, C.; Baumgart, M.; Bott, M. Pyruvate carboxylase variants enabling improved lysine production from glucose identified by biosensor-based high-throughput fluorescence-activated cell sorting screening. ACS Synth. Biol. 2019, 8, 274–281. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Upp, D.M.; Lewis, J.C.; Zhang, Y.-H.P.J. A high-throughput method for directed evolution of NAD(P)+-dependent dehydrogenases for the reduction of biomimetic nicotinamide analogues. ACS Catal. 2019, 9, 11709–11719. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, V.D.; Mohan, K.; Chappell, T.C.; Mays, Z.J.S.; Nair, N.U. Cheating the cheater: Suppressing false-positive enrichment during biosensor-guided biocatalyst engineering. ACS Synth. Biol. 2022, 11, 420–429. [Google Scholar] [CrossRef]
- Nasr, M.A.; Martin, V.J.J.; Kwan, D.H. Divergent directed evolution of a TetR-type repressor towards aromatic molecules. Nucleic Acids Res. 2023, 51, 7675–7690. [Google Scholar] [CrossRef]
- Du, H.; Liang, Y.; Li, J.; Yuan, X.; Tao, F.; Dong, C.; Shen, Z.; Sui, G.; Wang, P. Directed evolution of 4-hydroxyphenylpyruvate biosensors based on a dual selection system. Int. J. Mol. Sci. 2024, 25, 1533. [Google Scholar] [CrossRef]
- Liang, Y.; Luo, J.; Yang, C.; Guo, S.; Zhang, B.; Chen, F.; Su, K.; Zhang, Y.; Dong, Y.; Wang, Z.; et al. Directed evolution of the PobR allosteric transcription factor to generate a biosensor for 4-hydroxymandelic acid. World J. Microbiol. Biotechnol. 2022, 38, 104. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xu, S.; Li, S.; Tao, S.; Li, L.; Chen, S.; Wu, L. Directly evolved AlkS-based biosensor platform for monitoring and high-throughput screening of alkane production. ACS Synth. Biol. 2023, 12, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.P.; Pan, Y.; Niu, F.X.; Liao, Y.L.; Huang, M.; Liu, J.Z. Biosensor-assisted evolution for high-level production of 4-hydroxyphenylacetic acid in Escherichia coli. Metab. Eng. 2022, 70, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Li, N.; Zhou, S.; Zhang, L.; Xu, S.; Zhou, J. Improvement of chalcone synthase activity and high-efficiency fermentative production of (2S)-naringenin via in vivo biosensor-guided directed evolution. ACS Synth. Biol. 2024, 13, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Rugbjerg, P.; Sommer, M.O.A. Overcoming genetic heterogeneity in industrial fermentations. Nat. Biotechnol. 2019, 37, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bowen, C.H.; Liu, D.; Zhang, F. Exploiting nongenetic cell-to-cell variation for enhanced biosynthesis. Nat. Chem. Biol. 2016, 12, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Guan, A.; He, Z.; Wang, X.; Jia, Z.J.; Qin, J. Engineering the next-generation synthetic cell factory driven by protein engineering. Biotechnol. Adv. 2024, 73, 108366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Carothers, J.M.; Keasling, J.D. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 2012, 30, 354–359. [Google Scholar] [CrossRef]
- Zhou, S.; Yuan, S.F.; Nair, P.H.; Alper, H.S.; Deng, Y.; Zhou, J. Development of a growth coupled and multi-layered dynamic regulation network balancing malonyl-CoA node to enhance (2S)-naringenin biosynthesis in Escherichia coli. Metab. Eng. 2021, 67, 41–52. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Xu, Y.; Zhang, J.; Ma, L.; Qi, Q.; Wang, Q. Development of bifunctional biosensors for sensing and dynamic control of glycolysis flux in metabolic engineering. Metab. Eng. 2021, 68, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yang, G.; Gu, Y.; Sun, X.; Lu, Y.; Jiang, W. Developing an endogenous quorum-sensing based CRISPRi circuit for autonomous and tunable dynamic regulation of multiple targets in Streptomyces. Nucleic Acids Res. 2020, 48, 8188–8202. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J.L. Artificial intelligence in the prediction of protein-ligand interactions: Recent advances and future directions. Brief. Bioinform. 2022, 23, bbab476. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.X.; Bittrich, S.; Chao, H.Y.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold protein structure database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2023, 52, D368–D375. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Tang, X.; Zhang, X.R.; Ma, J.J.; Liu, F.; Jia, X.P.; Jiao, L.C. Semi-supervised multiscale dynamic graph convolution network for hyperspectral image classification. IEEE Trans. Neural Netw. Learn. Syst. 2024, 35, 6806–6820. [Google Scholar] [CrossRef] [PubMed]
- Chavez, M.R.; Butler, T.S.; Rekawek, P.; Heo, H.; Kinzler, W.L. Chat generative pre-trained transformer: Why we should embrace this technology. Am. J. Obstet. Gynecol. 2023, 228, 706–711. [Google Scholar] [CrossRef]
- Lee, G.S.; Kim, S.; Bae, S. Efficient design method for a forward-converter transformer based on a KNNGRUDNN model. IEEE Trans. Power Electron. 2023, 38, 73–78. [Google Scholar] [CrossRef]
- Bhatti, U.A.; Tang, H.; Wu, G.L.; Marjan, S.; Hussain, A. Deep learning with graph convolutional networks: An overview and latest applications in computational intelligence. Int. J. Intell. Syst. 2023, 2023, 8342104. [Google Scholar] [CrossRef]
- Yu, J.C.; Xu, T.Y.; Rong, Y.; Bian, Y.T.; Huang, J.Z.; He, R. Recognizing predictive substructures with subgraph information bottleneck. IEEE Trans. Pattern Anal. Mach. Intell. 2024, 46, 1650–1663. [Google Scholar] [CrossRef]
- Wei, Z.; Hua, K.; Wei, L.; Ma, S.; Jiang, R.; Zhang, X.; Li, Y.; Wong, W.H.; Wang, X. NeuronMotif: Deciphering cis-regulatory codes by layer-wise demixing of deep neural networks. Proc. Natl. Acad. Sci. USA 2023, 120, e2216698120. [Google Scholar] [CrossRef] [PubMed]
- Marques, G.; Agarwal, D.; de la Torre Díez, I. Automated medical diagnosis of COVID-19 through EfficientNet convolutional neural network. Appl. Soft Comput. 2020, 96, 106691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qin, Q.; Ye, Q.; Ruan, T. ST-Unet: Swin transformer boosted U-Net with cross-layer feature enhancement for medical image segmentation. Comput. Biol. Med. 2023, 153, 106516. [Google Scholar] [CrossRef] [PubMed]
- Singhal, K.; Azizi, S.; Tu, T.; Mahdavi, S.S.; Wei, J.; Chung, H.W.; Scales, N.; Tanwani, A.; Cole-Lewis, H.; Pfohl, S.; et al. Large language models encode clinical knowledge. Nature 2023, 620, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Schweidtmann, A.M. Deep reinforcement learning for process design: Review and perspective. Curr. Opin. Chem. Eng. 2024, 44, 101012. [Google Scholar] [CrossRef]
- Wang, D.; Gao, N.; Liu, D.R.; Li, J.N.; Lewis, F.L. Recent progress in reinforcement learning and adaptive dynamic programming for advanced control applications. IEEE-Caa J. Autom. Sin. 2024, 11, 18–36. [Google Scholar] [CrossRef]
- Kaufmann, E.; Bauersfeld, L.; Loquercio, A.; Mueller, M.; Koltun, V.; Scaramuzza, D. Champion-level drone racing using deep reinforcement learning. Nature 2023, 620, 982–987. [Google Scholar] [CrossRef]
- Chen, Y.B.; Mancini, M.; Zhu, X.T.; Akata, Z. Semi-supervised and unsupervised deep visual learning: A Survey. Ieee Trans. Pattern Anal. Mach. Intell. 2024, 46, 1327–1347. [Google Scholar] [CrossRef] [PubMed]
- Min, B.N.; Ross, H.; Sulem, E.; Ben Veyseh, A.P.; Nguyen, T.H.; Sainz, O.; Agirre, E.; Heintz, I.; Roth, D. Recent advances in natural language processing via large pre-trained language models: A Survey. ACM Comput. Surv. 2024, 56, 1–40. [Google Scholar] [CrossRef]
- Xue, Q.; Miao, P.; Miao, K.; Yu, Y.; Li, Z. An online automatic sorting system for defective Ginseng Radix et Rhizoma Rubra using deep learning. Chin. Herb. Med. 2023, 15, 447–456. [Google Scholar] [CrossRef]
- Klauschen, F.; Dippel, J.; Keyl, P.; Jurmeister, P.; Bockmayr, M.; Mock, A.; Buchstab, O.; Alber, M.; Ruff, L.; Montavon, G.; et al. Toward explainable artificial intelligence for precision pathology. Annu. Rev. Pathol.-Mech. Dis. 2024, 19, 541–570. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Shi, W.Q.; Isgut, M.; Zhong, Y.S.; Lais, P.; Gloster, L.; Sun, J.M.; Swain, A.; Giuste, F.; Wang, M.D. Integrating multi-omics data with EHR for precision medicine using advanced artificial intelligence. IEEE Rev. Biomed. Eng. 2024, 17, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.C.; Qian, Z.H.; Li, Q.Y.; Gao, Y.; Li, M.H. Assessment of pulmonary infectious disease treatment with Mongolian medicine formulae based on data mining, network pharmacology and molecular docking. Chin. Herb. Med. 2022, 14, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Zeng, X.X.; Zhao, Y.; Chen, R.S. AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct. Target. Ther. 2023, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Wang, J.J.; Williamson, D.F.K.; Chen, T.Y.; Lipkova, J.; Lu, M.Y.; Sahai, S.; Mahmood, F. Algorithmic fairness in artificial intelligence for medicine and healthcare. Nat. Biomed. Eng. 2023, 7, 719–742. [Google Scholar] [CrossRef]
- Zhang, W.D.; Li, Z.X.; Li, G.H.; Zhuang, P.X.; Hou, G.J.; Zhang, Q.; Li, C.Y. GACNet: Generate adversarial-driven cross-aware network for hyperspectral wheat variety identification. IEEE Trans. Geosci. Remote Sens. 2024, 62, 5503314. [Google Scholar] [CrossRef]
- Moghadam, P.Z.; Chung, Y.G.; Snurr, R.Q. Progress toward the computational discovery of new metal-organic framework adsorbents for energy applications. Nat. Energy 2024, 9, 121–133. [Google Scholar] [CrossRef]
- Bi, K.F.; Xie, L.X.; Zhang, H.H.; Chen, X.; Gu, X.T.; Tian, Q. Accurate medium-range global weather forecasting with 3D neural networks. Nature 2023, 619, 533–538. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.Z.; Liu, Y.L.; Jang, H. AlphaFold, allosteric, and orthosteric drug discovery: Ways forward. Drug Discov. Today 2023, 28, 103551. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bio-Element | Function | Challenge | Strategy | Accuracy | References | ||
|---|---|---|---|---|---|---|---|
| Traditional | AI | Traditional | AI | ||||
| Promoter | Rational design and activity prediction | Small library, vast sequence space | Experimental method | GAN, CNN | ns | 0.7 | [31] |
| DeepSEED (GAN, LSTM) | 0.78 | [47] | |||||
| Activity prediction | High prediction cost and low accuracy | CHIP-seq, RNA-seq | XGBoost | 0.88 | [48] | ||
| CHIP-seq, RNA-seq | iPro-GAN | 0.92 | [49] | ||||
| Enhancer | Rational design and activity prediction | Unclear motif syntax relationships, inadequate compatibility between motifs, and limited applicability | Experimental method | DeepSTARR (CNN), GAN | ns | 0.74 | [50] |
| Activity prediction | CHIP-seq, RNA-seq | iEnhancer-DCLA (CNN, BiLSTM, Attention) | 0.83 | [51] | |||
| RBS | Activity prediction | Demand for larger libraries, cumbersome experimental procedures, and complex thermodynamic analysis data | Experimental method | GPR, Bandit | ns | 34% high TIR | [52] |
| Ribosome loading, DNA methylation, NGS | CNN | 0.927 | [53] | ||||
| Protein | Rational design | Limited sequence space | ns | ProteinGAN (GAN) | ns | 0.88 | [54] |
| Rational design and activity prediction | Limited protein structure types and vast sequence space | Experimental method | WGAN, Rosetta | TM > 0.5 | [55] | ||
| Activity prediction | Low accuracy | AlphaFold 2 | TM > 0.78 | [56] | |||
| and limited accuracy for complex interactions | AlphaFold 3 | >0.8 | [57] | ||||
| Enzyme catalytic constant prediction | Low accuracy | DLKcat (CNN, GNN) | 0.71 (kcat) | [58] | |||
| UniKP (pretrained language models) | 0.85 (kcat), 0.73 (km), 0.81 (kcat/km) | [59] | |||||
| Enzyme function prediction | Small and imbalanced datasets | CLEAN (contrastive learning framework) | 0.86 | [60] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, N.; Yuan, Z.; Ma, Z.; Wu, Y.; Yin, L. AI-Assisted Rational Design and Activity Prediction of Biological Elements for Optimizing Transcription-Factor-Based Biosensors. Molecules 2024, 29, 3512. https://doi.org/10.3390/molecules29153512
Ding N, Yuan Z, Ma Z, Wu Y, Yin L. AI-Assisted Rational Design and Activity Prediction of Biological Elements for Optimizing Transcription-Factor-Based Biosensors. Molecules. 2024; 29(15):3512. https://doi.org/10.3390/molecules29153512
Chicago/Turabian StyleDing, Nana, Zenan Yuan, Zheng Ma, Yefei Wu, and Lianghong Yin. 2024. "AI-Assisted Rational Design and Activity Prediction of Biological Elements for Optimizing Transcription-Factor-Based Biosensors" Molecules 29, no. 15: 3512. https://doi.org/10.3390/molecules29153512
APA StyleDing, N., Yuan, Z., Ma, Z., Wu, Y., & Yin, L. (2024). AI-Assisted Rational Design and Activity Prediction of Biological Elements for Optimizing Transcription-Factor-Based Biosensors. Molecules, 29(15), 3512. https://doi.org/10.3390/molecules29153512