Abstract
The effect of H2 activation on the performance of CuFeOx catalyst for low-temperature CO oxidation was investigated. The characterizations of XRD, XPS, H2-TPR, O2-TPD, and in situ DRIFTS were employed to establish the relationship between physicochemical property and catalytic activity. The results showed that the CuFeOx catalyst activated with H2 at 100 °C displayed higher performance, which achieved 99.6% CO conversion at 175 °C. In addition, the H2 activation promoted the generation of Fe2+ species, and more oxygen vacancy could be formation with higher concentration of Oα species, which improved the migration rate of oxygen species in the reaction process. Furthermore, the reducibility of the catalyst was enhanced significantly, which increased the low-temperature activity. Moreover, the in situ DRIFTS experiments revealed that the reaction pathway of CO oxidation followed MvK mechanism at low temperature (<175 °C), and both MvK and L-H mechanism was involved at high temperature. The Cu+-CO and carbonate species were the main reactive intermediates, and the H2 activation increased the concentration of Cu+ species and accelerated the decomposition carbonate species, thus improving the catalytic performance effectively.
1. Introduction
The incomplete combustion of fossil fuels results in industrial flue gases containing certain concentrations of CO. For example, the low-temperature flue gas (80–180 °C) generated during the iron ore sintering process contains 0.5–2% CO [1,2]. The ability of CO to bind to hemoglobin is stronger than oxygen. The direct release of CO is detrimental to human health and causes various environmental issues, including the degradation of the ozone layer [3,4,5]. Catalytic oxidation could efficiently oxidize CO into nontoxic CO2 with minimal energy consumption, which is considered to be the most promising CO elimination technology at present [6,7]. Au, Pt, and other precious metal catalysts exhibit high catalytic activity [8]. Nevertheless, the elevated expense associated with precious metal catalysts poses a constraint on the widespread deployment of CO oxidation in large-scale industrial flue gas treatment. It is necessary to develop a catalytic material with low price and excellent performance for CO oxidation.
The Cu-based catalysts display a great development prospect in CO oxidation due to the unique CO adsorption and activation property [9,10]. However, the pure CuO catalyst has poor activity at low temperature. The catalyst of CuO combined with CeO2, Fe2O3, Co3O4, and MnO2 shows high catalytic performance at low temperatures [11,12,13,14,15]. Notably, the CuFeOx catalysts display exceptional performance in CO oxidation, attributed to the remarkable oxygen storage capacity and Fe2+/Fe3+ redox cycle inherent in Fe2O3 [16,17,18]. This suggests a potential for CuFeOx catalysts to supplant precious metal catalysts in CO removal from industrial flue gas. According to our previous research [19,20], the structure and oxidation state of the active components have significant effects on the catalytic performance. Previous studies have shown that the pretreatment and activation could adjust the structure and the content of oxidation states of active components of catalysts [21,22,23], which could effectively improve the catalytic performance. Wang et al. [21] reported that the H2 pretreated Pd/Fe2O3 catalyst exhibited higher catalytic activity due to the higher concentration of Fe2+ species and stronger reducibility. Wang et al. [22] explored the impact of H2 activation on the catalytic activity of the CuCeOx catalyst. The H2 pretreatment increased the amount of highly dispersed CuOx and oxygen vacancy, which enhanced the activity of CO oxidation. This showed us that the performance of the CuFeOx catalyst may be improved by H2 pretreatment. However, the effect of H2 pretreatment on the performance of the CuFeOx catalyst has seldom been explored in detail.
In this work, the CuFeOx catalyst was prepared and activated with H2 at different temperatures to investigate the influence of pretreatment on the performance. In addition, the XRD, BET, XPS, H2-TPR, and O2-TPD were characterized to establish the relationship between structure property and catalytic performance. Furthermore, the reaction mechanism was revealed by in situ DRIFTS studies. This work may provide a novel strategy for the improvement of performance of the CuFeOx catalyst.
2. Results and Discussion
2.1. Catalytic Performance
The activity of catalysts was measured and is displayed in Figure 1a. It was observed that the CO conversion increased with increasing temperature for all catalysts. As for pure Fe2O3, the CO conversion was only 41.7% at 250 °C. As expected, the activity was improved after the addition of Cu species, and 100% CO conversion could be achieved at 250 °C for the CuFeOx catalyst. In addition, the activity of CuFeOx and H2-activated catalysts was insignificantly different at low temperature, while the activities of CuFeOx-100 and CuFeOx-150 catalysts were higher than CuFeOx catalysts after 75 °C. The CuFeOx-100 catalyst displayed the highest activity with 99.6% CO conversion at 175 °C, which indicated that the appropriate activation temperature could effectively increase the catalytic performance. However, it could be noted that the CO conversion of the CuFeOx-200 catalyst was obviously lower than the CuFeOx catalyst, which may be due to the over-reduction decreasing the active species. The service life is another important factor in evaluating catalyst applications. The stability of the CuFeOx-100 catalyst was measured and is shown in Figure 1b. The reaction continued for 82 h at 175 °C, keeping the composition and airspeed of the gas mixture unchanged. It can be seen that the CO conversion almost did not decrease after 82 h, and displayed excellent stability.
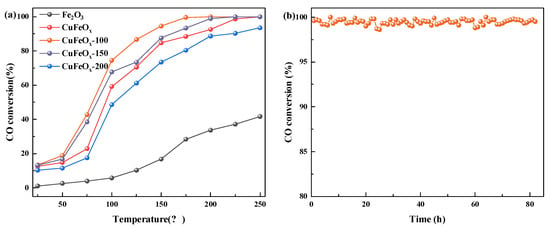
Figure 1.
(a) CO conversion of all catalysts. (b) The stability of CuFeOx-100 catalyst.
2.2. Structural and Textural Properties
The crystal structure of the catalysts was investigated with XRD and is depicted in Figure 2. The pure Fe2O3 displayed the diffraction peaks at 24.2°, 33.2°, 35.6°, 40.9°, 49.5°, 54.1°, 57.6°, 62.4°, 64.0°, 71.9°, and 75.5°, which corresponded to the crystallized α-Fe2O3 for (012), (104), (110), (113), (024), (116), (122), (214), (300), (101), and (220), respectively. The diffraction peaks of pure Fe2O3 presented with high crystallinity. As for the CuFeOx catalyst, new peaks attributed to CuO at 38.8°, 48.6°, and 61.6° appeared. In addition, the peak intensity of Fe2O3 decreased significantly compared with pure Fe2O3, indicating the formation of a strong interaction between Fe and Cu species, as a result of the smaller particle size and reduced crystallinity. For CuFeOx-100 and CuFeOx-150 catalysts, the peaks at 48.6° and 61.6° weakened and even disappeared, possibly attributable to the reduction of CuO to Cu2O and/or metallic Cu. The peaks assigned to metallic copper at 43.4° and 50.5° could be found on the CuFeOx-200 catalyst, which confirmed that part of the CuO species was reduced to metallic Cu species due to the H2 activation. Interestingly, the peaks of Fe2O3 at 35.7° on the H2-activated catalysts shifted to a lower 2θ degree compared to the CuFeOx catalyst. This demonstrated that the H2 activation promoted the formation of Fe2+ species and resulted in the lattice expansion, which suggested that the Fe2O3 also participated in the reduction process. In addition, no obvious diffraction peaks related to the CuFeOx phase were found.
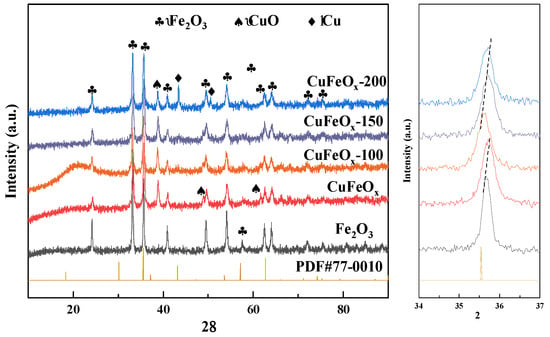
Figure 2.
XRD patterns of catalysts.
The N2 adsorption–desorption isotherms are displayed in Figure S1, and the physical property of the catalysts is shown in Table 1. As shown in Figure S1, all catalysts presented type IV isotherms and H3 type hysteresis loops, which suggests the existence of mesopores. The mesopores were conducive to the dispersion of active particles and the diffusion of reactive gases. The specific surface area, average pore diameter, and total pore volume of pure Fe2O3 was 9.85 m2/g, 14.96 nm, and 0.037 cm3/g, respectively. After the addition of the Cu species, the specific surface area and total pore volume of the CuFeOx catalyst increased slightly, while the average pore diameter was reduced. This suggests that the strong interaction among Fe and Cu species hindered the growth of particles, thereby enhancing the dispersion of active components, and more active sites could be provided for CO oxidation. In addition, the difference in specific surface area, average pore diameter, and total pore volume between CuFeOx and CuFeOx-100 catalysts could be negligible, which demonstrates that the influence of H2 activation on the surface structure of the catalyst was less. The CuFeOx and CuFeOx-100 catalysts had similar surface structure, while the activity of the CuFeOx-100 catalyst was obviously higher than the CuFeOx catalyst. This suggests that the surface physical properties were not the determining factor for the increased activity of the CuFeOx-100 catalyst. The morphological features of the catalysts were investigated with SEM, as displayed in Figure S2. The catalysts existed in irregular flakes and particles of various sizes. The surface morphology of the catalysts had no obvious change after H2 activation.

Table 1.
Catalysts’ BET specific surface area, pore volume, and average pore size.
2.3. Surface Chemical States
The effect of H2 activation on surface chemical composition on the catalysts was determined with XPS, as depicted in Figure 3. In addition, the atomic percentages of elements were calculated from the areas of the fitted peaks and were presented in Table 2. The Cu 2p of catalysts are depicted in Figure 3a, which could be separated into Cu 2p1/2 and Cu 2p3/2, respectively [24]. The two peaks of Cu 2p3/2 located at 933.4 and 934.8 eV corresponded to Cu+ and Cu2+ species, respectively [1,25]. In addition, the shake-up satellite peak at 942.3 eV was assigned to CuO. According to previous studies [9,26,27], the Cu+ species was the main adsorption and activation sites of CO, playing a crucial role in the CO oxidation process. It could be noted that the proportion of the CuFeOx catalyst was 10.2%, which was much lower than that of 17.5% on the CuFeOx-100 catalyst. This confirmed that the H2 activation could promote the generation of Cu+ species, thus increasing the CO conversion.
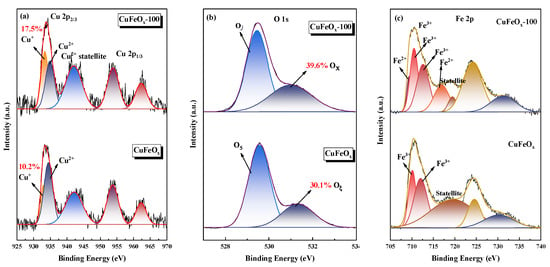
Figure 3.
XPS spectra of catalysts: (a) Cu 2p; (b) O 1s; and (c) Fe 2p.
Table 2.
XPS data of the catalysts.
The O 1s spectra are depicted in Figure 3b. There were two fitted peaks at 529.5 and 531.3 eV, which were attributed to the surface lattice oxygen species (O2−) and chemisorbed oxygen species (O2-), marked as Oβ and Oα, respectively [28]. Generally, the Oα species was formed on oxygen vacancy with higher mobility than Oβ species [4,29,30]. In addition, it was associated with charge unbalance, which was beneficial to the improvement of redox cycle, and an increase in low-temperature activity. It was found that the concentration of Oα species increased from 30.1% for the CuFeOx catalyst to 39.6% for the CuFeOx-100 catalyst, which implied that the H2 activation could accelerate the generation of oxygen vacancy. In addition, it was observed that the peak Oβ and Oα on the CuFeOx-100 catalyst shifted towards lower binding energy compared with the CuFeOx catalyst. This may be due to the H2 activation weakening the electron-withdrawing ability of Cu and/or Fe species and increasing the surrounding electron cloud density, which resulted in the decrease in binding energy.
For Fe 2p in Figure 3c, two main peaks at 710.8 and 724.0 eV for Fe 2p3/2 and Fe 2p1/2 were found on the CuFeOx catalyst [31,32], and a satellite peak at 719.4 eV for Fe3+ species was observed [33,34]. As for the CuFeOx-100 catalyst, peaks at 709.3 and 716.8 eV associated with Fe2+ species appeared, which confirmed that the H2 activation promoted the Fe3+ species reduced to Fe2+ species, in agreement with the XRD results. Previous studies had illustrated that the O2 could be dissociated to atomic O without a barrier by Fe2+ species [35,36]. The activity of CO oxidation was well correlated with the existence and concentration of Fe2+ species on the catalyst. Additionally, the coexistence of Fe2+ and Fe3+ species could establish the redox equilibrium of Cu+ + Fe3+↔Cu2+ + Fe2+ on the catalyst surface, which could facilitate the electron transfer between active components to improve the redox cycle. In summary, the H2 activation generated Fe2+ species, which effectively increased the activity of the CuFeOx catalyst.
2.4. Redox Properties
The H2-TPR was performed to measure the reducibility of catalysts and is shown in Figure 4a. As for the CuFeOx catalyst, the strong peak at 261.0 °C could be assigned to the coreduction of CuO to metallic Cu and Fe2O3 to Fe3O4, while the broad peaks in the range of 400 to 800 °C belonged to the sequential reduction of Fe3O4 to FeO and then to Fe. Compared to the pure Fe2O3 catalyst [12], the reduction temperature of Fe2O3 to Fe3O4 decreased to below 300 °C, indicating that the synergistic effect between Cu and Fe species significantly improved the redox capacity. That is, the Cu species as the active sites facilitated the overflow of H2 to Fe2O3, thus accelerating the reduction to Fe3O4 at low temperature. Additionally, a shoulder peak appeared on the H2-activated catalyst at lower temperature, which may be ascribed to the reduction of CuO or/and Cu2O to metallic Cu. This further confirmed that the H2 activation promoted the generation of more Cu2O. Moreover, the initial reduction temperature on the CuFeOx catalyst was 120 °C, while it shifted to 98 °C on the CuFeOx-100 catalyst. The improvement in initial redox property could effectively increase the catalytic activity at low temperature. The Fe species reduced at 400–800 °C had almost no catalytic activity, and activity tests were performed at low temperatures, so it was not involved in the reaction processes. The H2 consumption corresponding to the reduction peak below 400 °C was determined based on its integrated area, as depicted in Figure 4b. The total H2 consumption on the CuFeOx catalyst slightly decreased from 1645.2 μmol/g to 1639.6 and 1555.7 μmol/g on the CuFeOx-100 and CuFeOx-150 catalysts, respectively, while it significantly decreased to 1390.6 μmol/g on the CuFeOx-200 catalyst. The H2 activation at high temperature resulted in part of the active metal oxides being reduced to a metallic state, which may be the main reason for the decline in CO conversion of the CuFeOx-200 catalyst.
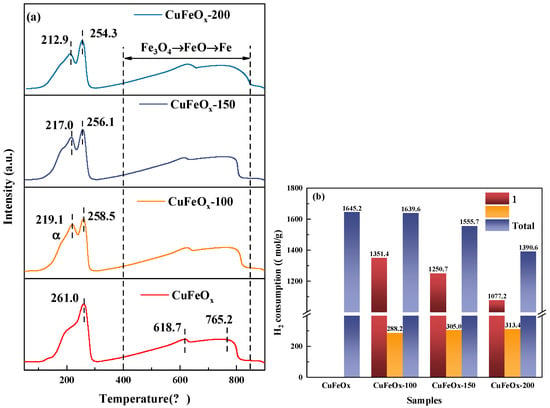
Figure 4.
H2-TPR profiles (a) and the amount of H2 consumption (b) of catalysts.
2.5. Temperature-Programmed Studies
The effect of H2 activation on the desorption of oxygen species was studied by O2-TPD, as exhibited in Figure 5. In Figure 5a, the desorption curves are categorized into three temperature regions [2,37]: (1) The surface chemisorbed oxygen (<200 °C), which is associated with the oxygen vacancy; (2) surface lattice oxygen (200–500 °C); and (3) bulk lattice oxygen (>500 °C). It can be noted that the desorption of surface chemisorbed oxygen on the H2-activated catalysts shifted to a lower-temperature position, which implies that its transfer ability was enhanced, and there was favorability for low-temperature CO oxidation. Additionally, the concentration of oxygen species was calculated by the peak area, as displayed in Figure 5b. Compared with the CuFeOx catalyst (15.0%), the proportion of surface chemisorbed oxygen species on the CuFeOx-100 (21.4%) and CuFeOx-150 (17.4%) catalysts obviously increased, which confirms that the H2 activation at a suitable temperature could produce more defects on the catalyst. In addition, the bulk lattice oxygen decreased from 19.2% on the CuFeOx catalyst to 16.5% and 13.3% on the CuFeOx-100 and CuFeOx-150 catalysts, respectively. This implied an enhancement in the migration of oxygen species from deeper layers to the surface, thereby improving catalytic performance effectively. It can be noted that the percentage of surface chemisorbed oxygen species declined on the CuFeOx-200 catalyst (12.7%) compared with the CuFeOx catalyst, which may be due to the overreduction of oxides to the metallic state, which resulted in the decrease in oxygen vacancy.
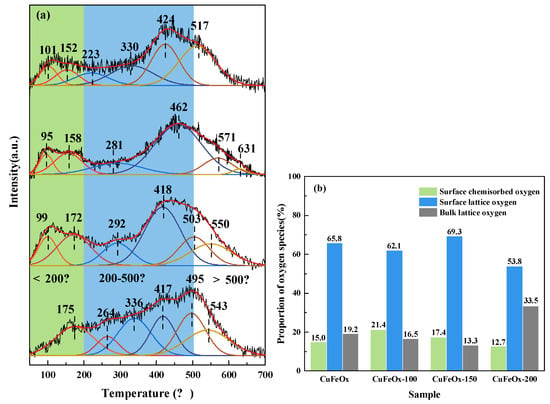
Figure 5.
(a) O2−TPD profiles and (b) proportion of oxygen species of catalysts.
2.6. Reaction Intermediates Analysis
2.6.1. In Situ DRIFT Spectra of CO Adsorption, Ar and O2 Purging
To investigate the active intermediates involved in the CO oxidation reaction, in situ DRIFT was performed. Firstly, the samples were purged in 1% CO/Ar from 25 to 175 °C with temperature rate of 5 min/°C, then switched with pure Ar for 5 min, and finally injected pure 15% O2/Ar with 5 min. As seen in Figure 6(a1) for the CuFeOx catalyst, the peaks at 2110 and 2172 cm−1 associated with Cu+-CO species could be found [38,39], and the intensity of Cu+-carbonyl species (Cu+-CO) strengthened with the increase in temperature initially and then decreased (Figure 6(c1)), which indicates that the Cu+ species was the main active site for CO adsorption and activation. Additionally, the peaks assigned to carbonate species (1014, 1103, and 1361 cm−1) [40], bicarbonate species (838 and 1245 cm−1), [41] and monodentate carbonates (1441 cm−1) [42] also declined with increasing temperature and were then enhanced, while the peak at 1605 cm−1, ascribed to bicarbonate species, decreased [43]. This illustrates that the carbonate species were the active intermediates in the CO oxidation process. In addition, the weak peaks at 2320 and 2356 cm−1 belonging to CO2 were found even at 25 °C [40], which proves that the CO oxidation can be conducted at ambient temperature, consistent with the results of activity testing. The catalysts were pretreated with pure Ar for 60 min at 300 °C, and the surface adsorbed oxygen was removed completely. Therefore, the CO2 originated from the reaction between the Cu+-CO species and surface lattice oxygen, and the process followed the MvK mechanism. As described in Figure 6(a2), the intensity of Cu+-CO species declined after purging with Ar at 175 °C. When O2 was introduced (Figure 6(a3)), the intensity of the Cu+-CO species declined and the peak at 1605 cm−1 (bicarbonate species) was enhanced, which illustrates that the consumed active oxygen species can be replenished with gaseous phase O2. As for the CuFeOx-100 catalyst in Figure 6(b1), the type of species was similar to the CuFeOx catalyst, while the intensity of the Cu+-CO and carbonate species was stronger (Figure 6(c1,c2)). This demonstrates that the quantities of Cu+ and active oxygen species on the CuFeOx-100 catalyst were higher than those of the CuFeOx catalyst, in agreement with the XPS and O2-TPD analysis. Furthermore, the intensity of Cu+-CO species began to reduce after 125 °C both for CuFeOx and CuFeOx-100 catalyst (Figure 6(c1,c2)), which suggests that the reaction had entered a phase of rapid progress. It was found that the reduction range of intensity for Cu+-CO species on the CuFeOx-100 catalyst was larger than that on the CuFeOx catalyst after 125 °C (Figure 6(c1)), which indicates that the Cu+-CO species was more active.
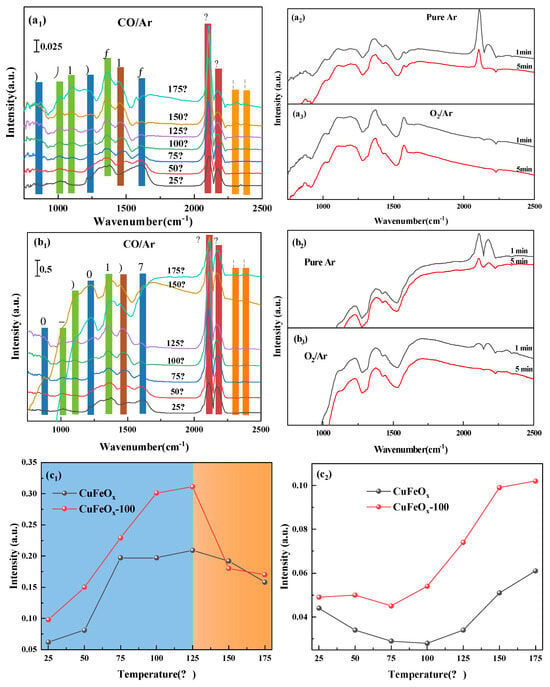
Figure 6.
In situ DRFITS spectra of CO adsorption, pure Ar purging, and pure O2 purging: CuFeOx (a1–a3) and CuFeOx−100 (b1–b3) catalysts. Behavior of Cu+−CO species at 2110 cm−1 (c1) and carbonate species at 1361 cm−1 (c2).( ♣ bicarbonate species, ♦ carbonate species, ♥ monodentate carbonates, ▲ Cu+-CO species, ■ CO2).
2.6.2. CO+O2 Coadsorption
The in situ DRFITS of CO+O2 coadsorption is displayed in Figure 7. As depicted in Figure 7a,b, the species and variation trend of intensity with temperature was similar in CO atmosphere both for CuFeOx and CuFeOx-100 catalysts. As illustrated in Figure 7c, the intensity of the Cu+-CO species on the CuFeOx and CuFeOx-100 catalysts was decreased to 100 and 75 °C compared with CO atmosphere, respectively, which proved that the O2-riched atmosphere was conducive to the CO oxidation. In addition, the Cu+-CO species on the CuFeOx-100 catalyst was stronger than the CuFeOx catalyst at 25 °C and lower at higher temperature, which further confirms that the Cu+-CO species with H2 activation was more active. As displayed in Figure 7d, the carbonate species both on CuFeOx and CuFeOx-100 catalysts was lower than in CO atmosphere, which implies that the presence of O2 can promote the decomposition of carbonate species. In contrast to the CO atmosphere (Figure 6(c2)), the strength of carbonate species on the CuFeOx-100 catalyst was higher than that on the CuFeOx catalyst (Figure 7d), indicating that carbonate species were more easily decomposed on the CuFeOx-100 catalyst in reaction to atmosphere. Interestingly, it was found that the intensity of the carbonate species started to decrease and was accompanied by the generation of CO2 sharply after 175 °C, which suggests that the CO oxidation process was conducted through the L-H mechanism.
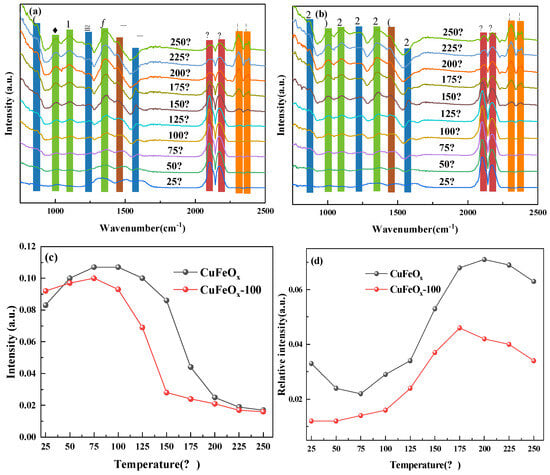
Figure 7.
In situ DRFITS spectra of CO+O2 coadsorption on CuFeOx (a) and CuFeOx−100 (b) catalyst. Behavior of Cu+−CO species at 2110 cm−1 (c) and carbonate species at 1361 cm−1 (d). ( ♣ bicarbonate species, ♦ carbonate species, ♥ monodentate carbonates, ▲ Cu+-CO species, ■ CO2).
3. Discussion
Based on the above results, the H2 activation with appropriate temperature can efficiently increase the activity of CuFeOx catalyst for CO oxidation (Figure 1a). XRD analysis (Figure 2) showed that the pure Fe2O3 had good crystallization and large grain size, and the addition of Cu species can decrease the crystallization. In addition, some peaks attributed to CuO disappeared with the H2 pretreatment at 100 and 150 °C, confirming that the CuO was reduced to Cu2O. After pretreatment at 200 °C, the metal Cu was produced. The specific surface area increased slightly with the introduction of Cu species (Table 1), which could promote the dispersion of active components and adsorption of reaction gas. Following H2 activation, there was minimal change observed in the specific surface area, average pore diameter, and pore volumes of the catalysts. This suggests that the surface physical structure may not be the primary factor contributing to the improvement in catalytic performance. Furthermore, the XPS results (Figure 4) illustrate that the percentage of Cu+ species increased on the H2-activated catalyst, which could offer more active sites for CO, and more Oα species were produced with the H2 pretreatment, which proved that more oxygen vacancy was formed. This could facilitate the activation of oxygen species and the circulation of the reaction. In addition, the Fe2+ species was generated, in agreement with the XRD results, which was beneficial to the dissociation of O2 to oxygen atoms. Moreover, the H2-TPR analysis indicated that the moderate H2 activation could enhance the reducibility of the CuFeOx catalyst, which could increase the catalytic activity for CO oxidation. However, the H2 activation at high temperature led to the decrease in H2 consumption and redox property. The O2-TPD results indicate that H2 activation promoted the migration of oxygen species from deeper layers to the surface. As a result, the concentration of both surface chemisorbed oxygen and surface lattice oxygen increased, potentially providing more active oxygen species for CO oxidation.
In situ DRIFTS experiments were performed to investigate the reaction mechanism. The results indicate that the predominant reaction pathway followed the MvK mechanism at low temperatures (<175 °C). The specific procedure was as follows: (1) Firstly, the CO combined with Cu+ to form Cu+-CO species (Equation (1)). The H2 activation increased the quantity of Cu+ species, which enhanced the reaction procedure of Equation (1). Subsequently, the Cu+-CO species reacted with surface lattice oxygen to directly generate CO2 and leave the oxygen vacancies, as displayed in Equation (2). Finally, the oxygen vacancies were resupplemented by gas O2, and the cyclic process was completed (Equation (3)).
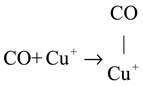
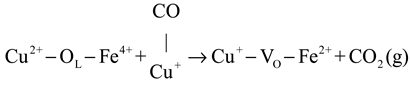
CO oxidation on the CuFeOx catalyst at high temperatures (>175 °C) proceeded via both the MvK and the L-H mechanism, with the latter described in Equations (4)–(6). The gas O2 was captured by oxygen vacancy and formed chemisorbed oxygen species, as shown in Equation (4). The H2 soft pretreatment increased the concentration of oxygen vacancy on the CuFeOx catalyst, which promoted the process of Equation (4). Then, the chemisorbed oxygen species reacted with Cu+-CO species (Equation (1)) to generate carbonate species, as exhibited in Equation (5). The carbonate species decomposed into CO2, as described in Equation (6). The H2 activation accelerated the decomposition of carbonate species, which alleviated the accumulation of carbonate occupying the active sites and improved the efficiency of CO oxidation. Based on the above, a possible reaction model is proposed and is illustrated in Scheme 1.
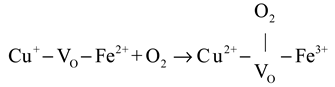
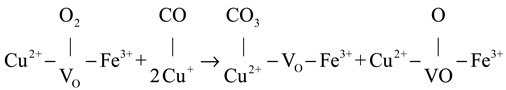
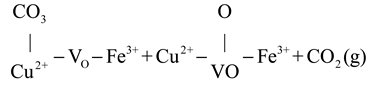
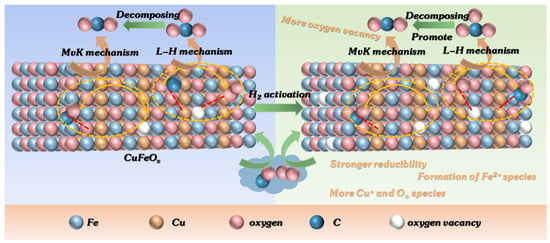
Scheme 1.
Mechanism model of CO oxidation on CuFeOx and CuFeOx-100 catalysts.
4. Materials and Methods
4.1. Preparation of Catalyst
The catalysts were synthesized using the coimpregnation method. Cu(NO3)2·3H2O and Fe(NO3)3·9H2O were dissolved in ionized water with the molar rate of Cu:Fe = 1:2. Then, the solution was stirred by the magnetic force and heating in a water bath until the water was completely evaporated. Subsequently, the solid was dried for 24 h at 80 °C. Finally, it was calcined at 450 °C for 4 h. The obtained sample was donated as a CuFeOx catalyst. In the H2 activation stage, the tube furnace was vacuumed, and then CuFeOx was heated to 100 °C under a N2 protective atmosphere. Finally, 10% H2/He was pretreated for 0.5 h. The obtained sample was marked as the CuFeOx-100 catalyst, and the samples pretreated with 10% H2/He at 150 and 200 °C were marked as the CuFeOx-150 and CuFeOx-200 catalyst, respectively.
4.2. Catalytic Performance Test
The catalytic activity was evaluated on a fixed-bed quartz reactor with an inner diameter of 8 mm, and 300 mg of catalyst was used for each test. A type K thermocouple was inserted into the catalyst bed to monitor the reaction temperature. The composition of the simulated gas mixture was 1% CO, 10% O2, and N2 balance. The total gas flow rate was maintained at 300 mL/min, while the reaction temperature was incrementally increased from 25 to 300 °C at a heating rate of 5 °C/min. Test points were recorded at 25 °C intervals, and the CO concentration at each point remained constant for 30 min. Gas concentrations at the inlet and outlet were measured using an online gas chromatograph. The CO conversion rate was determined by employing Equation (7):
where the [CO]in and [CO]out represent the concentration of CO at the inlet and outlet, respectively.
4.3. Catalyst Characterization
The physicochemical properties of catalysts were charactered with XRD, BET, XPS, H2-TPR, O2-TPD, and in situ DRIFTS, and can be found in the supporting information (SI).
5. Conclusions
In this work, the effect of H2 activation on the performance of the CuFeOx catalyst for low-temperature CO oxidation was investigated. It was found that the soft H2 pretreatment could effectively improve the activity of the CuFeOx catalyst, and the catalyst activated at 100 °C displayed the highest performance, which corresponded to 99.6% CO conversion at 175 °C. The influence of H2 activation on the surface physical structure was negligible. The H2 activation enhanced the reducibility of CuFeOx catalyst, thus improving the low-temperature activity. In addition, the H2 activation generated the Fe2+ species, which was beneficial to the dissociation of O2. Furthermore, the amount of Oα species and oxygen vacancy increased after H2 activation, which increased the cycle efficiency of the CO oxidation process. Moreover, the migration of oxygen species from the deep layer to the surface was promoted, which could provide more active oxygen species for CO oxidation. The in situ DRFITS demonstrated that the CO oxidation pathway mainly involved the MvK mechanism at low temperature, and both MvK and L-H mechanisms at high temperature. The Cu+-CO and carbonate species were the main active intermediates, and the H2 activation increased the amount of Cu+ species and accelerated the decomposition of carbonate species, which increased the activity of the CuFeOx catalyst.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29143347/s1, Figure S1: N2 adsorption–desorption isotherms. Figure S2: The SEM profiles of catalysts.
Author Contributions
Methodology, Y.S. and S.R.; Software, Z.S.; Investigation, Z.S., X.X. and Hao Meng; Resources, Y.S. and W.N.; Data curation, Y.S. and H.M.; Writing—original draft, Z.S.; Writing—review & editing, S.R.; Visualization, X.X. and W.N.; Supervision, W.N.; Project administration, H.M. and S.R.; Funding acquisition, X.X. All authors have read and agreed to the published version of the manuscript.
Funding
The present work was financially supported by Shaanxi Province Innovation Capability Support Plan Project (2023-CX-TD-53), Shaanxi Province Key Research and Development Plan Project (2024GX-YBXM-422) and Cultivation of excellent doctoral dissertation (2023XYBPY0011).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Shen, Z.H.; Xing, X.D.; Wang, S.X.; Zheng, Z.Y.; Lv, M. Low temperature CO oxidation from sintering flue gas on CuO-CeO2/AC-Fe catalyst. Catal. Today 2023, 423, 113988. [Google Scholar] [CrossRef]
- Wang, J.; Xing, Y.; Su, W.; Li, K.; Zhang, W. Bifunctional Mn-Cu-CeOx/γ-Al2O3 catalysts for low-temperature simultaneous removal of NOx and CO. Fuel 2022, 321, 124050. [Google Scholar] [CrossRef]
- Xu, J.C.; Wang, S.; Zhang, C.Y.; Zhang, J.; Xu, L.H.; Shi, Y.; Xu, G.Q.; Yao, S.L. Exploring the role of oxygen species on /3-and γ-MnO2 catalyst under in-plasma catalysis configuration at different temperature in CO oxidation using operand TPR-DRIFTS-MS. J. Alloys Compd. 2024, 978, 173462. [Google Scholar] [CrossRef]
- Waikar, J.; More, P. Oxygen deficient Ce doped CO supported on alumina catalyst for low-temperature CO oxidation in presence of H2O and SO2. Fuel 2023, 331, 125880. [Google Scholar] [CrossRef]
- Cai, J.Y.; Yu, Z.H.; Fan, X.; Li, J. Effect of TiO2 calcination pretreatment on the performance of Pt/TiO2 catalyst for CO Oxidation. Molecules 2022, 27, 3875. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.H.; Xing, X.D.; Wang, S.X.; Ren, S.; Lv, M.; Zheng, Z.Y.; Jiang, X. Removal of CO in flue gas by catalytic oxidation: A review. Z. Phys. Chem. 2024, 238. [Google Scholar] [CrossRef]
- Feng, C.L.; Liu, X.L.; Zhu, T.Y.; Tian, M.K. Catalytic oxidation of CO on noble metal-based catalysts. Environ. Sci. Pollut. Res. 2021, 28, 24847–24871. [Google Scholar] [CrossRef] [PubMed]
- Khalafallah, D.; Miao, J.; Zhi, M.; Hong, Z. Structuring graphene quantum dots anchored CuO for high-performance hybrid supercapacitors. J. Taiwan Inst. Chem. Eng. 2021, 122, 168–175. [Google Scholar] [CrossRef]
- Xu, L.L.; Yang, X.; Shi, Y.Y.; Chen, M.D.; Xue, Y.Y.; Wu, C.E.; Qiu, J.; Cheng, G.; Wang, N.; Xu, J.X.; et al. CO oxidation over the Cu2O/CuO hollow sphere heterojunction catalysts with enhanced low-temperature activities. Int. J. Hydrogen Energy 2023, 48, 24845–24859. [Google Scholar] [CrossRef]
- Cui, Y.; Song, H.K.; Shi, Y.Y.; Ge, P.X.; Chen, M.D.; Xu, L.L. Enhancing the low-temperature CO oxidation over CuO-based α-MnO2 nanowire catalysts. Nanomaterials 2022, 12, 2083. [Google Scholar] [CrossRef]
- Gong, S.W.; Zhu, G.Q.; Xie, X.Q.; Rao, F.; Zhu, L.J.; An, Y.R.; Shi, X.J.; Huang, Y.; Liu, P.; Hojamberdiev, M. Engineering the CuO/MnO2 interface with multivalent conversion for low-temperature and efficient CO oxidation. Appl. Surf. Sci. 2024, 652, 159326. [Google Scholar] [CrossRef]
- Bulavchenko, O.A.; Pochtar, A.A.; Gerasimov, E.Y.; Fedorov, A.V.; Chesalov, Y.A.; Saraev, A.A.; Yakovlev, V.A.; Kaichev, V.V. Chemical and texture promoters in Cu-Fe-Al oxide nanocomposite catalysts for combustion of solid fuel gasification products. Appl. Catal. A 2020, 590, 117364. [Google Scholar] [CrossRef]
- Wang, L.P.; Li, Q.; Liu, X.; Li, C.; Zhao, Z.Z.; Diao, S.T.; Cao, D.F.; Xiang, D.C.; Wu, C.N.; Liu, K. Improved CO-PROX selectivity of CuO/CeO2 catalysts by decorating with lanthanum via surface Cuξ+ redox site. Appl. Surf. Sci. 2024, 649, 159087. [Google Scholar] [CrossRef]
- Kerkar, R.D.; Salker, A.V. Highly active nano-composite of cobalt-copper-manganese oxides for room temperature CO oxidation. Appl. Nanosci. 2021, 11, 2861–2867. [Google Scholar] [CrossRef]
- Zhao, F.; Shi, Y.Y.; Xu, L.L.; Chen, M.D.; Xue, Y.Y.; Wu, C.E.; Qiu, J.; Cheng, G.; Xu, J.X.; Hu, X. Designing highly efficient Cu2O-CuO heterojunction CO oxidation catalysts: The roles of the support type and Cu2O-CuO interface effect. Nanomaterials 2022, 12, 3020. [Google Scholar] [CrossRef]
- Rezaei, P.; Rezaei, M.; Meshkani, F. Low temperature CO oxidation over mesoporous iron and copper mixed oxides nanopowders synthesized by a simple one-pot solid-state method. Process Saf. Environ. Prot. 2018, 119, 379–388. [Google Scholar] [CrossRef]
- Amini, E.; Rezaei, M. Preparation of mesoporous Fe-Cu mixed metal oxide nanopowder as active and stable catalyst for low-temperature CO oxidation. Chin. J. Catal. 2015, 36, 1711–1718. [Google Scholar] [CrossRef]
- Yeste, M.P.; Vidal, H.; García-Cabeza, A.L.; Hernández-Garrido, J.C.; Guerra, F.M.; Cifredo, G.A.; González-Leal, J.M.; Gatica, J.M. Low temperature prepared copper-iron mixed oxides for the selective CO oxidation in the presence of hydrogen. Appl. Catal. A 2018, 552, 58–69. [Google Scholar] [CrossRef]
- Shen, Z.H.; Xing, X.D.; She, Y.; Wang, S.X.; Lv, M.; Li, J.X.; Li, H.Z. Revealing of K and SO2 poisoning mechanism on CuCeOx catalyst for low-temperature CO oxidation. Chem. Eng. J. 2024, 485, 149373. [Google Scholar] [CrossRef]
- Shen, Z.H.; Xing, X.D.; Wang, S.X.; Lv, M.; Zheng, Z.Y.; Li, J.X.; Li, H.Z. High activity of CuO/#-Fe2O3 for low temperature CO oxidation: Effect of support crystal types in catalyst design. J. Energy Inst. 2023, 110, 101339. [Google Scholar]
- Wang, L.; Pu, C.; Xu, L.; Cai, Y.; Guo, Y.; Guo, Y.; Lu, G. Effect of supports over Pd/Fe2O3 on CO oxidation at low temperature. Fuel Process. Technol. 2017, 160, 152–157. [Google Scholar] [CrossRef]
- Wang, W.W.; Du, P.P.; Zou, S.H.; He, H.Y.; Wang, R.X.; Jin, Z.; Shi, S.; Huang, Y.Y.; Si, R.; Song, Q.S.; et al. Highly dispersed copper oxide clusters as active species in copper-ceria catalyst for preferential oxidation of carbon monoxide. ACS Catal. 2015, 5, 2088–2099. [Google Scholar] [CrossRef]
- Niu, L.; Liu, X.; Wen, X.; Yang, Y.; Xu, J.; Li, Y. Effect of potassium promoter on phase transformation during H2 pretreatment of a Fe2O3 Fischer Tropsch synthesis catalyst precursor. Catal. Today 2020, 343, 101–111. [Google Scholar] [CrossRef]
- Cam, T.S.; Omarov, S.O.; Chebanenko, M.I.; Sklyarova, A.S.; Nevedomskiy, V.N.; Popkov, V.I. One step closer to the low-temperature CO oxidation over non-noble CuO/CeO2 nanocatalyst: The effect of CuO loading. J. Environ. Chem. Eng. 2021, 9, 105373. [Google Scholar] [CrossRef]
- Khalafallah, D.; Zhi, M.; Hong, Z. Rational engineering of hierarchical mesoporous CuxFeySe battery-type electrodes for asymmetric hybrid supercapacitors. Ceram. Int. 2021, 47, 29081–29090. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Fan, L.P.; Liao, W.Q.; Zhao, F.Y.; Tang, C.; Zhang, J.; Feng, M.; Lu, J.Q. Structure sensitivity of CuO in CO oxidation over CeO2-CuO/Cu2O catalysts. J. Catal. 2022, 405, 333–345. [Google Scholar] [CrossRef]
- Tang, X.Y.; Tariq, N.U.; Wang, J.C.; Cheng, Y.L.; Wang, Y.L.; Yuan, Z.G.; Liu, B.D. CuO/TiO2/Ti monolithic catalysts for low-temperature CO oxidation. ACS Appl. Nano Mater. 2023, 7, 809–817. [Google Scholar] [CrossRef]
- Gong, X.F.; Xu, J.C.; Zhang, T.T.; Sun, Y.; Fang, S.Y.; Li, N.; Zhu, J.L.; Wu, Z.L.; Li, J.; Gao, E.H.; et al. DRIFTS-MS investigation of low-temperature CO oxidation on Cu-doped manganese oxide prepared using nitrate aerosol decomposition. Molecules 2023, 28, 3511. [Google Scholar] [CrossRef]
- Xu, J.C.; Wu, Z.L.; Gao, E.R.; Zhu, J.L.; Yao, S.L.; Li, J. Revealing the role of oxygen vacancies on ?-MnO2 of different morphologies in CO oxidation using operando DRIFTS-MS. Appl. Surf. Sci. 2023, 618, 156643. [Google Scholar] [CrossRef]
- Shan, R.T.; Sheng, Z.T.; Hu, S.; Xiao, H.F.; Zhang, Y.H.; Zhang, J.H.; Wang, L.; Zhang, C.B.; Li, J.L. Enhancing oxidation reaction over Pt-MnO2 catalyst by activation of surface oxygen. J. Environ. Sci. 2023, 134, 117–125. [Google Scholar] [CrossRef]
- Teng, W.X.; Li, J.L.; Dai, X.Y.; Chen, Y.F.; Wu, H.M.; Liu, W.Z.; Ren, S.; Yang, J.; Liu, Q.C. Promoting the NH3-SCR performance of Fe2O3-TiO2 catalyst through regulation of the exposed crystal facets of Fe2O3. Appl. Surf. Sci. 2024, 647, 158938. [Google Scholar] [CrossRef]
- Wang, G.W.; Zhang, Y.K.; Lu, J.M.; Liu, Y.J.; Yang, M.; Peng, G.J.; Jia, L.J.; Wang, H.M.; Xia, F.T.; Zhang, Q.L. The improving effect of SO42- on environmental-friendly SO42-/a-Fe2O3 catalyst for NH3-SCR. J. Energy Inst. 2023, 110, 101347. [Google Scholar] [CrossRef]
- Li, X.Y.; Chen, J.; Lu, C.M.; Luo, G.Q.; Yao, H. Performance of Mo modified γ-Fe2O3 catalyst for selective catalytic reduction of NOx with ammonia: Presence of arsenic in flue gas. Fuel 2021, 294, 120552. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Li, J.; Cai, J.Y.; Li, S.Y.; Fan, X.; Song, L.Y.; Guo, R.X.; Liu, J.S. Enhancement of low-temperature activity of γ-Fe2O3-modified V2O5-MoO3/TiO2 catalysts for selective catalytic reduction of NOx with NH3. J. Environ. Chem. Eng. 2024, 12, 112589. [Google Scholar] [CrossRef]
- Fu, Q.; Li, W.X.; Yao, Y.; Liu, H.; Su, H.Y.; Ma, D.; Gu, X.K.; Chen, L.; Wang, Z.; Zhang, H.J.S. Interface-confined ferrous centers for catalytic oxidation. Science 2010, 328, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Qiao, B.; Liu, J.; Huang, Y.; Wang, A.; Li, L.; Zhang, W.; Allard, L.F.; Wang, X.; Zhang, T. Design of a highly active Ir/Fe(OH)x catalyst: Versatile application of Pt-group metals for the preferential oxidation of carbon monoxide. Angew. Chem., Int. Ed. 2012, 51, 2920–2924. [Google Scholar] [CrossRef] [PubMed]
- Gui, R.; Xiao, J.; Gao, Y.; Li, Y.; Zhu, T.; Wang, Q. Simultaneously achieving selective catalytic reduction of NOx with NH3 and catalytic oxida tion of CO with O2 over one finely optimized bifunctional catalyst Mn2Cu1Al1Ox at low temperatures. Appl. Catal., B 2022, 306, 121104. [Google Scholar] [CrossRef]
- Qiu, Z.H.; Guo, X.L.; Mao, J.X.; Zhou, R.X. Insights into the structure-performance relationship of CuOx-CeO2 catalysts for preferential oxidation of CO: Investigation on thermally induced copper migration process. Appl. Surf. Sci. 2022, 600, 154100. [Google Scholar] [CrossRef]
- Hou, X.L.; Zhao, Q.Y.; Liu, X.L.; Chong, M.B.; Cheng, D.G.; Chen, F.Q.; Zhan, X.L. CeO2 crystal plane engineering to modulate CuO-CeO2 interactions and modify the CO-PROX reaction pathway. Chem. Eng. J. 2024, 480, 147982. [Google Scholar] [CrossRef]
- Zeng, Y.; Rong, W.; Zhang, S.; Zhong, Q.; Zhong, Z. Ti species regulated CuO/CeO2 catalyst for efficient simultaneously NH3-SCR denitration and CO oxidation under oxygen-rich conditions. Fuel 2024, 358, 130277. [Google Scholar] [CrossRef]
- Wang, L.; Peng, H.; Shi, S.L.; Hu, Z.; Zhang, B.Z.; Ding, S.M.; Wang, S.H.; Chen, C. Metal-organic framework derived hollow CuO/CeO2 nano-sphere: To expose more highly dispersed Cu-O-Ce interface for enhancing preferential CO oxidation. Appl. Surf. Sci. 2022, 573, 151611. [Google Scholar] [CrossRef]
- Lu, J.C.; Wang, J.; Zou, Q.; He, D.D.; Zhang, L.M.; Xu, Z.Z.; He, S.F.; Luo, Y.M. Unravelling the nature of the active species as well as the doping effect over Cu/Ce-based catalyst for carbon monoxide preferential oxidation. ACS Catal. 2019, 9, 2177–2195. [Google Scholar] [CrossRef]
- Liu, Q.; Mi, J.; Chen, X.; Wang, S.; Chen, J.; Li, J. Effects of phosphorus modification on the catalytic properties and performance of CuCeZr mixed metal catalyst for simultaneous removal of CO and NOx. Chem. Eng. J. 2021, 423, 130228. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).