
Visible-Light-Mediated Ring-Opening Geminal Dibromination of Alkenes via Alkoxy Radicals Enabled by Electron Donor–Acceptor Complex


Abstract
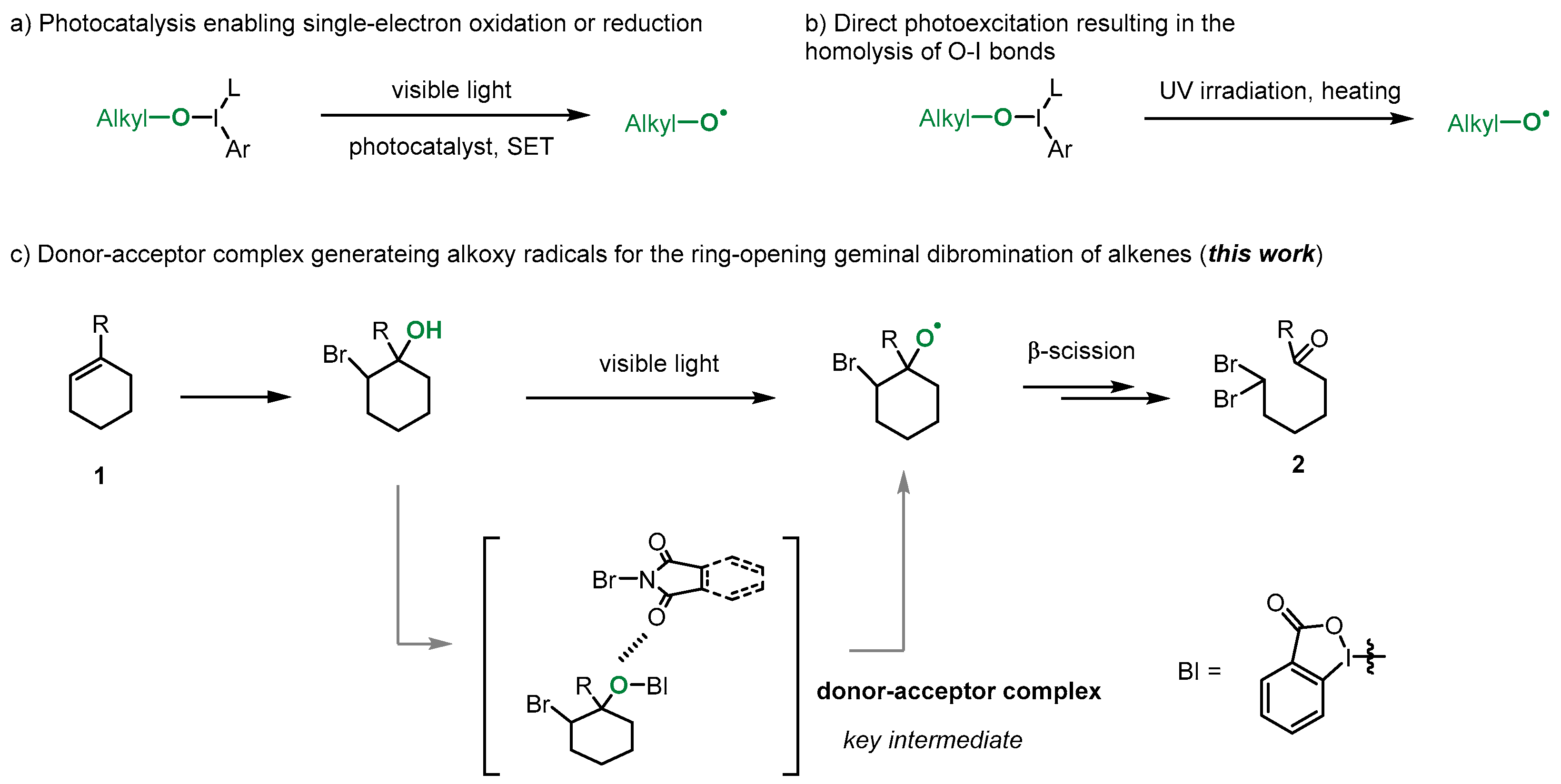
1. Introduction
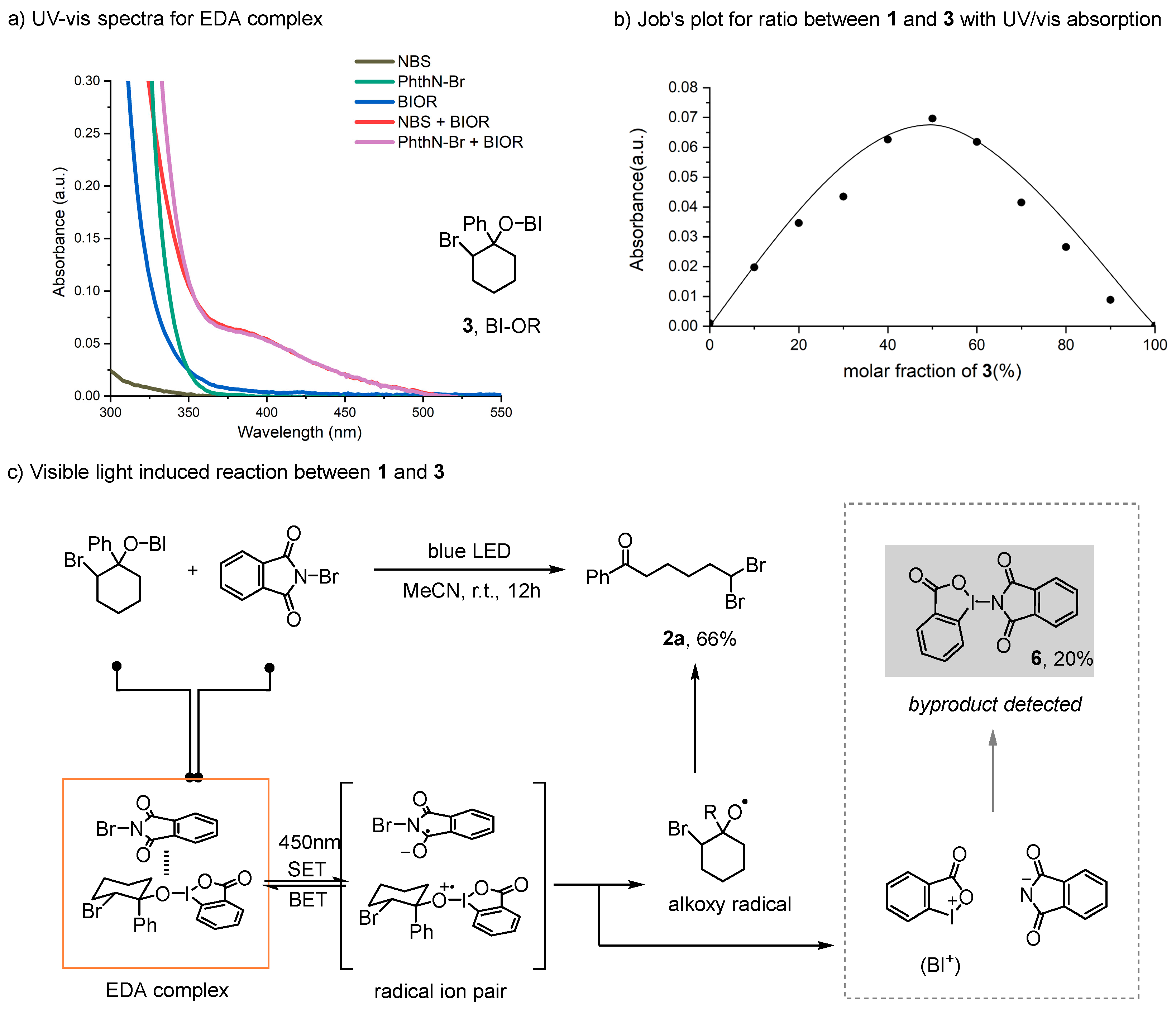
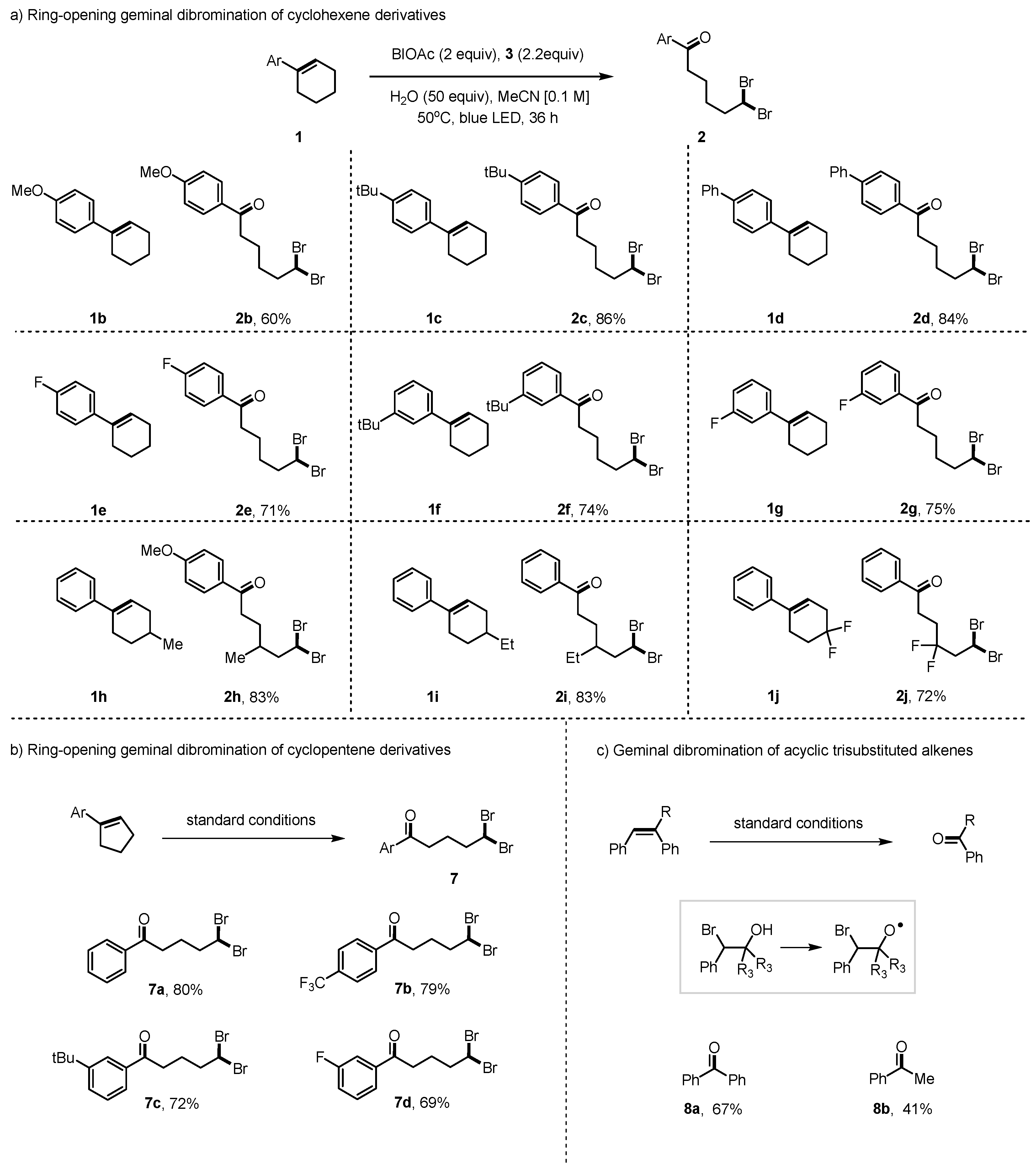
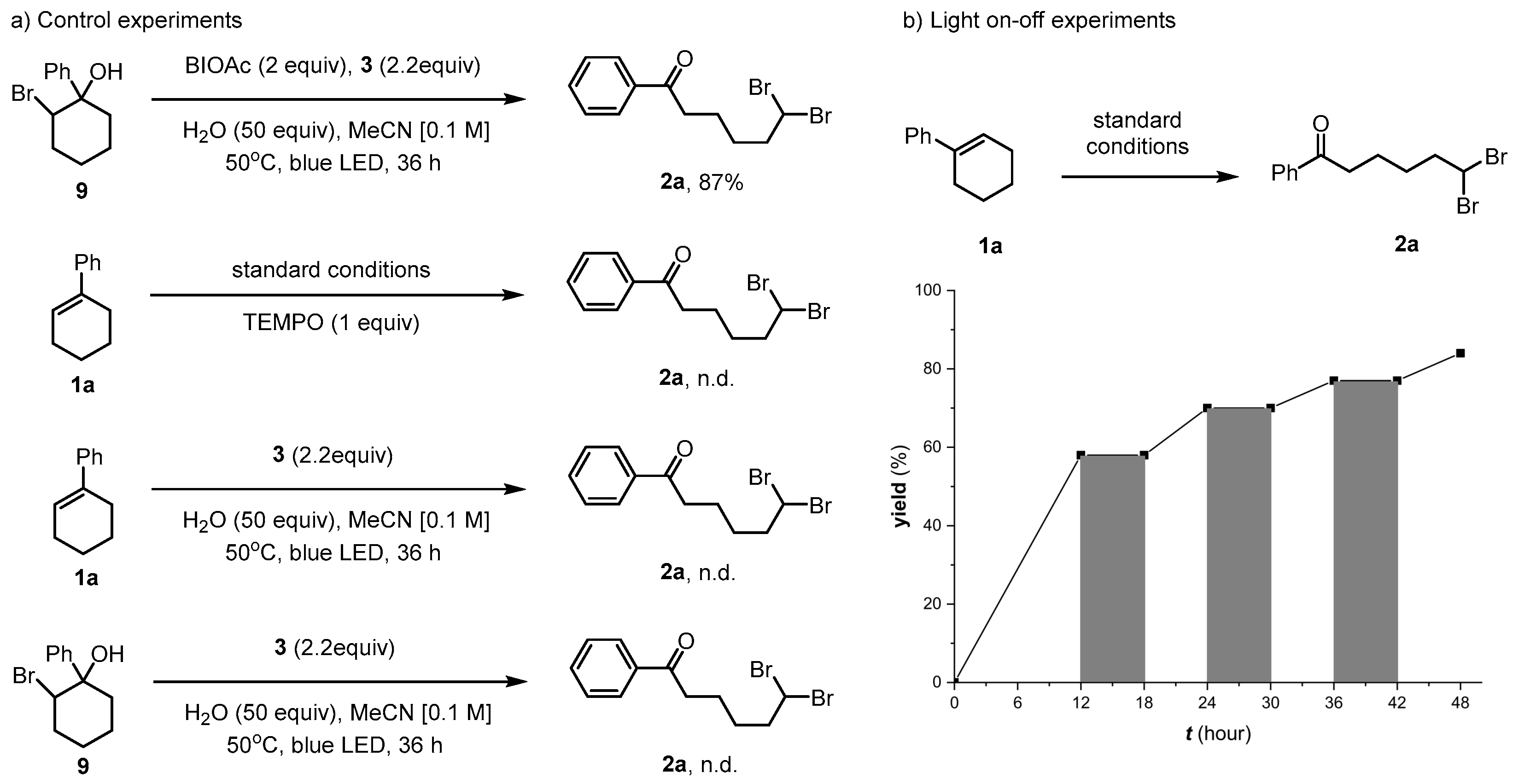
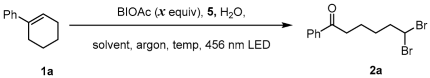
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Photocatalytic Oxidation Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosokha, S.V.; Kochi, J.K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41, 641–653. [Google Scholar] [CrossRef]
- Foster, R. Electron donor-acceptor complexes. J. Phys. Chem. 1980, 84, 2135–2141. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Mazzarella, D.; Melchiorre, P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-Q.; Majumder, S.; Yang, M.-H.; Guo, S.-R. Recent advances in catalyst-free photochemical reactions via electron-donor-acceptor (EDA) complex process. Tetrahedron Lett. 2020, 61, 151506. [Google Scholar] [CrossRef]
- Silvi, M.; Melchiorre, P. Enhancing the potential of enantioselective organocatalysis with light. Nature 2018, 554, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.G.S.; de MLima, T.; Duarte, M.; Jurberg, I.D.; Paixão, M.W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. [Google Scholar] [CrossRef]
- Arceo, E.; Jurberg, I.D.; Álvarez-Fernández, A.; Melchiorre, P. Photochemical activity of a key donor–acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nat. Chem. 2013, 5, 750–756. [Google Scholar] [CrossRef]
- Murphy, J.J.; Bastida, D.; Paria, S.; Fagnoni, M.; Melchiorre, P. Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals. Nature 2016, 532, 218–222. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Xu, R.; Chen, Y. Donor–Acceptor Complex Enables Alkoxyl Radical Generation for Metal-Free C(sp3)–C(sp3) Cleavage and Allylation/Alkenylation. Angew. Chem. Int. Ed. 2017, 56, 12619–12623. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Li, D.; Chen, Y. Metal-Free C(sp3)–H Allylation via Aryl Carboxyl Radicals Enabled by Donor–Acceptor Complex. Org. Lett. 2018, 20, 3296–3299. [Google Scholar] [CrossRef]
- Davies, J.; Booth, S.G.; Essafi, S.; Dryfe, R.A.W.; Leonori, D. Visible-Light-Mediated Generation of Nitrogen-Centered Radicals: Metal-Free Hydroimination and Iminohydroxylation Cyclization Reactions. Angew. Chem. Int. Ed. 2015, 54, 14017–14021. [Google Scholar] [CrossRef]
- Quint, V.; Morlet-Savary, F.; Lohier, J.-F.; Lalevée, J.; Gaumont, A.-C.; Lakhdar, S. Metal-Free, Visible Light-Photocatalyzed Synthesis of Benzo[b]phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yu, S. Hydrotrifluoromethylation of Unactivated Alkenes and Alkynes Enabled by an Electron-Donor–Acceptor Complex of Togni’s Reagent with a Tertiary Amine. Org. Lett. 2016, 18, 2962–2965. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, A.; Pradeilles, J.; Wang, Y.; Mutsuga, T.; Myers, E.L.; Aggarwal, V.K. Photoinduced decarboxylative borylation of carboxylic acids. Science 2017, 357, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Spell, M.L.; Deveaux, K.; Bresnahan, C.G.; Bernard, B.L.; Sheffield, W.; Kumar, R.; Ragains, J.R. A Visible-Light-Promoted O-Glycosylation with a Thioglycoside Donor. Angew. Chem. Int. Ed. 2016, 55, 6515–6519. [Google Scholar] [CrossRef]
- Sandfort, F.; Strieth-Kalthoff, F.; Klauck, F.J.R.; James, M.J.; Glorius, F. Deaminative Borylation of Aliphatic Amines Enabled by Visible Light Excitation of an Electron Donor–Acceptor Complex. Chem. Eur. J. 2018, 24, 17210–17214. [Google Scholar] [CrossRef]
- Sun, J.; He, Y.; An, X.-D.; Zhang, X.; Yu, L.; Yu, S. Visible-light-induced iminyl radical formation via electron-donor–acceptor complexes: A photocatalyst-free approach to phenanthridines and quinolines. Org. Chem. Front. 2018, 5, 977–981. [Google Scholar] [CrossRef]
- Gray, P.; Williams, A. The Thermochemistry and Reactivity Of Alkoxyl Radicals. Chem. Rev. 1959, 59, 239–328. [Google Scholar] [CrossRef]
- Batt, L. Reactions of alkoxy and alkyl peroxy radicals. Int. Rev. Phy. Chem. 1987, 6, 53–90. [Google Scholar] [CrossRef]
- Ren, R.; Zhu, C. Radical-Mediated Ring-Opening Functionalization of Cyclobutanols: A Shortcut to γ-Substituted Ketones. Synlett 2016, 27, 1139–1144. [Google Scholar] [CrossRef]
- Tsui, E.; Wang, H.; Knowles, R.R. Catalytic generation of alkoxy radicals from unfunctionalized alcohols. Chem. Sci. 2020, 11, 11124–11141. [Google Scholar] [CrossRef] [PubMed]
- Concepción, J.I.; Francisco, C.G.; Hernández, R.; Salazar, J.A.; Suárez, E. Intramolecular hydrogen abstraction. Iodosobenzene diacetate, an efficient and convenient reagent for alkoxy radical generation. Tetrahedron Lett. 1984, 25, 1953–1956. [Google Scholar] [CrossRef]
- Lee, J.; Oh, J.; Jin, S.-J.; Choi, J.-R.; Atwood, J.L.; Cha, J.K. Fragmentation of Alkoxy Radicals and Oxidative Elimination of Alicyclic Iodides. J. Org. Chem. 1994, 59, 6955–6964. [Google Scholar] [CrossRef]
- Zhao, P.; Incarvito, C.D.; Hartwig, J.F. Direct Observation of β-Aryl Eliminations from Rh(I) Alkoxides. J. Am. Chem. Soc. 2006, 128, 3124–3125. [Google Scholar] [CrossRef] [PubMed]
- Ilangovan, A.; Saravanakumar, S.; Malayappasamy, S. γ-Carbonyl Quinones: Radical Strategy for the Synthesis of Evelynin and Its Analogues by C–H Activation of Quinones Using Cyclopropanols. Org. Lett. 2013, 15, 4968–4971. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Fan, X.; Yu, J.; Zhu, C. Silver-Catalyzed Ring-Opening Strategy for the Synthesis of β- and γ-Fluorinated Ketones. J. Am. Chem. Soc. 2015, 137, 3490–3493. [Google Scholar] [CrossRef]
- Lu, S.-C.; Li, H.-S.; Xu, S.; Duan, G.-Y. Silver-catalyzed C2-selective direct alkylation of heteroarenes with tertiary cycloalkanols. Org. Biomol. Chem. 2017, 15, 324–327. [Google Scholar] [CrossRef]
- Yamamoto, K.; Toguchi, H.; Kuriyama, M.; Watanabe, S.; Iwasaki, F.; Onomura, O. Electrophotochemical Ring-Opening Bromination of tert-Cycloalkanols. J. Org. Chem. 2021, 86, 16177–16186. [Google Scholar] [CrossRef]
- Chang, L.; An, Q.; Duan, L.; Feng, K.; Zuo, Z. Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122, 2429–2486. [Google Scholar] [CrossRef]
- Hu, A.; Guo, J.-J.; Pan, H.; Zuo, Z. Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 2018, 361, 668–672. [Google Scholar] [CrossRef]
- Guo, J.-J.; Hu, A.; Chen, Y.; Sun, J.; Tang, H.; Zuo, Z. Photocatalytic C–C Bond Cleavage and Amination of Cycloalkanols by Cerium(III) Chloride Complex. Angew. Chem. Int. Ed. 2016, 55, 15319–15322. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Chen, Y. Visible-light-induced alkoxyl radical generation for inert chemical bond cleavage/functionalization. Chem. Commun. 2018, 54, 6105–6112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Zhang, F.; Hu, C.; Chen, Y. Generation of Alkoxyl Radicals by Photoredox Catalysis Enables Selective C(sp3)–H Functionalization under Mild Reaction Conditions. Angew. Chem. Int. Ed. 2016, 55, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, D.; Liu, S.; Ge, Y.; Lan, Y.; Chen, Y. Visible-Light-Induced Alkoxyl Radicals Enable α-C(sp3)-H Bond Allylation. iScience 2020, 23, 100755. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Shao, Y.; Wu, S.; Liu, P.; Li, J.; Qin, H.; Zhang, Y.; Xue, X.-S.; Chen, Y. Distal Amidoketone Synthesis Enabled by Dimethyl Benziodoxoles via Dual Copper/Photoredox Catalysis. ACS Catal. 2023, 13, 3749–3756. [Google Scholar] [CrossRef]
- Cao, Z.; Wang, X.; Wu, X.; Zhu, C. Iodobenzene-catalyzed photochemical heteroarylation of alcohols by rupture of inert C–H and C–C bonds. Tetrahedron Chem. 2022, 4, 100031. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Tang, N.; Wu, Z.; Wang, D.; Ji, M.; Xu, Y.; Wang, M.; Zhu, C. Metal-free alcohol-directed regioselective heteroarylation of remote unactivated C(sp3)–H bonds. Nat. Commun. 2018, 9, 3343. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Harms, K.; Meggers, E. Catalytic Asymmetric C–H Functionalization under Photoredox Conditions by Radical Translocation and Stereocontrolled Alkene Addition. Angew. Chem. Int. Ed. 2016, 55, 13495–13498. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Liu, C.; Fan, J.; Wang, M.; Yan, X.; Huang, M.; Cai, S. Photocatalytic dehydrogenated etherification of 2-aryl benzylic alcohols. Green Chem. 2022, 24, 7442–7447. [Google Scholar] [CrossRef]
- Hu, X.; Li, G.-X.; He, G.; Chen, G. Minisci C–H alkylation of N-heteroarenes with aliphatic alcohols via β-scission of alkoxy radical intermediates. Org. Chem. Front. 2019, 6, 3205–3209. [Google Scholar] [CrossRef]
- Li, G.-X.; Hu, X.; He, G.; Chen, G. Photoredox-mediated remote C(sp3)–H heteroarylation of free alcohols. Chem. Sci. 2019, 10, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-L.; Wang, Y.; Wang, Z.; Dou, B.; Wang, J. Ring-opening iodination and bromination of unstrained cycloalkanols through β-scission of alkoxy radicals. Chem. Commun. 2020, 56, 5002–5005. [Google Scholar] [CrossRef] [PubMed]
- Rivero, A.R.; Fodran, P.; Ondrejková, A.; Wallentin, C.-J. Alcohol Etherification via Alkoxy Radicals Generated by Visible-Light Photoredox Catalysis. Org. Lett. 2020, 22, 8436–8440. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ishida, N. β-Scission of Alkoxy Radicals in Synthetic Transformations. Chem. Lett. 2017, 46, 1692–1700. [Google Scholar] [CrossRef]
- Jia, K.; Pan, Y.; Chen, Y. Selective Carbonyl–C(sp3) Bond Cleavage to Construct Ynamides, Ynoates, and Ynones by Photoredox Catalysis. Angew. Chem. Int. Ed. 2017, 56, 2478–2481. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Zhang, F.; Huang, H.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)–C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc. 2016, 138, 1514–1517. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Chen, Y. Selective C−H Acyloxylation of Sulfides/Disulfides Enabled by Hypervalent Iodine Reagents. Adv. Synth. Catal. 2023, 365, 2690–2696. [Google Scholar] [CrossRef]
- Wu, S.; Li, J.; He, R.; Jia, K.; Chen, Y. Terminal Trifluoromethylation of Ketones via Selective C–C Cleavage of Cycloalkanols Enabled by Hypervalent Iodine Reagents. Org. Lett. 2021, 23, 9204–9209. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Mathew, T.; Hoole, D.; Esteves, P.M.; Wang, Q.; Rasul, G.; Olah, G.A. N-Halosuccinimide/BF3–H2O, Efficient Electrophilic Halogenating Systems for Aromatics. J. Am. Chem. Soc. 2004, 126, 15770–15776. [Google Scholar] [CrossRef]
- Bartoli, S.; Cipollone, A.; Squarcia, A.; Madami, A.; Fattori, D. Electrophilic Bromination of meta-Substituted Anilines with N-Bromosuccin imide: Regioselectivity and Solvent Effect. Synthesis 2009, 2009, 1305–1308. [Google Scholar]
- Rogers, D.A.; Brown, R.G.; Brandeburg, Z.C.; Ko, E.Y.; Hopkins, M.D.; LeBlanc, G.; Lamar, A.A. Organic Dye-Catalyzed, Visible-Light Photoredox Bromination of Arenes and Heteroarenes Using N-Bromosuccinimide. ACS Omega 2018, 3, 12868–12877. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Toh, R.W.; Shi, X.; Wang, T.; Cong, X.; Wu, J. Photo-mediated selective deconstructive geminal dihalogenation of trisubstituted alkenes. Nat. Commun. 2020, 11, 4462. [Google Scholar] [CrossRef] [PubMed]
- Narender, M.; Reddy, M.S.; Nageswar, Y.V.D.; Rao, K.R. Aqueous phase synthesis of vic-halohydrins from olefins and N-halosuccinimides in the presence of β-cyclodextrin. J. Mol. Catal. A Chem. 2006, 258, 10–14. [Google Scholar] [CrossRef]
- Inamoto, K.; Yamada, T.; Kato, S.; Kikkawa, S.; Kondo, Y. Facile deprotection of dithioacetals by using a novel 1,4-benzoquinone/cat. NaI system. Tetrahedron 2013, 44, 9192. [Google Scholar] [CrossRef]
- Yadav, D.K.; Lokhande, R.S.; Pitale, S.M.; Janwadkar, S.P.; Navarkar, P.S.; Rana, P.K. Study of New Selective Reagent Acetophenone 2′, 4′-Dihydroxy Semicarbazone for Extractive Spectrophotometric Determination of Vanadium. World J. Anal. Chem. 2014, 2, 10–14. [Google Scholar] [CrossRef]
- Jia, K.; Li, J.; Chen, Y. Selective P−C(sp3) Bond Cleavage and Radical Alkynylation of α-Phosphorus Alcohols by Photoredox Catalysis. Chem. Eur. J. 2018, 24, 3174. [Google Scholar] [CrossRef]
- Kiyokawa, K.; Kosaka, T.; Kojima, T.; Minakata, S. Synthesis and Structure of Hypervalent Iodine(III) Reagents Containing Phthalimidate and Application to Oxidative Amination Reactions. Angew. Chem. Int. Ed. Engl. 2015, 54, 13719–13723. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | x | Solvent | Temperature | Yield(%) |
|---|---|---|---|---|
| 1 | 1 | MeCN | r.t. | 25 |
| 2 | 1.5 | MeCN | r.t. | 39 |
| 3 | 2 | MeCN | r.t. | 54 |
| 4 2 | 2 | MeCN | r.t. | 52 |
| 5 | 2 | MeCN | 50 | 76 |
| 6 | 2 | DCM | 50 | 46 |
| 7 | 2 | PhCl | 50 | 8 |
| 8 | 2 | Acetone | 50 | ND |
| 9 | 2 | THF | 50 | ND |
| 10 | 2 | EtOAc | 50 | 24 |
| 11 3 | 2 | MeCN | 50 | 83 |
| 12 4 | 2 | MeCN | 50 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, R.; Wang, Y.; Zhang, J.; Wu, C.; Zhang, Z.; Zhang, D. Visible-Light-Mediated Ring-Opening Geminal Dibromination of Alkenes via Alkoxy Radicals Enabled by Electron Donor–Acceptor Complex. Molecules 2024, 29, 3281. https://doi.org/10.3390/molecules29143281
Wei R, Wang Y, Zhang J, Wu C, Zhang Z, Zhang D. Visible-Light-Mediated Ring-Opening Geminal Dibromination of Alkenes via Alkoxy Radicals Enabled by Electron Donor–Acceptor Complex. Molecules. 2024; 29(14):3281. https://doi.org/10.3390/molecules29143281
Chicago/Turabian StyleWei, Rong, Yuan Wang, Juantao Zhang, Chunsheng Wu, Zhenhua Zhang, and Duo Zhang. 2024. "Visible-Light-Mediated Ring-Opening Geminal Dibromination of Alkenes via Alkoxy Radicals Enabled by Electron Donor–Acceptor Complex" Molecules 29, no. 14: 3281. https://doi.org/10.3390/molecules29143281
APA StyleWei, R., Wang, Y., Zhang, J., Wu, C., Zhang, Z., & Zhang, D. (2024). Visible-Light-Mediated Ring-Opening Geminal Dibromination of Alkenes via Alkoxy Radicals Enabled by Electron Donor–Acceptor Complex. Molecules, 29(14), 3281. https://doi.org/10.3390/molecules29143281