
EMPDTA: An End-to-End Multimodal Representation Learning Framework with Pocket Online Detection for Drug–Target Affinity Prediction

Abstract
1. Introduction
2. Results
2.1. Pocket Detection Performance
2.2. Multimodal Models Achieve Better Performance Than Single Modal Models
2.3. Joint Training with Fine-Tuning Demonstrates Superior Performance
2.4. Comparison with State-of-the-Art Methods
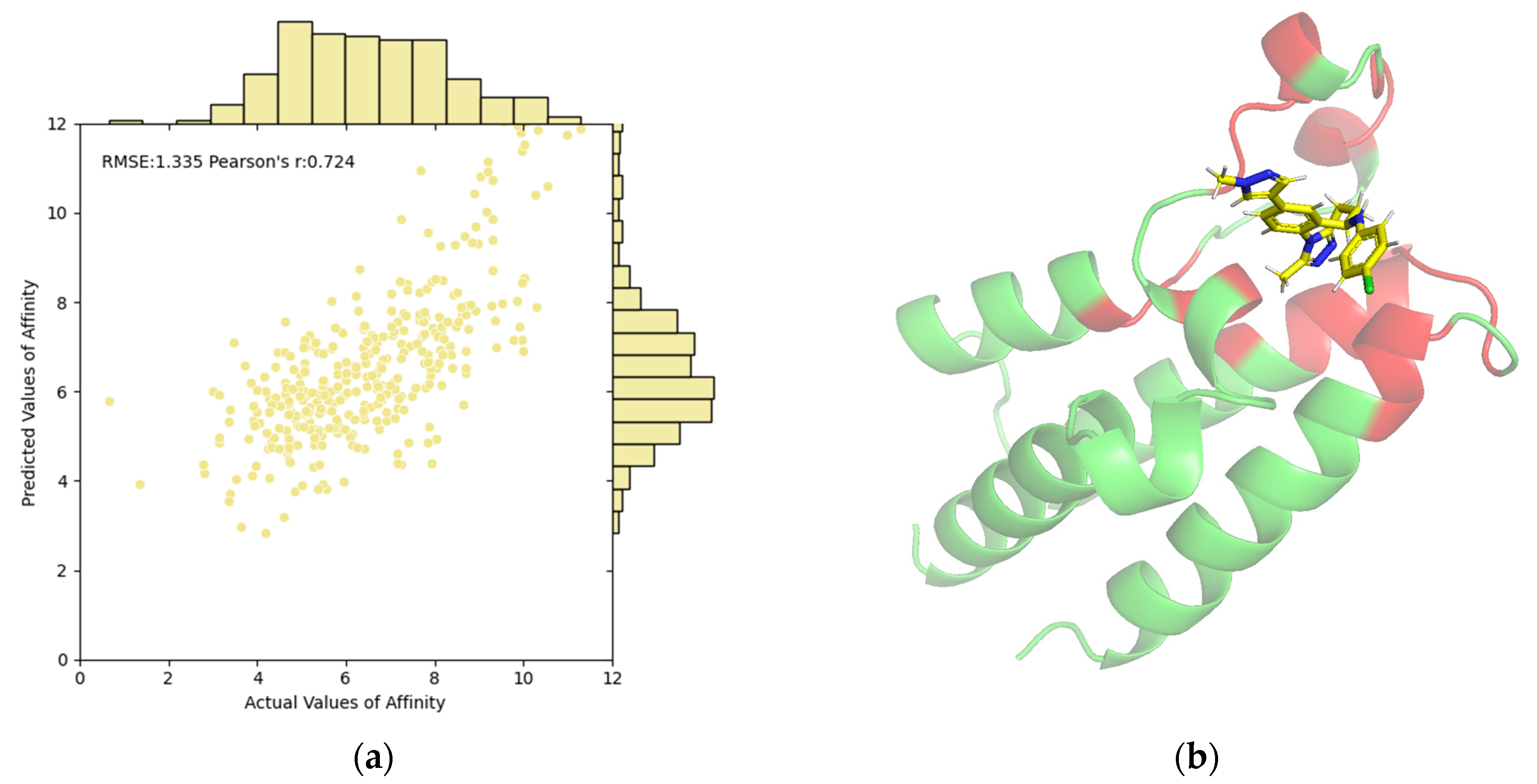
2.5. Model Interpretability on Both Affinity and Pocket Prediction
3. Materials and Methods
3.1. Dataset Construction
- The Davis and Filtered Davis datasets. The Davis dataset comprises 30,056 drug–target pairs with affinity values () among 72 drugs and 442 targets [24]. The Filtered Davis is derived from the Davis dataset, excluding pairs with no observed binding [21]. Consequently, the Filtered Davis dataset contains 72 drugs and 379 unique targets, forming 9125 interactions.
- KIBA dataset. KIBA incorporates a comprehensive combination of the inhibition constant (), dissociation constant (), and half-maximal inhibitory concentration (IC50) as affinity values [25]. It consists of 2111 drugs and 229 targets, forming 118,254 interactions.
- PDBbind dataset. PDBbind (v2020) comprises experimentally measured structures of 19,443 protein–ligand complexes with binding affinities [26].
3.2. Problem Formulation
3.3. Notation and Preprocessing
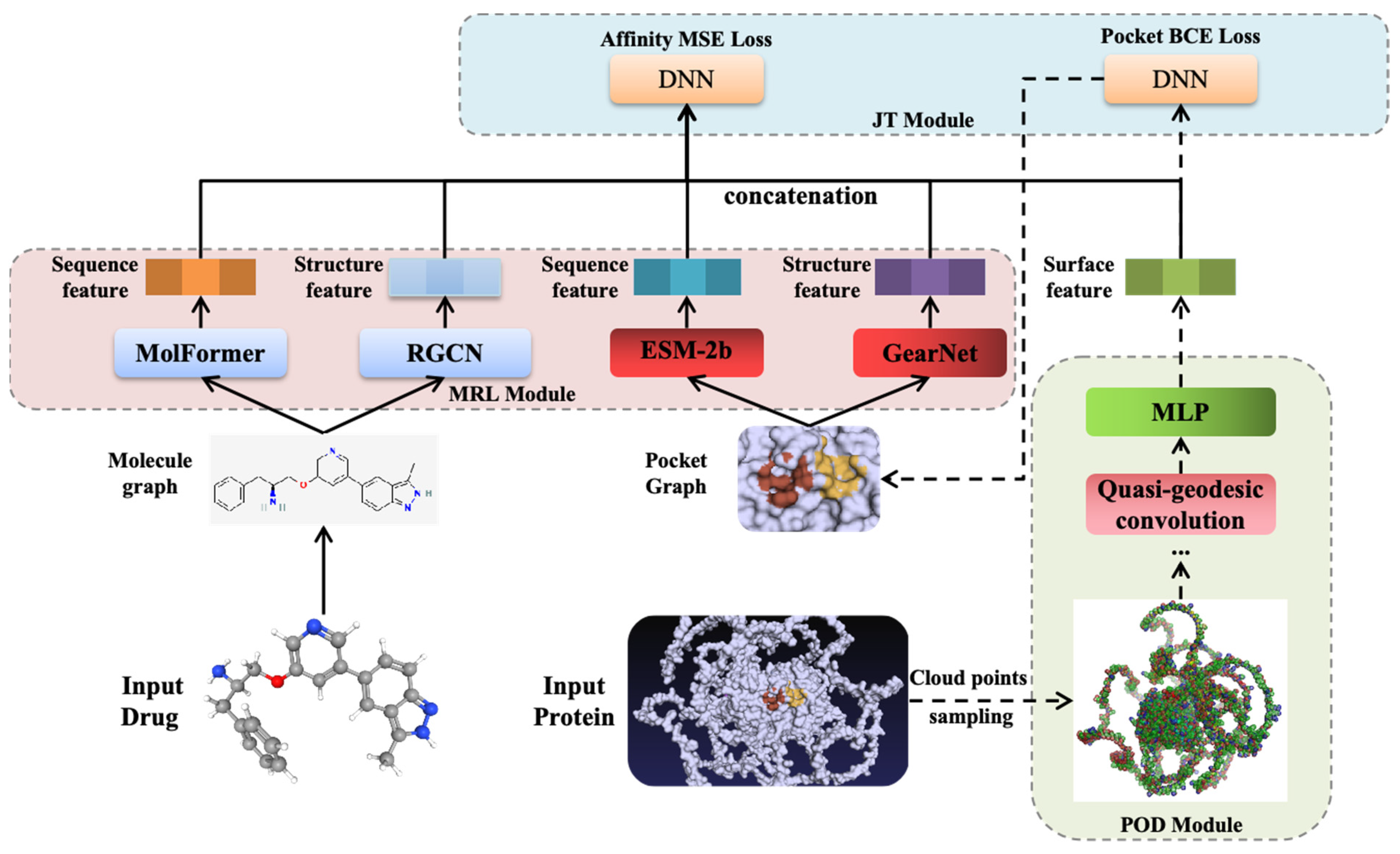
3.4. Model Architecture of EMPDTA
3.5. Pocket Online Detection Module
3.5.1. Cloud Points Sampling
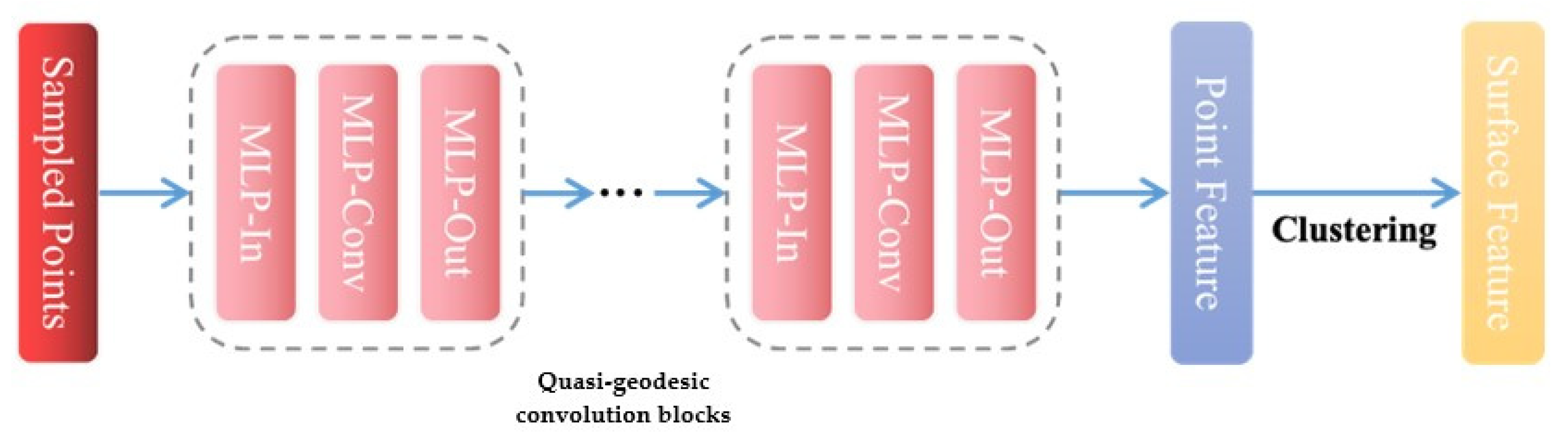
3.5.2. Quasi-Geodesic Convolution
3.6. Multimodal Representation Learning Module
3.6.1. Sequence Modality
3.6.2. Structure Modality
3.6.3. Surface Modality
3.7. Joint Training Module
3.8. Model Training and Evaluation
- SimBoost [10] leverages features of drugs, targets, and drug–target pairs, using gradient-boosting regression trees as the prediction model.
- DeepDTA [11] is an innovative method using two branches of CNN blocks to encode drug SMILES strings and protein sequences.
- MDeePred [21] feeds multi-channel protein features into a CNN and fingerprint-based molecule vectors into a fully connected neural network (FNN).
- GraphDTA [12] introduces molecular graphs into DTA prediction, marking a pioneering approach.
- DeepGLSTM [22] employs three blocks of graph convolutional networks (GCN) for drug molecules and bidirectional LSTM for protein sequences.
- MFR-DTA [23] proposes a novel architecture that includes BioMLP/CNN blocks, an Elem-feature fusion block, and a Mix–Decoder block to extract drug–target interaction (DTI) information and predict binding regions simultaneously.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pei, Q.; Gao, K.; Wu, L.; Zhu, J.; Xia, Y.; Xie, S.; Qin, T.; He, K.; Liu, T.-Y.; Yan, R. FABind: Fast and Accurate Protein-Ligand Binding. Adv. Neural Inf. Process. Syst. 2023, 36, 55963–55980. [Google Scholar]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J. Artificial Intelligence in the Prediction of Protein–Ligand Interactions: Recent Advances and Future Directions. Brief. Bioinform. 2022, 23, bbab476. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Krivák, R.; Hoksza, D. P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J. Cheminform. 2018, 10, 39. [Google Scholar] [CrossRef]
- Huang, B.; Schroeder, M. LIGSITEcsc: Predicting Ligand Binding Sites Using the Connolly Surface and Degree of Conservation. BMC Struct. Biol. 2006, 6, 19. [Google Scholar] [CrossRef]
- Lu, W.; Wu, Q.; Zhang, J.; Rao, J.; Li, C.; Zheng, S. TANKBind: Trigonometry-Aware Neural NetworKs for Drug-Protein Binding Structure Prediction. Adv. Neural Inf. Process. Syst. 2022, 35, 7236–7249. [Google Scholar]
- Pahikkala, T.; Airola, A.; Pietilä, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward More Realistic Drug-Target Interaction Predictions. Brief Bioinform. 2015, 16, 325–337. [Google Scholar] [CrossRef]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A Read-across Approach for Predicting Drug–Target Binding Affinities Using Gradient Boosting Machines. J. Cheminformatics 2017, 9, 24. [Google Scholar] [CrossRef]
- Öztürk, H.; Olmez, E.; Özgür, A. DeepDTA: Deep Drug–Target Binding Affinity Prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Nguyen, T.; Le, H.; Quinn, T.P.; Nguyen, T.; Le, T.D.; Venkatesh, S. GraphDTA: Predicting Drug–Target Binding Affinity with Graph Neural Networks. Bioinformatics 2021, 37, 1140–1147. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Ackloo, S.; Al-awar, R.; Amaro, R.E.; Arrowsmith, C.H.; Azevedo, H.; Batey, R.A.; Bengio, Y.; Betz, U.A.K.; Bologa, C.G.; Chodera, J.D.; et al. CACHE (Critical Assessment of Computational Hit-Finding Experiments): A Public–Private Partnership Benchmarking Initiative to Enable the Development of Computational Methods for Hit-Finding. Nat. Rev. Chem. 2022, 6, 287–295. [Google Scholar] [CrossRef]
- Li, X.; Jacobson, M.P.; Friesner, R.A. High-Resolution Prediction of Protein Helix Positions and Orientations. Proteins: Struct. Funct. Bioinform. 2004, 55, 368–382. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Batzner, S.; Musaelian, A.; Sun, L.; Geiger, M.; Mailoa, J.P.; Kornbluth, M.; Molinari, N.; Smidt, T.E.; Kozinsky, B. E(3)-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials. Nat. Commun. 2022, 13, 2453. [Google Scholar] [CrossRef]
- Roche, R.; Moussad, B.; Shuvo, M.H.; Bhattacharya, D. E(3) Equivariant Graph Neural Networks for Robust and Accurate Protein-Protein Interaction Site Prediction. PLoS Comput. Biol. 2023, 19, e1011435. [Google Scholar] [CrossRef]
- D’Souza, S.; Prema, K.V.; Balaji, S.; Shah, R. Deep Learning-Based Modeling of Drug–Target Interaction Prediction Incorporating Binding Site Information of Proteins. Interdiscip. Sci. Comput. Life Sci. 2023, 15, 306–315. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Cetin Atalay, R.; Cansen Kahraman, D.; Doğan, T.; Martin, M.; Atalay, V. MDeePred: Novel Multi-Channel Protein Featurization for Deep Learning-Based Binding Affinity Prediction in Drug Discovery. Bioinformatics 2021, 37, 693–704. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ghosh, M.; Basuchowdhuri, P. DeepGLSTM: Deep Graph Convolutional Network and LSTM Based Approach for Predicting Drug-Target Binding Affinity. In Proceedings of the 2022 SIAM International Conference on Data Mining (SDM) Proceedings; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 2022; pp. 729–737. [Google Scholar]
- Hua, Y.; Song, X.; Feng, Z.; Wu, X. MFR-DTA: A Multi-Functional and Robust Model for Predicting Drug-Target Binding Affinity and Region. Bioinformatics 2023, 39, btad056. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgård, S.; Ciceri, P.; Wodicka, L.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making Sense of Large-Scale Kinase Inhibitor Bioactivity Data Sets: A Comparative and Integrative Analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the Basis for Developing Protein-Ligand Interaction Scoring Functions. Acc. Chem. Res. 2017, 50, 302–309. [Google Scholar] [CrossRef]
- Abdollahi, N.; Tonekaboni, S.; Huang, J.J.C.; Wang, B.; MacKinnon, S. NodeCoder: A Graph-Based Machine Learning Platform to Predict Active Sites of Modeled Protein Structures. arXiv 2023, arXiv:2302.03590. [Google Scholar]
- Zhu, Z.; Shi, C.; Zhang, Z.; Liu, S.; Xu, M.; Yuan, X.; Zhang, Y.; Chen, J.; Cai, H.; Lu, J.; et al. TorchDrug: A Powerful and Flexible Machine Learning Platform for Drug Discovery. arXiv 2022, arXiv:2202.08320. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, M.; Jamasb, A.; Chenthamarakshan, V.; Lozano, A.; Das, P.; Tang, J. Protein Representation Learning by Geometric Structure Pretraining. arXiv 2022, arXiv:2203.06125. [Google Scholar]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large Language Models Generate Functional Protein Sequences across Diverse Families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef]
- Sverrisson, F.; Feydy, J.; Correia, B.E.; Bronstein, M.M. Fast End-to-End Learning on Protein Surfaces. In Proceedings of the 2021 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), Nashville, TN, USA, 20–25 June 2021; pp. 15267–15276. [Google Scholar] [CrossRef]
- Charlier, B.; Feydy, J.; Glaunès, J.; Collin, F.-D.; Durif, G. Kernel Operations on the GPU, with Autodiff, without Memory Overflows. J. Mach. Learn. Res. 2020, 22, 1–6. [Google Scholar]
- Ross, J.; Belgodere, B.; Chenthamarakshan, V.; Padhi, I.; Mroueh, Y.; Das, P. Large-Scale Chemical Language Representations Capture Molecular Structure and Properties. Nat. Mach. Intell. 2022, 4, 1256–1264. [Google Scholar] [CrossRef]
- Klicpera, J.; Groß, J.; Günnemann, S. Directional Message Passing for Molecular Graphs. arXiv 2020, arXiv:2003.03123. [Google Scholar]
- Li, S.; Zhou, J.; Xu, T.; Dou, D.; Xiong, H. GeomGCL: Geometric Graph Contrastive Learning for Molecular Property Prediction. Proc. AAAI Conf. Artif. Intell. 2021, 36, 4541–4549. [Google Scholar] [CrossRef]
- Schlichtkrull, M.; Kipf, T.; Bloem, P.; van den Berg, R.; Titov, I.; Welling, M. Modeling Relational Data with Graph Convolutional Networks. In Proceedings of The Semantic Web: 15th International Conference, ESWC 2018, Proceedings 15, Heraklion, Crete, Greece, 3–7 June 2018; Springer International Publishing: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Loshchilov, I.; Hutter, F. Decoupled Weight Decay Regularization 2019. In Proceedings of the 7th International Conference on Learning Representations, New Orleans, LA, USA, 6–9 May 2019. [Google Scholar]
- Smith, T.F.; Waterman, M.S. Identification of Common Molecular Subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Airola, A.; Pahikkala, T. Fast Kronecker Product Kernel Methods via Generalized Vec Trick. IEEE Trans. Neural Netw. Learn. Syst. 2018, 29, 3374–3387. [Google Scholar] [CrossRef]
- Cichońska, A.; Ravikumar, B.; Allaway, R.J.; Wan, F.; Park, S.; Isayev, O.; Li, S.; Mason, M.; Lamb, A.; Tanoli, Z.; et al. Crowdsourced Mapping of Unexplored Target Space of Kinase Inhibitors. Nat. Commun. 2021, 12, 3307. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Method | Davis | KIBA | PDBbind | Params | |||
|---|---|---|---|---|---|---|---|
| AUROC↑ | AUPRC↑ | AUROC↑ | AUPRC↑ | AUROC↑ | AUPRC↑ | ||
| P2Rank | 0.8455 | 0.2055 | 0.8606 | 0.2290 | 0.6236 | 0.3749 | offline |
| CNN | 0.9344 | 0.7695 | 0.9320 | 0.6944 | 0.7996 | 0.5615 | 0.4 M |
| GCN | 0.9148 | 0.5377 | 0.9144 | 0.5332 | 0.8065 | 0.5413 | 0.8 M |
| GearNet | 0.9448 | 0.7948 | 0.9513 | 0.8197 | 0.8919 | 0.6838 | 4.6 M |
| POD | 0.9427 | 0.6667 | 0.9351 | 0.6454 | 0.9116 | 0.7085 | 0.03 M |
| Modal | Drug | Target | Affinity Metrics | |||||
|---|---|---|---|---|---|---|---|---|
| Seq | Str | Seq | Str | Sur | RMSE↓ | CI↑ | Spearman↑ | |
| Single | √ | — | √ | — | — | 0.469 | 0.746 | 0.524 |
| — | √ | — | √ | — | 0.529 | 0.732 | 0.467 | |
| Multi | √ | √ | √ | √ | — | 0.458 | 0.749 | 0.533 |
| √ | √ | √ | √ | √ | 0.454 | 0.750 | 0.543 | |
| Training Method | Affinity Metrics | Pocket Metrics | Time↓ * | ||||
|---|---|---|---|---|---|---|---|
| RMSE↓ | CI↑ | Spearman↑ | AUROC↑ | AUPRC↑ | |||
| Single | Fine-tune | 0.667 (0.010) | 0.751 (0.005) | 0.679 (0.011) | 0.911 (0.004) | 0.314 (0.011) | 105 s |
| Single | Normal | 0.675 (0.005) | 0.746 (0.004) | 0.667 (0.009) | 0.519 (0.129) | 0.039 (0.016) | 236 s |
| Multi | Fine-tune | 0.663 (0.006) | 0.752 (0.003) | 0.681 (0.008) | 0.964 (0.003) | 0.803 (0.007) | 175 s |
| Multi | Normal | 0.673 (0.007) | 0.747 (0.002) | 0.670 (0.004) | 0.971 (0.007) | 0.763 (0.050) | 224 s |
| Method | RMSE↓ | CI↑ | Spearman↑ |
|---|---|---|---|
| CGKronRLS a | 0.769 (0.010) | 0.740 (0.003) | 0.643 (0.008) |
| MDeePred a | 0.742 (0.009) | 0.733 (0.004) | 0.618 (0.009) |
| DeepDTA a | 0.931 (0.015) | 0.653 (0.005) | 0.430 (0.013) |
| EMPDTA | 0.663 (0.006) | 0.752 (0.003) | 0.681 (0.008) |
| Method | Davis | KIBA | ||||
|---|---|---|---|---|---|---|
| MSE↓ | CI↑ | rm2↑ | MSE↓ | CI↑ | rm2↑ | |
| KronRLS [9] | 0.379 | 0.871 | 0.407 | 0.411 | 0.782 | 0.342 |
| SimBoost [10] | 0.282 | 0.872 | 0.644 | 0.222 | 0.836 | 0.629 |
| DeepDTA [11] | 0.261 | 0.878 | 0.630 | 0.194 | 0.863 | 0.673 |
| GraphDTA [12] | 0.229 | 0.893 | — | 0.147 | 0.882 | — |
| DeepGLSTM [22] | 0.232 | 0.895 | 0.680 | 0.133 | 0.897 | 0.792 |
| MFR-DTA [23] | 0.221 | 0.905 | 0.705 | 0.136 | 0.898 | 0.789 |
| EMPDTA | 0.218 | 0.891 | 0.689 | 0.142 | 0.878 | 0.763 |
| Methods | RMSE↓ | Pearson↑ | Spearman↑ | MAE↓ |
|---|---|---|---|---|
| TransCPI b | 1.741 | 0.576 | 0.540 | 1.404 |
| MONN b | 1.438 | 0.624 | 0.589 | 1.143 |
| PIGNet b | 2.640 | 0.511 | 0.489 | 2.110 |
| IGN b | 1.433 | 0.698 | 0.641 | 1.169 |
| HOLOPROT b | 1.546 | 0.602 | 0.571 | 1.208 |
| STAMPDPI b | 1.658 | 0.545 | 0.411 | 1.325 |
| TANKBind b | 1.346 | 0.726 | 0.703 | 1.070 |
| EMPDTA | 1.335 | 0.724 | 0.698 | 1.069 |
| Dataset | Affinity Metrics | Pocket Metrics | |||
|---|---|---|---|---|---|
| RMSE↓ | CI↑ | Spearman↑ | AUROC↑ | AUPRC↑ | |
| Filtered Davis | 0.663 | 0.752 | 0.681 | 0.964 | 0.803 |
| Davis | 0.468 | 0.891 | 0.701 | 0.987 | 0.876 |
| KIBA | 0.385 | 0.871 | 0.865 | 0.964 | 0.686 |
| PDBbind | 1.335 | 0.755 | 0.698 | 0.874 | 0.581 |
| Datasets | Affinity-Related | Pocket-Related | |||
|---|---|---|---|---|---|
| Drugs | Targets | Interactions | Pocket Ration | Average residues | |
| Davis | 68 | 442 | 30,056 | 1.36% | 790 |
| Filtered Davis | 68 | 379 | 9125 | 1.36% | 790 |
| KIBA | 2111 | 229 | 118,254 | 1.79% | 729 |
| PDBbind * | 15,689 | 12,828 | 19,350 | 9.51% | 692 |
| name | GLY | ALA | THR | SER | CYS | ASN | ASP | PRO | GLN | VAL |
| Radius | 1.46 | 1.54 | 1.61 | 1.64 | 1.80 | 1.83 | 1.88 | 1.88 | 1.96 | 1.99 |
| name | ILE | LEU | HIS | MET | GLU | LYS | ARG | PHE | TYR | TRP |
| Radius | 1.99 | 2.02 | 2.02 | 2.05 | 2.07 | 2.17 | 2.17 | 2.18 | 2.19 | 2.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, D.; Xie, J. EMPDTA: An End-to-End Multimodal Representation Learning Framework with Pocket Online Detection for Drug–Target Affinity Prediction. Molecules 2024, 29, 2912. https://doi.org/10.3390/molecules29122912
Huang D, Xie J. EMPDTA: An End-to-End Multimodal Representation Learning Framework with Pocket Online Detection for Drug–Target Affinity Prediction. Molecules. 2024; 29(12):2912. https://doi.org/10.3390/molecules29122912
Chicago/Turabian StyleHuang, Dingkai, and Jiang Xie. 2024. "EMPDTA: An End-to-End Multimodal Representation Learning Framework with Pocket Online Detection for Drug–Target Affinity Prediction" Molecules 29, no. 12: 2912. https://doi.org/10.3390/molecules29122912
APA StyleHuang, D., & Xie, J. (2024). EMPDTA: An End-to-End Multimodal Representation Learning Framework with Pocket Online Detection for Drug–Target Affinity Prediction. Molecules, 29(12), 2912. https://doi.org/10.3390/molecules29122912