
Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review

Abstract
1. Introduction
2. Tau Protein and Neurodegenerative Diseases
2.1. Alzheimer’s Disease (AD)
2.2. Parkinson’s Disease (PD)
2.3. Huntington’s Disease (HD)
3. Post-Translational Modifications of Tau Protein
3.1. Phosphorylation of Tau Protein
3.2. Acetylation of Tau Protein
3.3. Glycosylation of Tau Protein
3.4. Methylation of Tau Protein
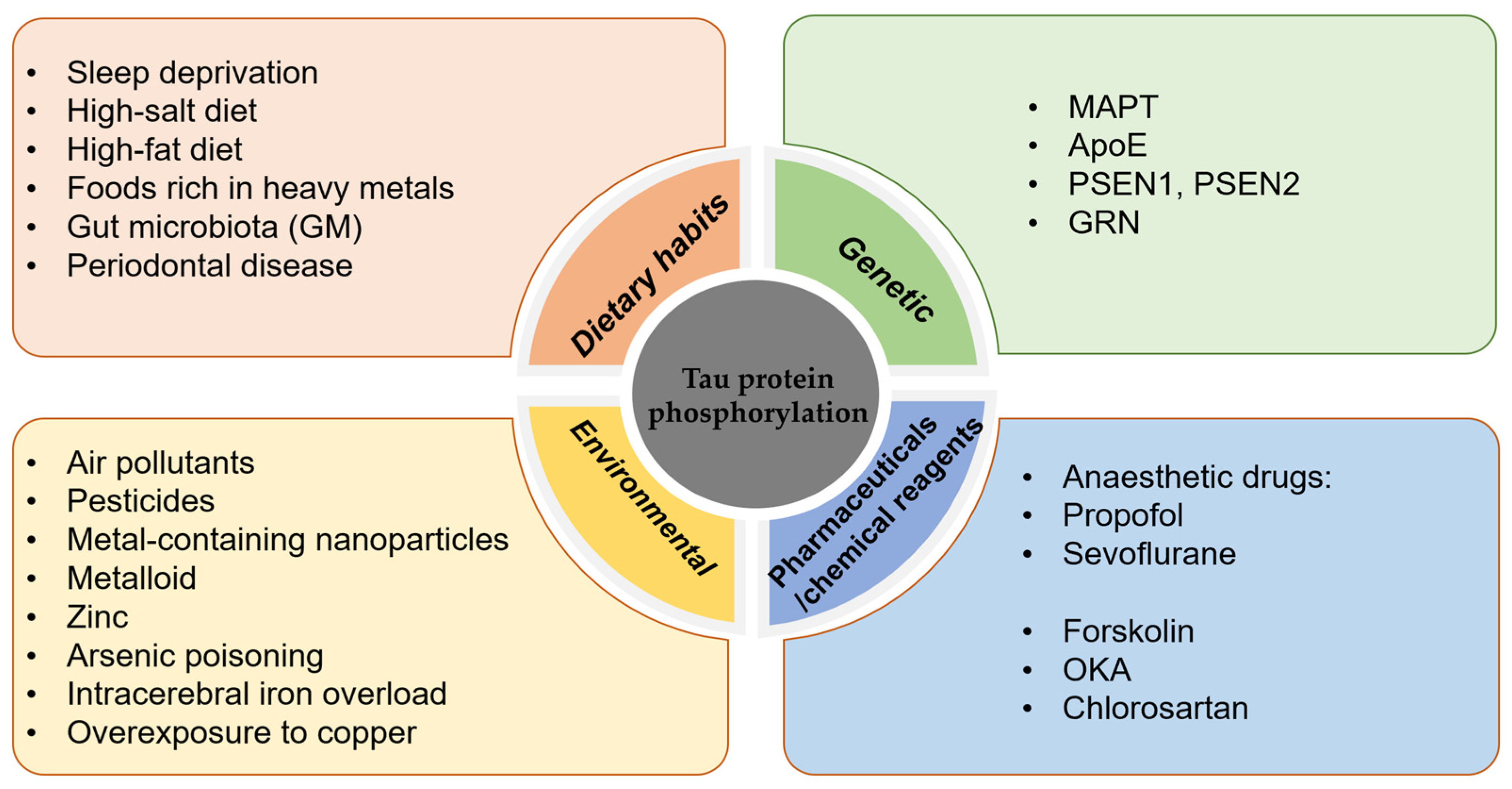
4. Factors Influencing the Hyperphosphorylation of Tau Protein
4.1. Dietary Habits
4.2. Genetic Factors
4.2.1. MAPT
4.2.2. ApoE
4.2.3. Others
4.3. Environmental/Chemical Factors
5. Progress in the Development of Therapeutic Drugs Targeting Tau Protein
5.1. Drugs Targeting Post-Translational Modifications of Tau Protein
5.2. Tau Protein Aggregation Inhibitors
5.3. Others
6. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Alonso, A.d.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Gorath, M.; Stahnke, T.; Mronga, T.; Goldbaum, O.; Richter-Landsberg, C. Developmental changes of tau protein and mRNA in cultured rat brain oligodendrocytes. Glia 2001, 36, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Waheed, Z.; Choudhary, J.; Jatala, F.H.; Fatimah, N.A.; Zerr, I.; Zafar, S. The Role of Tau Proteoforms in Health and Disease. Mol. Neurobiol. 2023, 60, 5155–5166. [Google Scholar] [CrossRef] [PubMed]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef]
- Brunden, K.R.; Trojanowski, J.Q.; Smith, A.B., 3rd; Lee, V.M.; Ballatore, C. Microtubule-stabilizing agents as potential therapeutics for neurodegenerative disease. Bioorganic Med. Chem. 2014, 22, 5040–5049. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, A.; Panda, D. Regulation of neuronal microtubule dynamics by tau: Implications for tauopathies. Int. J. Biol. Macromol. 2019, 133, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Lee, V.M.; Trojanowski, J.Q. Tau and axonopathy in neurodegenerative disorders. Neuromolecular Med. 2002, 2, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Qiang, L. Tau: It’s Not What You Think. Trends Cell Biol. 2019, 29, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol. 2018, 28, 2181–2189.e2184. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, E.H.; Hejab, N.M.A.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-atomic model of microtubule-tau interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Alonso, A.D.; Beharry, C.; Corbo, C.P.; Cohen, L.S. Molecular mechanism of prion-like tau-induced neurodegeneration. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buée, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Glynn, C.; Rodriguez, J.A.; Hyman, B.T. The structural line between prion and “prion-like”: Insights from prion protein and tau. Curr. Opin. Neurobiol. 2024, 86, 102857. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Madero-Pérez, J.; Swaney, D.L.; Chang, T.S.; Moritz, M.; Konrad, C.; Ward, M.E.; Stevenson, E.; Hüttenhain, R.; Kauwe, G.; et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 2022, 185, 712–728.e714. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Alvir, R.V.; Alvir, R.V.; Bunquin, L.E.; Pradeepkiran, J.A.; Reddy, P.H. A partial reduction of VDAC1 enhances mitophagy, autophagy, synaptic activities in a transgenic Tau mouse model. Aging Cell 2022, 21, e13663. [Google Scholar] [CrossRef]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Dermaut, B.; Kumar-Singh, S.; Rademakers, R.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Tau is central in the genetic Alzheimer-frontotemporal dementia spectrum. Trends Genet. 2005, 21, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Jeans, A.F. Hebbian plasticity: The elusive missing link at the heart of Alzheimer’s disease pathogenesis? Neural Regen. Res. 2023, 18, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xiao, Q.; Luo, C.; Zhao, Y.; Pu, D.; Zhao, K.; Chen, J.; Wang, M.; Liao, Z. High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp. Cell Res. 2017, 359, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, S.L.; Pi, L.H.; Shi, X.J.; Ma, L.R.; Chen, Z.; Qu, M.L.; Li, X.; Nie, S.D.; Liao, D.F.; et al. High glucose induces formation of tau hyperphosphorylation via Cav-1-mTOR pathway: A potential molecular mechanism for diabetes-induced cognitive dysfunction. Oncotarget 2017, 8, 40843–40856. [Google Scholar] [CrossRef] [PubMed]
- Haukedal, H.; Freude, K.K. Implications of Glycosylation in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 625348. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R.; et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 2016, 90, 245–260. [Google Scholar] [CrossRef]
- Stancu, I.C.; Lodder, C.; Botella Lucena, P.; Vanherle, S.; Gutierrez de Rave, M.; Terwel, D.; Bottelbergs, A.; Dewachter, I. The NLRP3 inflammasome modulates tau pathology and neurodegeneration in a tauopathy model. Glia 2022, 70, 1117–1132. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet. Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W. Theodore Cooper Memorial Lecture. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: A new perspective. Hypertension 1995, 25, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.H.; Wu, S.Y.; Yu, M.; Huang, S.H.; Lee, C.W.; Jiang, M.J.; Lin, P.Y.; Yang, T.T.; Kuo, Y.M. Hypertension Accelerates Alzheimer’s Disease-Related Pathologies in Pigs and 3xTg Mice. Front. Aging Neurosci. 2018, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Yamamoto, T.; Kimura, S.; Mino, T.; Iwamoto, T. Brain white matter lesions and postoperative cognitive dysfunction: A review. J. Anesth. 2019, 33, 336–340. [Google Scholar] [CrossRef]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Højrup, P.; Gliemann, J.; Jakes, R. α-Synuclein Binds to Tau and Stimulates the Protein Kinase A-catalyzed Tau Phosphorylation of Serine Residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef]
- Pan, L.; Li, C.; Meng, L.; Tian, Y.; He, M.; Yuan, X.; Zhang, G.; Zhang, Z.; Xiong, J.; Chen, G.; et al. Tau accelerates alpha-synuclein aggregation and spreading in Parkinson’s disease. Brain 2022, 145, 3454–3471. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Meng, L.; He, M.; Zhang, Z. Tau in the Pathophysiology of Parkinson’s Disease. J. Mol. Neurosci. 2021, 71, 2179–2191. [Google Scholar] [CrossRef]
- Fernández-Nogales, M.; Lucas, J.J. Altered Levels and Isoforms of Tau and Nuclear Membrane Invaginations in Huntington’s Disease. Front. Cell. Neurosci. 2020, 13, 574. [Google Scholar] [CrossRef]
- Mees, I.; Nisbet, R.M.; Hannan, A.J.; Renoir, T. Implications of Tau Dysregulation in Huntington’s Disease and Potential for New Therapeutics. J. Huntington’s Dis. 2023, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Noël, A.; Julien, C.; Cisbani, G.; Milot-Rousseau, P.; Morin, F.; Dickler, M.; Goupil, C.; Bezeau, F.; Poitras, I.; et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Avila, J. Tauopathies. Cell Mol. Life Sci. 2007, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Khera, A.; Alajangi, H.K.; Sharma, A.; Jaiswal, P.K.; Singh, G.; Barnwal, R.P. Role of Tau in Various Tauopathies, Treatment Approaches, and Emerging Role of Nanotechnology in Neurodegenerative Disorders. Mol. Neurobiol. 2023, 60, 1690–1720. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Shen, F.; Regmi, D.; Petersen, K.; Karim, M.R.U.; Du, D. Tau liquid-liquid phase separation: At the crossroads of tau physiology and tauopathy. J. Cell. Physiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, J.; Wang, X.; Su, Z.; Gao, M.; Huang, Y. Liquid—Liquid phase separation of tau: Driving forces, regulation, and biological implications. Neurobiol. Dis. 2023, 183, 106167. [Google Scholar] [CrossRef] [PubMed]
- Ainani, H.; Bouchmaa, N.; Ben Mrid, R.; El Fatimy, R. Liquid-liquid phase separation of protein tau: An emerging process in Alzheimer’s disease pathogenesis. Neurobiol. Dis. 2023, 178, 106011. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, G.A.P.; Cordeiro, Y.; Silva, J.L.; Vieira, T. Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases. Adv. Protein Chem. Struct. Biol. 2019, 118, 289–331. [Google Scholar] [CrossRef]
- Ferreon, J.C.; Jain, A.; Choi, K.J.; Tsoi, P.S.; MacKenzie, K.R.; Jung, S.Y.; Ferreon, A.C. Acetylation Disfavors Tau Phase Separation. Int. J. Mol. Sci. 2018, 19, 1360. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, B.; Chen, X.; Chu, L. Liquid-liquid phase separation in Alzheimer’s disease. J. Mol. Med. 2024, 102, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [PubMed]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020, 12, eaau5732. [Google Scholar] [CrossRef] [PubMed]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Prabakaran, S.; Cantrelle, F.X.; Chambraud, B.; Gunawardena, J.; Lippens, G.; Landrieu, I. Characterization of Neuronal Tau Protein as a Target of Extracellular Signal-regulated Kinase. J. Biol. Chem. 2016, 291, 7742–7753. [Google Scholar] [CrossRef] [PubMed]
- Stefanoska, K.; Gajwani, M.; Tan, A.R.P.; Ahel, H.I.; Asih, P.R.; Volkerling, A.; Poljak, A.; Ittner, A. Alzheimer’s disease: Ablating single master site abolishes tau hyperphosphorylation. Sci. Adv. 2022, 8, eabl8809. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Yao, X.Q.; Zhang, X.X.; Yin, Y.Y.; Liu, B.; Luo, D.J.; Liu, D.; Chen, N.N.; Ni, Z.F.; Wang, X.; Wang, Q.; et al. Glycogen synthase kinase-3beta regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not Src. Biochem. J. 2011, 437, 335–344. [Google Scholar] [CrossRef]
- Sontag, J.M.; Schuhmacher, D.; Taleski, G.; Jordan, A.; Khan, S.; Hoffman, A.; Gomez, R.J.; Mazalouskas, M.D.; Hanks, S.K.; Spiller, B.W.; et al. A new paradigm for regulation of protein phosphatase 2A function via Src and Fyn kinase-mediated tyrosine phosphorylation. J. Biol. Chem. 2022, 298, 102248. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Gu, J.; Yin, X.; Jin, N.; Xie, S.; Wang, Y.; Chang, H.; Qian, W.; Shi, J.; et al. Cross talk between PI3K-AKT-GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiol. Aging 2015, 36, 188–200. [Google Scholar] [CrossRef]
- Arif, M.; Kazim, S.F.; Grundke-Iqbal, I.; Garruto, R.M.; Iqbal, K. Tau pathology involves protein phosphatase 2A in parkinsonism-dementia of Guam. Proc. Natl. Acad. Sci. USA 2014, 111, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Martin-Avila, A.; Medina-Tamayo, J.; Ibarra-Sanchez, A.; Vazquez-Victorio, G.; Castillo-Arellano, J.I.; Hernandez-Mondragon, A.C.; Rivera, J.; Madera-Salcedo, I.K.; Blank, U.; Macias-Silva, M.; et al. Protein Tyrosine Kinase Fyn Regulates TLR4-Elicited Responses on Mast Cells Controlling the Function of a PP2A-PKCalpha/beta Signaling Node Leading to TNF Secretion. J. Immunol. 2016, 196, 5075–5088. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Belrose, J.; Trepanier, C.H.; Lei, G.; Jackson, M.F.; MacDonald, J.F. Fyn, a potential target for Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kabir, F.; Atkinson, R.; Cook, A.L.; Phipps, A.J.; King, A.E. The role of altered protein acetylation in neurodegenerative disease. Front. Aging Neurosci. 2023, 14, 1025473. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.; Claiborn, K.C.; Gan, L. Regulation of Tau Homeostasis and Toxicity by Acetylation. Adv. Exp. Med. Biol. 2019, 1184, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.K.; Vázquez-Rosa, E.; Koh, Y.; Dhar, M.; Chaubey, K.; Cintrón-Pérez, C.J.; Barker, S.; Miller, E.; Franke, K.; Noterman, M.F.; et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021, 184, 2715–2732.e2723. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Rivière, G.; Hebestreit, A.; de Opakua, A.I.; Vorberg, I.M.; Andreas, L.B.; Zweckstetter, M. Acetylation discriminates disease-specific tau deposition. Nat. Commun. 2023, 14, 5919. [Google Scholar] [CrossRef] [PubMed]
- KPark, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef]
- Kim, M.-S.; Mun, Y.-S.; Lee, S.-E.; Cho, W.-Y.; Han, S.-H.; Kim, D.-H.; Yoon, S.-Y. Tau acetylation at K280 regulates tau phosphorylation. Int. J. Neurosci. 2022, 133, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Liu, F. Pathological Changes of Tau Related to Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 931–944. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Hu, Y.; Zhao, J.-N.; Huang, C.-H.; Li, T.; Zhang, B.-G.; He, Y.; Wu, Y.-Q.; Zhang, Z.-J.; et al. Acetylated tau exacerbates learning and memory impairment by disturbing with mitochondrial homeostasis. Redox Biol. 2023, 62, 102697. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.; McClean, P.L.; Smyth, T.; Doherty, M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines 2019, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Losev, Y.; Frenkel-Pinter, M.; Abu-Hussien, M.; Viswanathan, G.K.; Elyashiv-Revivo, D.; Geries, R.; Khalaila, I.; Gazit, E.; Segal, D. Differential effects of putative N-glycosylation sites in human Tau on Alzheimer’s disease-related neurodegeneration. Cell Mol. Life Sci. 2021, 78, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lang, M. New insight into protein glycosylation in the development of Alzheimer’s disease. Cell Death Discov. 2023, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, P.; Kang, H.; Lee, B. Glycosylation and behavioral symptoms in neurological disorders. Transl. Psychiatry 2023, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Takatani, T.; Yeh, K.; Hsu, J.W.; King, M.R. Targeting Underglycosylated MUC1 for the Selective Capture of Highly Metastatic Breast Cancer Cells Under Flow. Cell Mol. Bioeng. 2013, 6, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Kontaxi, C.; Piccardo, P.; Gill, A.C. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A.; et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef]
- Rathert, P.; Dhayalan, A.; Murakami, M.; Zhang, X.; Tamas, R.; Jurkowska, R.; Komatsu, Y.; Shinkai, Y.; Cheng, X.; Jeltsch, A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 2008, 4, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Balmik, A.A.; Chinnathambi, S. Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun. Signal. 2021, 19, 51. [Google Scholar] [CrossRef]
- Xia, Y.; Bell, B.M.; Giasson, B.I. Tau Lysine Pseudomethylation Regulates Microtubule Binding and Enhances Prion-like Tau Aggregation. Int. J. Mol. Sci. 2023, 24, 8286. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Liu, H.; Lu, W.; Kotzbauer, P.T.; Bateman, R.J.; Lucey, B.P. Sleep Deprivation Affects Tau Phosphorylation in Human Cerebrospinal Fluid. Ann. Neurol. 2020, 87, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Hu, M.T.M. Sleep Disturbance as Potential Risk and Progression Factor for Parkinson’s Disease. J. Park. Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef]
- Faraco, G.; Brea, D.; Garcia-Bonilla, L.; Wang, G.; Racchumi, G.; Chang, H.; Buendia, I.; Santisteban, M.M.; Segarra, S.G.; Koizumi, K.; et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat. Neurosci. 2018, 21, 240–249. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Z.H.; Kang, S.S.; Liu, X.; Xia, Y.; Chan, C.B.; Ye, K. High-fat diet-induced diabetes couples to Alzheimer’s disease through inflammation-activated C/EBPbeta/AEP pathway. Mol. Psychiatry 2022, 27, 3396–3409. [Google Scholar] [CrossRef]
- Elahi, M.; Motoi, Y.; Shimonaka, S.; Ishida, Y.; Hioki, H.; Takanashi, M.; Ishiguro, K.; Imai, Y.; Hattori, N. High-fat diet-induced activation of SGK1 promotes Alzheimer’s disease-associated tau pathology. Hum. Mol. Genet. 2021, 30, 1693–1710. [Google Scholar] [CrossRef]
- Hantikainen, E.; Roos, E.; Bellocco, R.; D’Antonio, A.; Grotta, A.; Adami, H.O.; Ye, W.; Trolle Lagerros, Y.; Bonn, S. Dietary fat intake and risk of Parkinson disease: Results from the Swedish National March Cohort. Eur. J. Epidemiol. 2022, 37, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Lee, P.C.; Hansen, J.; Lassen, C.F.; Ketzel, M.; Sørensen, M.; Raaschou-Nielsen, O. Traffic-Related Air Pollution and Parkinson’s Disease in Denmark: A Case-Control Study. Environ. Health Perspect. 2016, 124, 351–356. [Google Scholar] [CrossRef]
- Tanner, C.M.; Chen, B.; Wang, W.; Peng, M.; Liu, Z.; Liang, X.; Kao, L.C.; Gilley, D.W.; Goetz, C.G.; Schoenberg, B.S. Environmental factors and Parkinson’s disease: A case-control study in China. Neurology 1989, 39, 660–664. [Google Scholar] [CrossRef]
- Tong, S.; Yang, L.; Gong, H.; Wang, L.; Li, H.; Yu, J.; Li, Y.; Deji, Y.; Nima, C.; Zhao, S.; et al. Bioaccumulation characteristics, transfer model of heavy metals in soil-crop system and health assessment in plateau region, China. Ecotoxicol. Environ. Saf. 2022, 241, 113733. [Google Scholar] [CrossRef]
- Marizzoni, M.; Mirabelli, P.; Mombelli, E.; Coppola, L.; Festari, C.; Lopizzo, N.; Luongo, D.; Mazzelli, M.; Naviglio, D.; Blouin, J.L.; et al. A peripheral signature of Alzheimer’s disease featuring microbiota-gut-brain axis markers. Alzheimers Res. Ther. 2023, 15, 101. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Sisodia, S.S.; Vassar, R.J. The gut microbiome in Alzheimer’s disease: What we know and what remains to be explored. Mol. Neurodegener. 2023, 18, 9. [Google Scholar] [CrossRef]
- Seo, D.O.; O’Donnell, D.; Jain, N.; Ulrich, J.D.; Herz, J.; Li, Y.; Lemieux, M.; Cheng, J.; Hu, H.; Serrano, J.R.; et al. ApoE isoform- and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science 2023, 379, eadd1236. [Google Scholar] [CrossRef] [PubMed]
- Filippini, T.; Adani, G.; Malavolti, M.; Garuti, C.; Cilloni, S.; Vinceti, G.; Zamboni, G.; Tondelli, M.; Galli, C.; Costa, M.; et al. Dietary Habits and Risk of Early-Onset Dementia in an Italian Case-Control Study. Nutrients 2020, 12, 3682. [Google Scholar] [CrossRef]
- Glans, I.; Sonestedt, E.; Nägga, K.; Gustavsson, A.M.; González-Padilla, E.; Borne, Y.; Stomrud, E.; Melander, O.; Nilsson, P.M.; Palmqvist, S.; et al. Association Between Dietary Habits in Midlife With Dementia Incidence Over a 20-Year Period. Neurology 2023, 100, e28–e37. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Gao, X.; Na, M.; Kris-Etherton, P.M.; Mitchell, D.C.; Jensen, G.L. Dietary Pattern, Diet Quality, and Dementia: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. J. Alzheimers Dis. 2020, 78, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Wu, Y.T.; Chang, Y.C. Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimers Res. Ther. 2017, 9, 56. [Google Scholar] [CrossRef]
- Sparks Stein, P.; Steffen, M.J.; Smith, C.; Jicha, G.; Ebersole, J.L.; Abner, E.; Dawson, D., 3rd. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 196–203. [Google Scholar] [CrossRef]
- Botelho, J.; Leira, Y.; Viana, J.; Machado, V.; Lyra, P.; Aldrey, J.M.; Pias-Peleteiro, J.M.; Blanco, J.; Sobrino, T.; Mendes, J.J. The Role of Inflammatory Diet and Vitamin D on the Link between Periodontitis and Cognitive Function: A Mediation Analysis in Older Adults. Nutrients 2021, 13, 924. [Google Scholar] [CrossRef] [PubMed]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef]
- Haditsch, U.; Roth, T.; Rodriguez, L.; Hancock, S.; Cecere, T.; Nguyen, M.; Arastu-Kapur, S.; Broce, S.; Raha, D.; Lynch, C.C.; et al. Alzheimer’s Disease-Like Neurodegeneration in Porphyromonas gingivalis Infected Neurons with Persistent Expression of Active Gingipains. J. Alzheimers Dis. 2020, 75, 1361–1376. [Google Scholar] [CrossRef] [PubMed]
- Kanagasingam, S.; von Ruhland, C.; Welbury, R.; Singhrao, S.K. Antimicrobial, Polarizing Light, and Paired Helical Filament Properties of Fragmented Tau Peptides of Selected Putative Gingipains. J. Alzheimers Dis. 2022, 89, 1279–1291. [Google Scholar] [CrossRef]
- Farrugia, C.; Stafford, G.P.; Murdoch, C. Porphyromonas gingivalis Outer Membrane Vesicles Increase Vascular Permeability. J. Dent. Res. 2020, 99, 1494–1501. [Google Scholar] [CrossRef]
- Gong, T.; Chen, Q.; Mao, H.; Zhang, Y.; Ren, H.; Xu, M.; Chen, H.; Yang, D. Outer membrane vesicles of Porphyromonas gingivalis trigger NLRP3 inflammasome and induce neuroinflammation, tau phosphorylation, and memory dysfunction in mice. Front. Cell Infect. Microbiol. 2022, 12, 925435. [Google Scholar] [CrossRef]
- Olsen, I.; Singhrao, S.K. Interaction between genetic factors, Porphyromonas gingivalis and microglia to promote Alzheimer’s disease. J. Oral. Microbiol. 2020, 12, 1820834. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, X.; Yan, X.; Mizutani, S.; Kashiwazaki, H.; Ni, J.; Wu, Z. GSK3beta is involved in promoting Alzheimer’s disease pathologies following chronic systemic exposure to Porphyromonas gingivalis lipopolysaccharide in amyloid precursor protein(NL-F/NL-F) knock-in mice. Brain Behav. Immun. 2021, 98, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Qiu, W.; Zhu, X.; Li, X.; Xie, Z.; Carreras, I.; Dedeoglu, A.; Van Dyke, T.; Han, Y.W.; Karimbux, N.; et al. The Periodontal Pathogen Fusobacterium nucleatum Exacerbates Alzheimer’s Pathogenesis via Specific Pathways. Front. Aging Neurosci. 2022, 14, 912709. [Google Scholar] [CrossRef]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef]
- Plachokova, A.S.; Gjaltema, J.; Hagens, E.R.C.; Hashemi, Z.; Knüppe, T.B.A.; Kootstra, T.J.M.; Visser, A.; Bloem, B.R. Periodontitis: A Plausible Modifiable Risk Factor for Neurodegenerative Diseases? A Comprehensive Review. Int. J. Mol. Sci. 2024, 25, 4504. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Leveille, E.; Ross, O.A.; Gan-Or, Z. Tau and MAPT genetics in tauopathies and synucleinopathies. Park. Relat. Disord. 2021, 90, 142–154. [Google Scholar] [CrossRef]
- Sarnowski, C.; Ghanbari, M.; Bis, J.C.; Logue, M.; Fornage, M.; Mishra, A.; Ahmad, S.; Beiser, A.S.; Boerwinkle, E.; Bouteloup, V.; et al. Meta-analysis of genome-wide association studies identifies ancestry-specific associations underlying circulating total tau levels. Commun. Biol. 2022, 5, 336. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L.; et al. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef]
- Nazarian, A.; Philipp, I.; Culminskaya, I.; He, L.; Kulminski, A.M. Inter- and intra-chromosomal modulators of the APOE varepsilon2 and varepsilon4 effects on the Alzheimer’s disease risk. Geroscience 2023, 45, 233–247. [Google Scholar] [CrossRef]
- Pereira, J.B.; Harrison, T.M.; La Joie, R.; Baker, S.L.; Jagust, W.J. Spatial patterns of tau deposition are associated with amyloid, ApoE, sex, and cognitive decline in older adults. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2155–2164. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama 1997, 278, 1349–1356. [Google Scholar]
- Than, S.; Moran, C.; Beare, R.; Vincent, A.J.; Collyer, T.A.; Wang, W.; Callisaya, M.L.; Thomson, R.; Phan, T.G.; Fornito, A.; et al. Interactions Between Age, Sex, Menopause, and Brain Structure at Midlife: A UK Biobank Study. J. Clin. Endocrinol. Metab. 2021, 106, 410–420. [Google Scholar] [CrossRef]
- Buckley, R.F.; Scott, M.R.; Jacobs, H.I.L.; Schultz, A.P.; Properzi, M.J.; Amariglio, R.E.; Hohman, T.J.; Mayblyum, D.V.; Rubinstein, Z.B.; Manning, L.; et al. Sex Mediates Relationships Between Regional Tau Pathology and Cognitive Decline. Ann. Neurol. 2020, 88, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Carrière, I.; Carcaillon, L.; Dartigues, J.F.; Auriacombe, S.; Rouaud, O.; Berr, C.; Ritchie, K.; Scarabin, P.Y.; Ancelin, M.L. Estrogen receptor polymorphisms and incident dementia: The prospective 3C study. Alzheimer’s Dement. 2013, 10, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Taxier, L.R.; Philippi, S.M.; York, J.M.; LaDu, M.J.; Frick, K.M. The detrimental effects of APOE4 on risk for Alzheimer’s disease may result from altered dendritic spine density, synaptic proteins, and estrogen receptor alpha. Neurobiol. Aging 2022, 112, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Woody, S.K.; Chhibber, A. Estrogen receptor β in Alzheimer’s disease: From mechanisms to therapeutics. Ageing Res. Rev. 2015, 24, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.N.M.; Hornberger, M.; Ritchie, C.W.; Minihane, A.M. Hormone replacement therapy is associated with improved cognition and larger brain volumes in at-risk APOE4 women: Results from the European Prevention of Alzheimer’s Disease (EPAD) cohort. Alzheimers Res. Ther. 2023, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, X.; Chaput, D.; Shin, M.-K.; Koh, Y.; Gan, L.; Pieper, A.A.; Woo, J.-A.A.; Kang, D.E. X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 2022, 185, 3913–3930.e3919. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Ge, Y.J.; Tan, C.C.; Cao, X.P.; Tan, L.; Xu, W. The Proportion of APOE4 Carriers Among Non-Demented Individuals: A Pooled Analysis of 389,000 Community-Dwellers. J. Alzheimers Dis. 2021, 81, 1331–1339. [Google Scholar] [CrossRef]
- Ham, S.; Kim, T.K.; Lee, S.; Tang, Y.-P.; Im, H.-I. MicroRNA Profiling in Aging Brain of PSEN1/PSEN2 Double Knockout Mice. Mol. Neurobiol. 2017, 55, 5232–5242. [Google Scholar] [CrossRef] [PubMed]
- Mirnics, K.; Norstrom, E.M.; Garbett, K.; Choi, S.; Zhang, X.; Ebert, P.; Sisodia, S.S. Molecular signatures of neurodegeneration in the cortex of PS1/PS2 double knockout mice. Mol. Neurodegener. 2008, 3, 14. [Google Scholar] [CrossRef]
- Koga, S.; Zhou, X.; Murakami, A.; Fernandez De Castro, C.; Baker, M.C.; Rademakers, R.; Dickson, D.W. Concurrent tau pathologies in frontotemporal lobar degeneration with TDP-43 pathology. Neuropathol. Appl. Neurobiol. 2021, 48, e12778. [Google Scholar] [CrossRef]
- Logan, T.; Simon, M.J.; Rana, A.; Cherf, G.M.; Srivastava, A.; Davis, S.S.; Low, R.L.Y.; Chiu, C.-L.; Fang, M.; Huang, F.; et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 2021, 184, 4651–4668.e4625. [Google Scholar] [CrossRef] [PubMed]
- Tyler, C.R.; Allan, A.M. The Effects of Arsenic Exposure on Neurological and Cognitive Dysfunction in Human and Rodent Studies: A Review. Curr. Env. Health Rep. 2014, 1, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Ariafar, S.; Makhdoomi, S.; Mohammadi, M. Arsenic and Tau Phosphorylation: A Mechanistic Review. Biol. Trace Elem. Res. 2023, 201, 5708–5720. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Jing, X.P.; Zhou, X.W.; Wang, X.L.; Yang, Y.; Sun, X.Y.; Qiu, M.; Cao, F.Y.; Lu, Y.M.; Liu, R.; et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol. Aging 2013, 34, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Liuyang, Z.Y.; Lei, P.; Yan, X.; Shentu, Y.P.; Liang, J.W.; Zhou, H.; Pei, L.; Xiong, Y.; Hou, T.Y.; et al. Zinc induces CDK5 activation and neuronal death through CDK5-Tyr15 phosphorylation in ischemic stroke. Cell Death Dis. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Kalionis, B.; Murthi, P.; Xia, S.; Guan, Y. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front. Neurol. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Laferla, F.M. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 2009, 108, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Mateo, D.; Marquès, M.; Torrente, M. Metals linked with the most prevalent primary neurodegenerative dementias in the elderly: A narrative review. Environ. Res. 2023, 236, 116722. [Google Scholar] [CrossRef] [PubMed]
- Newell, M.E.; Babbrah, A.; Aravindan, A.; Rathnam, R.; Kiernan, R.; Driver, E.M.; Bowes, D.A.; Halden, R.U. Prevalence rates of neurodegenerative diseases versus human exposures to heavy metals across the United States. Sci. Total Environ. 2024, 928, 172260. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wu, L.; Su, Q.; Ji, X.; Zhou, J.; Wu, S.; Tang, Y.; Li, H. Neurotoxic effects of heavy metal pollutants in the environment: Focusing on epigenetic mechanisms. Environ. Pollut. 2024, 345, 123563. [Google Scholar] [CrossRef]
- Zheng, H.B.; Fu, Y.T.; Wang, G.; Sun, L.H.; Fan, Y.Y.; Yang, T.W. Hyperphosphorylation of protein Tau in hippocampus may cause cognitive dysfunction of propofol-anesthetized rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Le Freche, H.; Brouillette, J.; Fernandez-Gomez, F.J.; Patin, P.; Caillierez, R.; Zommer, N.; Sergeant, N.; Buee-Scherrer, V.; Lebuffe, G.; Blum, D.; et al. Tau phosphorylation and sevoflurane anesthesia: An association to postoperative cognitive impairment. Anesthesiology 2012, 116, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, Y.; Tan, H.; Boukhali, M.; Khatri, A.; Yu, Y.; Hua, F.; Liu, L.; Li, M.; Yang, G.; et al. Tau Contributes to Sevoflurane-induced Neurocognitive Impairment in Neonatal Mice. Anesthesiology 2020, 133, 595–610. [Google Scholar] [CrossRef]
- Wang, H.H.; Li, Y.; Li, A.; Yan, F.; Li, Z.L.; Liu, Z.Y.; Zhang, L.; Zhang, J.; Dong, W.R.; Zhang, L. Forskolin Induces Hyperphosphorylation of Tau Accompanied by Cell Cycle Reactivation in Primary Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. Molecular and cellular mechanism of okadaic acid (OKA)-induced neurotoxicity: A novel tool for Alzheimer’s disease therapeutic application. Mol. Neurobiol. 2014, 50, 852–865. [Google Scholar] [CrossRef]
- Congdon, E.E.; Ji, C.; Tetlow, A.M.; Jiang, Y.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease: Current status and future directions. Nat. Rev. Neurol. 2023, 19, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Alzforum. Antisense Therapy Stifles CSF Tau in Mild Alzheimer’s Disease. 2021. Available online: https://www.alzforum.org/news/conference-coverage/antisense-therapy-stifles-csftau-mild-alzheimers-disease-0 (accessed on 22 May 2024).
- Chohan, M.O.; Khatoon, S.; Iqbal, I.G.; Iqbal, K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006, 580, 3973–3979. [Google Scholar] [CrossRef] [PubMed]
- Vivash, L.; Bertram, K.L.; Malpas, C.B.; Marotta, C.; Harding, I.H.; Kolbe, S.; Fielding, J.; Clough, M.; Lewis, S.J.G.; Tisch, S.; et al. Sodium selenate as a disease-modifying treatment for progressive supranuclear palsy: Protocol for a phase 2, randomised, double-blind, placebo-controlled trial. BMJ Open 2021, 11, e055019. [Google Scholar] [CrossRef]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmüller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef]
- Duthie, A.; van Aalten, L.; MacDonald, C.; McNeilly, A.; Gallagher, J.; Geddes, J.; Lovestone, S.; Sutherland, C. Recruitment, Retainment, and Biomarkers of Response; A Pilot Trial of Lithium in Humans With Mild Cognitive Impairment. Front. Mol. Neurosci. 2019, 12, 163. [Google Scholar] [CrossRef]
- Small, G.W.; Siddarth, P.; Li, Z.; Miller, K.J.; Ercoli, L.; Emerson, N.D.; Martinez, J.; Wong, K.P.; Liu, J.; Merrill, D.A.; et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr. Psychiatry 2018, 26, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P.; et al. FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Immune, A. AC Immune’s Alzheimer’s Disease Vaccine-Candidate ACI-35.030 Selected for Further Development. Available online: https://ir.acimmune.com/news-releases/news-release-details/ac-immunes-alzheimers-disease-vaccine-candidate-aci-35030 (accessed on 22 May 2024).
- Tai, H.C.; Ma, H.T.; Huang, S.C.; Wu, M.F.; Wu, C.L.; Lai, Y.T.; Li, Z.L.; Margolin, R.; Intorcia, A.J.; Serrano, G.E.; et al. The Tau Oligomer Antibody APNmAb005 Detects Early-Stage Pathological Tau Enriched at Synapses and Rescues Neuronal Loss in Long-Term Treatments. BioRxiv. Available online: https://www.biorxiv.org/content/10.1101/2022.06.24.497452v1 (accessed on 22 May 2024).
- Courade, J.P.; Angers, R.; Mairet-Coello, G.; Pacico, N.; Tyson, K.; Lightwood, D.; Munro, R.; McMillan, D.; Griffin, R.; Baker, T.; et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018, 136, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Helboe, L.; Rosenqvist, N.; Volbracht, C.; Pedersen, L.; Pedersen, J.T.; Christensen, S.; Egebjerg, J.; Christoffersen, C.T.; Bang-Andersen, B.; Beach, T.G.; et al. Highly Specific and Sensitive Target Binding by the Humanized pS396-Tau Antibody hC10.2 Across a Wide Spectrum of Alzheimer’s Disease and Primary Tauopathy Postmortem Brains. J. Alzheimers Dis. 2022, 88, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Eisai. Eisai Presents Data Showing Quantification of Tau Microtubule Binding Region in Cerebrospinal Fluid and the Identification of a Target Engagement Biomarker for the New Anti-Tau Antibody E2814 at Alzheimer’s Association International Conference (AAIC). Eisai, 2019. Available online: https://www.eisai.com/news/2019/news201955.html (accessed on 22 May 2024).
- Alzforum. Therapeutics: BIIB076. Alzforum. Available online: https://www.alzforum.org/therapeutics/biib076 (accessed on 22 May 2024).
- Boxer, A.L.; Qureshi, I.; Ahlijanian, M.; Grundman, M.; Golbe, L.I.; Litvan, I.; Honig, L.S.; Tuite, P.; McFarland, N.R.; O’Suilleabhain, P.; et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: A randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet. Neurol. 2019, 18, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Alzforum. Therapeutics: MK-2214. Alzforum. Available online: https://www.alzforum.org/therapeutics/mk-2214 (accessed on 22 May 2024).
- Alzforum. Therapeutics: PRX005. Alzforum. Available online: https://www.alzforum.org/therapeutics/prx005 (accessed on 22 May 2024).
- Alzforum. Therapeutics: JNJ-63733657. Alzforum. Available online: https://www.alzforum.org/therapeutics/jnj-63733657 (accessed on 22 May 2024).
- Wiseman, A.L.; Briggs, C.A.; Peritt, A.; Kapecki, N.; Peterson, D.A.; Shim, S.S.; Stutzmann, G.E. Lithium Provides Broad Therapeutic Benefits in an Alzheimer’s Disease Mouse Model. J. Alzheimers Dis. 2023, 91, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, G.U.; Huppertz, H.J.; Wagenpfeil, S.; Andres, M.V.; Belloch, V.; Leon, T.; Del Ser, T.; Investigators, T.M. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov. Disord. 2014, 29, 479–487. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef]
- Tang, S.J.; Fesharaki-Zadeh, A.; Takahashi, H.; Nies, S.H.; Smith, L.M.; Luo, A.; Chyung, A.; Chiasseu, M.; Strittmatter, S.M. Fyn kinase inhibition reduces protein aggregation, increases synapse density and improves memory in transgenic and traumatic Tauopathy. Acta Neuropathol. Commun. 2020, 8, 96. [Google Scholar] [CrossRef]
- Wadhwa, P.; Jain, P.; Jadhav, H.R. Glycogen Synthase Kinase 3 (GSK3): Its Role and Inhibitors. Curr. Top. Med. Chem. 2020, 20, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef]
- Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann. Neurol. 2020, 88, 183–194. [Google Scholar] [CrossRef]
- Stevenson, M.; Varghese, R.; Hebron, M.L.; Liu, X.; Ratliff, N.; Smith, A.; Turner, R.S.; Moussa, C. Inhibition of discoidin domain receptor (DDR)-1 with nilotinib alters CSF miRNAs and is associated with reduced inflammation and vascular fibrosis in Alzheimer’s disease. J. Neuroinflammation 2023, 20, 116. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, X.; Gao, W.; Wang, Q.; Zhang, L.; Li, Y.; Li, L.; Zhang, L. Cornel Iridoid Glycoside Inhibits Tau Hyperphosphorylation via Regulating Cross-Talk Between GSK-3beta and PP2A Signaling. Front. Pharmacol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Saletti, P.G.; Casillas-Espinosa, P.M.; Lisgaras, C.P.; Mowrey, W.B.; Li, Q.; Liu, W.; Brady, R.D.; Ali, I.; Silva, J.; Yamakawa, G.; et al. Tau Phosphorylation Patterns in the Rat Cerebral Cortex After Traumatic Brain Injury and Sodium Selenate Effects: An Epibios4rx Project 2 Study. J. Neurotrauma 2023, 11, 1154. [Google Scholar] [CrossRef] [PubMed]
- van Eersel, J.; Ke, Y.D.; Liu, X.; Delerue, F.; Kril, J.J.; Gotz, J.; Ittner, L.M. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. USA 2010, 107, 13888–13893. [Google Scholar] [CrossRef]
- Wang, H.; Sui, H.; Zheng, Y.; Jiang, Y.; Shi, Y.; Liang, J.; Zhao, L. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3beta pathway. Nanoscale 2019, 11, 7481–7496. [Google Scholar] [CrossRef]
- Li, H.Q.; Ip, S.P.; Yuan, Q.J.; Zheng, G.Q.; Tsim, K.K.W.; Dong, T.T.X.; Lin, G.; Han, Y.; Liu, Y.; Xian, Y.F.; et al. Isorhynchophylline ameliorates cognitive impairment via modulating amyloid pathology, tau hyperphosphorylation and neuroinflammation: Studies in a transgenic mouse model of Alzheimer’s disease. Brain Behav. Immun. 2019, 82, 264–278. [Google Scholar] [CrossRef]
- Song, H.-L.; Kim, N.-Y.; Park, J.; Kim, M.I.; Jeon, Y.-N.; Lee, S.-J.; Cho, K.; Shim, Y.-L.; Lee, K.-H.; Mun, Y.-S.; et al. Monoclonal antibody Y01 prevents tauopathy progression induced by lysine 280–acetylated tau in cell and mouse models. J. Clin. Investig. 2023, 133, e156537. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bharti; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef]
- Harrington, C.R.; Storey, J.M.D.; Clunas, S.; Harrington, K.A.; Horsley, D.; Ishaq, A.; Kemp, S.J.; Larch, C.P.; Marshall, C.; Nicoll, S.L.; et al. Cellular Models of Aggregation-dependent Template-directed Proteolysis to Characterize Tau Aggregation Inhibitors for Treatment of Alzheimer Disease. J. Biol. Chem. 2015, 290, 10862–10875. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Ono, K.; Yamada, M. REVIEW: Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar] [CrossRef]
- Li, L.; Wang, F.; Jia, X.; Yao, L.; Liu, Y. Research Mechanism and Progress of the Natural Compound Curcumin in Treating Alzheimer’s Disease. Mini Rev. Med. Chem. 2023, 24, 1590–1601. [Google Scholar] [CrossRef]
- Yanamandra, K.; Patel, T.K.; Jiang, H.; Schindler, S.; Ulrich, J.D.; Boxer, A.L.; Miller, B.L.; Kerwin, D.R.; Gallardo, G.; Stewart, F.; et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci. Transl. Med. 2017, 9, eaal2029. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 3954. [Google Scholar] [CrossRef]
- Tang, J.J.; Huang, L.F.; Deng, J.L.; Wang, Y.M.; Guo, C.; Peng, X.N.; Liu, Z.; Gao, J.M. Cognitive enhancement and neuroprotective effects of OABL, a sesquiterpene lactone in 5xFAD Alzheimer’s disease mice model. Redox Biol. 2022, 50, 102229. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Bai, K.; Yang, X.; Sun, C.; Ji, Y.; Zhou, J.; Zhang, H.; Ding, Y. “Drug-Carrier” Synergy Therapy for Amyloid-β Clearance and Inhibition of Tau Phosphorylation via Biomimetic Lipid Nanocomposite Assembly. Adv. Sci. 2022, 9, e2106072. [Google Scholar] [CrossRef]
- Abskharon, R.; Pan, H.; Sawaya, M.R.; Seidler, P.M.; Olivares, E.J.; Chen, Y.; Murray, K.A.; Zhang, J.; Lantz, C.; Bentzel, M.; et al. Structure-based design of nanobodies that inhibit seeding of Alzheimer’s patient-extracted tau fibrils. Proc. Natl. Acad. Sci. USA 2023, 120, e2300258120. [Google Scholar] [CrossRef]
- Wasielewska, J.M.; Chaves, J.C.S.; Johnston, R.L.; Milton, L.A.; Hernández, D.; Chen, L.; Song, J.; Lee, W.; Leinenga, G.; Nisbet, R.M.; et al. A sporadic Alzheimer’s blood-brain barrier model for developing ultrasound-mediated delivery of Aducanumab and anti-Tau antibodies. Theranostics 2022, 12, 6826–6847. [Google Scholar] [CrossRef]
- Su, J.; Xiao, Y.; Wei, L.; Lei, H.; Sun, F.; Wang, W.; Yin, J.; Xiong, R.; Li, S.; Zhang, P.; et al. Generation of tau dephosphorylation-targeting chimeras for the treatment of Alzheimer’s disease and related tauopathies. Sci. Bull. 2024, 69, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Basheer, N.; Smolek, T.; Hassan, I.; Liu, F.; Iqbal, K.; Zilka, N.; Novak, P. Does modulation of tau hyperphosphorylation represent a reasonable therapeutic strategy for Alzheimer’s disease? From preclinical studies to the clinical trials. Mol. Psychiatry 2023, 28, 2197–2214. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.J.; Jeong, H.R.; Park, J.H.; Moon, M.; Hoe, H.S. Inhibiting EGFR/HER-2 ameliorates neuroinflammatory responses and the early stage of tau pathology through DYRK1A. Front. Immunol. 2022, 13, 903309. [Google Scholar] [CrossRef]
- Mummery, C.J.; Borjesson-Hanson, A.; Blackburn, D.J.; Vijverberg, E.G.B.; De Deyn, P.P.; Ducharme, S.; Jonsson, M.; Schneider, A.; Rinne, J.O.; Ludolph, A.C.; et al. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: A phase 1b, randomized, placebo-controlled trial. Nat. Med. 2023, 29, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Hu, Z.; An, E.; Okeke, I.; Zheng, S.; Luo, X.; Gong, A.; Jaime-Figueroa, S.; Crews, C.M. Modulation of Phosphoprotein Activity by Phosphorylation Targeting Chimeras (PhosTACs). ACS Chem. Biol. 2021, 16, 2808–2815. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, P.H.; Li, W.; Douglas, T.; Hines, J.; Liu, Y.; Crews, C.M. Targeted Dephosphorylation of Tau by Phosphorylation Targeting Chimeras (PhosTACs) as a Therapeutic Modality. J. Am. Chem. Soc. 2023, 145, 4045–4055. [Google Scholar] [CrossRef]
- Hu, Z.; Crews, C.M. Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. Chembiochem A Eur. J. Chem. Biol. 2022, 23, e202100270. [Google Scholar] [CrossRef]

{kind=link}
| Drug Type | Drug Name (Ongoing Clinical Trials) |
|---|---|
| Reducing tau expression | NIO752, BIIB080 [157] |
| Phosphatase modifiers | Memantine [158], Sodium selenate [159] |
| Kinase inhibitors | Tideglusib [160], Lithium [161] |
| Tau aggregation inhibitors | Curcumin [162] |
| Active immunisation | AADvac1 [163], ACI-35 [164] |
| Passive immunisation | APNmAb005 [165], Bepranemab [166], Lu AF87908 [167], E2814 [168], BIIB076 [169], BIIB092 [170], MK-2214 [171], PRX005 [172], JNJ-63733657 [173] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Zhi, W.; Wang, L. Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review. Molecules 2024, 29, 2812. https://doi.org/10.3390/molecules29122812
Yang J, Zhi W, Wang L. Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review. Molecules. 2024; 29(12):2812. https://doi.org/10.3390/molecules29122812
Chicago/Turabian StyleYang, Jiakai, Weijia Zhi, and Lifeng Wang. 2024. "Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review" Molecules 29, no. 12: 2812. https://doi.org/10.3390/molecules29122812
APA StyleYang, J., Zhi, W., & Wang, L. (2024). Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review. Molecules, 29(12), 2812. https://doi.org/10.3390/molecules29122812