
Graphene Oxide Strengthens Gelatine through Non-Covalent Interactions with Its Amorphous Region



Abstract

1. Introduction
2. Results and Discussion
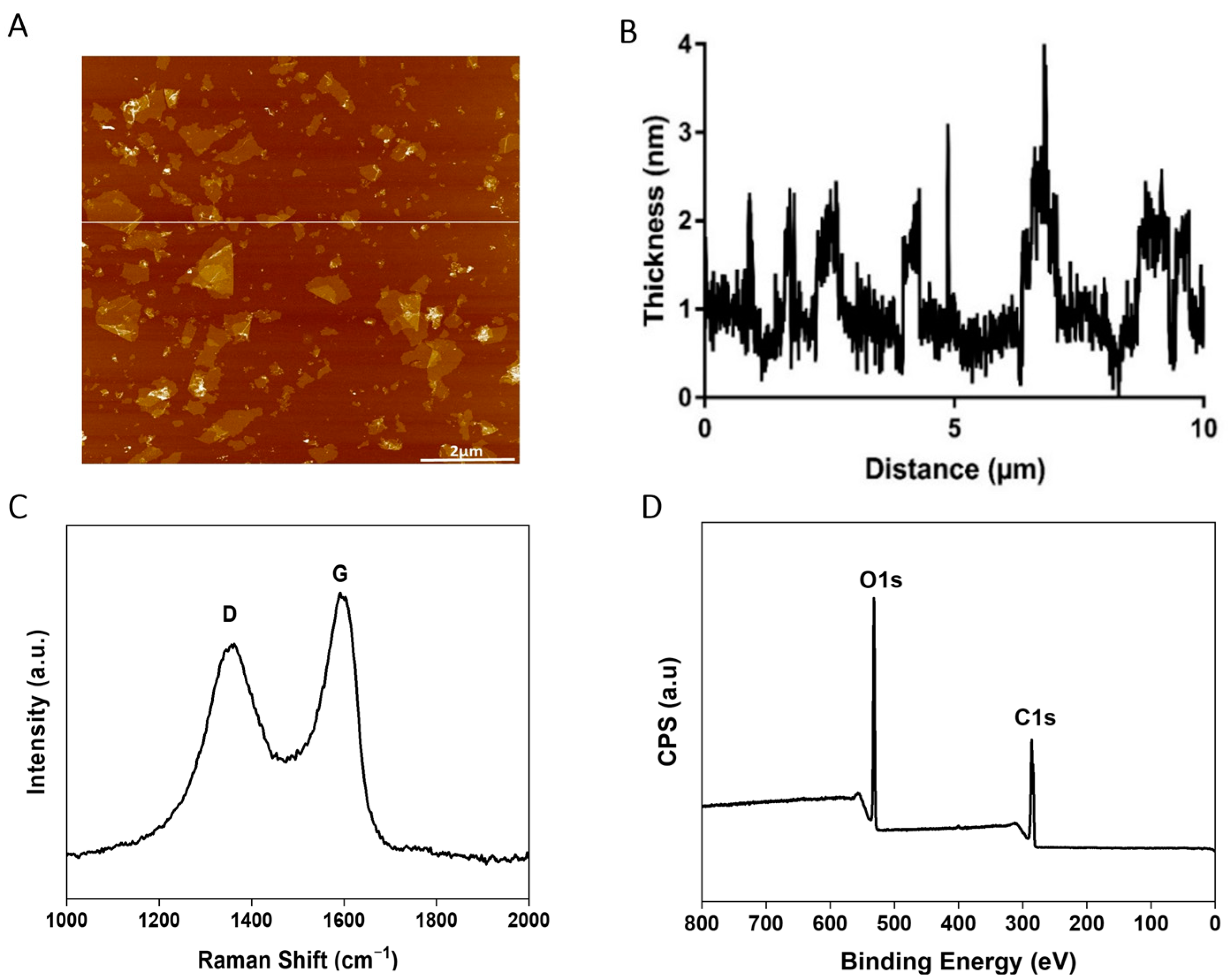
2.1. Characterizations of GO Sample
2.2. Effects of GO on the Mechanical Property of Gelatine-GO Composite Films
2.3. Effects of GO on the Cytotoxicity of HEK-293 Kidney Cells
2.4. Physicochemical Characterization of GO–gelatine Composites
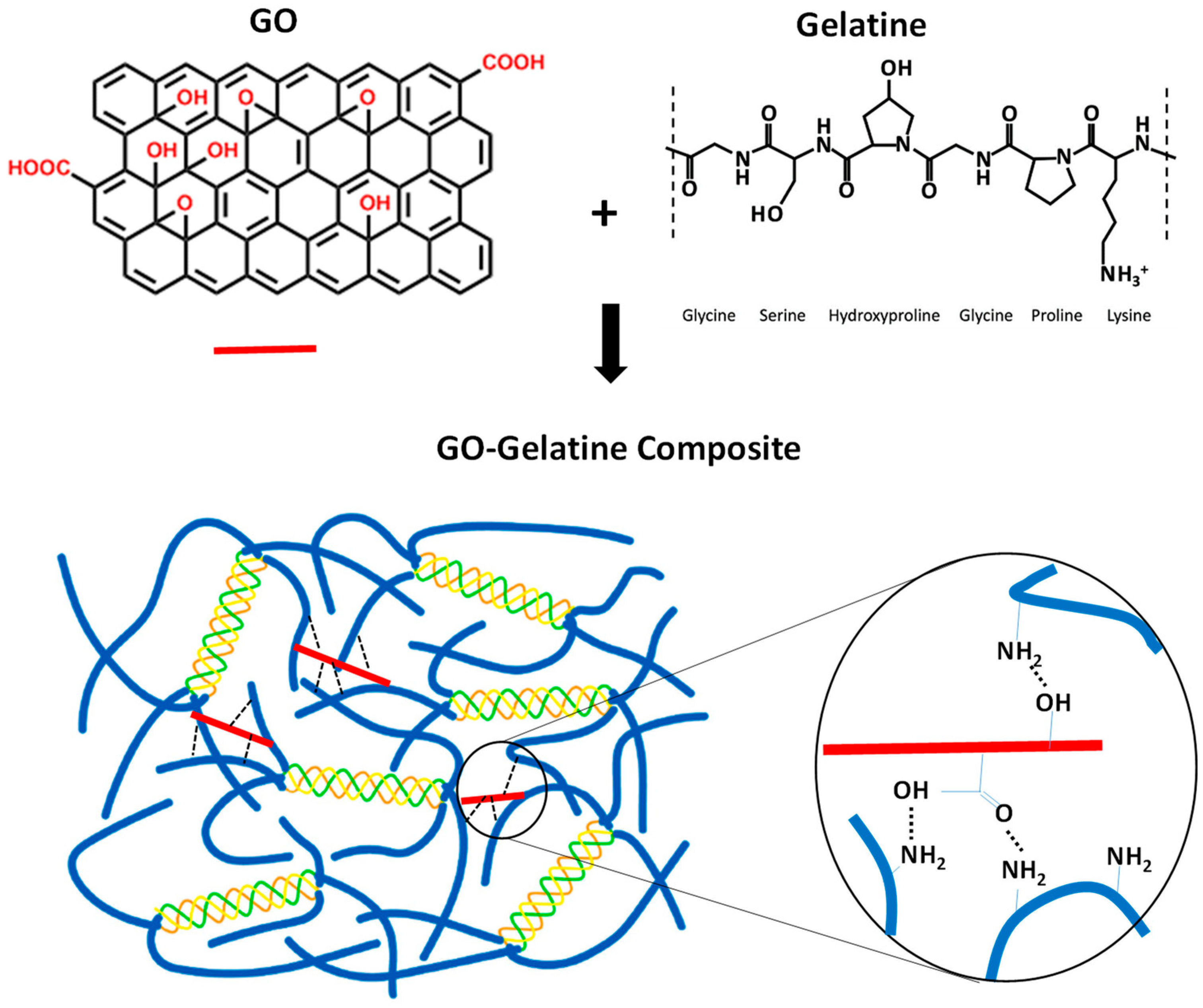
2.5. Mechanism for How GO Improves the Mechanical Property of Gelatine
3. Materials and Methods
3.1. Materials
3.2. GO Synthesis
3.3. Preparation of GO, Gelatine, and GO–gelatine Nanocomposite-Coated Substrates for Biocompatibility Studies
3.4. Preparation of Gelatine and GO–gelatine Nanocomposite Films for Material Characterisations
3.5. Cell Culture
3.6. Cell Viability Assay
3.7. RealTime-GloTM Annexin V Apoptosis-Necrosis Assay
3.8. Circular Dichroism (CD) Measurement
3.9. Physicochemical Material Characterizations of GO/Gelatine Films
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Langer, R.; Vacanti Joseph, P. Tissue Engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Persidis, A. Tissue engineering. Nat. Biotechnol. 1999, 17, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.L.; Holmes, D.F. Collagenous Extracellular Matrix Biomaterials for Tissue Engineering: Lessons from the Common Sea Urchin Tissue. Int. J. Mol. Sci. 2017, 18, 901. [Google Scholar] [CrossRef]
- Xing, H.; Lee, H.; Luo, L.; Kyriakides, T.R. Extracellular Matrix-Derived Biomaterials in Engineering Cell Function. Biotechnol. Adv. 2020, 42, 107421. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. A Review of the Effects of Collagen Treatment in Clinical Studies. Polymers 2021, 13, 3868. [Google Scholar] [CrossRef] [PubMed]
- Djagny, K.B.; Wang, Z.; Xu, S. Gelatin: A Valuable Protein for Food and Pharmaceutical Industries: Review. Crit. Rev. Food Sci. Nutr. 2001, 41, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-J.; Ou, Y.-C. The micro patterning of glutaraldehyde (GA)-crosslinked gelatin and its application to cell-culture. Lab A Chip 2005, 5, 979–984. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, S.; Xu, C.; Wei, B.; Zhang, J.; Xu, Y.; Zhu, B.; Cao, Y.; Wu, X.; Xiong, Z.; et al. A New Method of Gelatin Modified Collagen and Viscoelastic Study of Gelatin-Collagen Composite Hydrogel. Macromol. Res. 2020, 28, 861–868. [Google Scholar] [CrossRef]
- Su, K.; Wang, C. Recent advances in the use of gelatin in biomedical research. Biotechnol. Lett. 2015, 37, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Ikada, Y. Protein release from gelatin matrices. Adv. Drug Deliv. Rev. 1998, 31, 287–301. [Google Scholar] [CrossRef]
- Davidenko, N.; Schuster, C.F.; Bax, D.V.; Farndale, R.W.; Hamaia, S.; Best, S.M.; Cameron, R.E. Evaluation of cell binding to collagen and gelatin: A study of theeffect of 2D and 3D architecture and surface chemistry. J. Mater. Sci. Mater. Med. 2016, 27, 148. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.B.; Kim, D.; Kim, D.; Park, H.; Lee, S.-H. Engineering and Functionalization of Gelatin Biomaterials: From Cell Culture to Medical Applications. Tissue Eng. Part B Rev. 2020, 26, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Kavoosi, G.; Dadfar, S.M.M.; Dadfar, S.M.A.; Ahmadi, F.; Niakosari, M. Investigation of gelatin/multi-walled carbon nanotube nanocomposite films as packaging materials. Food Sci. Nutr. 2014, 2, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Curvello, R.; Raghuwanshi, V.S.; Garnier, G. Engineering nanocellulose hydrogels for biomedical applications. Adv. Colloid Interface Sci. 2019, 267, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, B.; Mohanty, M.; Umashankar, P.R.; Jayakrishnan, A. Evaluation of an in situ forming hydrogel wound dressing based on oxidized alginate and gelatin. Biomaterials 2005, 26, 6335–6342. [Google Scholar] [CrossRef] [PubMed]
- Oun, A.A.; Rhim, J.-W. Preparation and characterization of sodium carboxymethyl cellulose/cotton linter cellulose nanofibril composite films. Carbohydr. Polym. 2015, 127, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Poursamar, S.A.; Lehner, A.N.; Azami, M.; Ebrahimi-Barough, S.; Samadikuchaksaraei, A.; Antunes, A.P.M. The effects of crosslinkers on physical, mechanical, and cytotoxic properties of gelatin sponge prepared via in-situ gas foaming method as a tissue engineering scaffold. Mater. Sci. Eng. C 2016, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, H.; Rasmussen, K.; Sokull-Klüttgen, B. Regulatory Aspects of Nanomaterials in the EU. Chem. Ing. Tech. 2017, 89, 224–231. [Google Scholar] [CrossRef]
- Ghosal, K.; Sarkar, K. Biomedical Applications of Graphene Nanomaterials and Beyond. ACS Biomater. Sci. Eng. 2018, 4, 2653–2703. [Google Scholar] [CrossRef]
- Chung, C.; Kim, Y.-K.; Shin, D.; Ryoo, S.-R.; Hong, B.H.; Min, D.-H. Biomedical Applications of Graphene and Graphene Oxide. Acc. Chem. Res. 2013, 46, 2211–2224. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Feng, W.; Wang, Z. Biomedical applications of chitosan-graphene oxide nanocomposites. iScience 2022, 25, 103629. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Gai, Y.; Wang, W.; Zhao, Y. Green synthesis of biocompatiable chitosan–graphene oxide hybrid nanosheet by ultrasonication method. Ultrason. Sonochem. 2016, 32, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Balapanuru, J.; Yang, J.-X.; Xiao, S.; Bao, Q.; Jahan, M.; Polavarapu, L.; Wei, J.; Xu, Q.-H.; Loh, K.P. A Graphene Oxide–Organic Dye Ionic Complex with DNA-Sensing and Optical-Limiting Properties. Angew. Chem. Int. Ed. 2010, 49, 6549–6553. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Pan, Y.; Ping, Y.; Sahoo, N.G.; Wu, T.; Li, L.; Li, J.; Gan, L.H. Chitosan-Functionalized Graphene Oxide as a Nanocarrier for Drug and Gene Delivery. Small 2011, 7, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wen, H.; Shi, Q.; Zheng, G. Study on immobilization of (+) γ-lactamase using a new type of epoxy graphene oxide carrier. Process Biochem. 2016, 51, 270–276. [Google Scholar] [CrossRef]
- Soozanipour, A.; Taheri-Kafrani, A. Chapter Fourteen—Enzyme Immobilization on Functionalized Graphene Oxide Nanosheets: Efficient and Robust Biocatalysts. In Methods in Enzymology; Kumar, C.V., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 609, pp. 371–403. [Google Scholar]
- Yang, X.; Tu, Y.; Li, L.; Shang, S.; Tao, X.-M. Well-Dispersed Chitosan/Graphene Oxide Nanocomposites. ACS Appl. Mater. Interfaces 2010, 2, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Ramazani, S.; Karimi, M. Aligned poly(ε-caprolactone)/graphene oxide and reduced graphene oxide nanocomposite nanofibers: Morphological, mechanical and structural properties. Mater. Sci. Eng. C 2015, 56, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, X.; Chen, S.; Li, H.; Fu, X.; Wu, Q.; Li, S.; Li, Y.; Su, B.-L.; Ruoff, R.S. One-pot fabrication of FePt/reduced graphene oxide composites as highly active and stable electrocatalysts for the oxygen reduction reaction. Carbon 2014, 68, 755–762. [Google Scholar] [CrossRef]
- Qi, Y.Y.; Tai, Z.X.; Sun, D.F.; Chen, J.T.; Ma, H.B.; Yan, X.B.; Liu, B.; Xue, Q.J. Fabrication and characterization of poly(vinyl alcohol)/graphene oxide nanofibrous biocomposite scaffolds. J. Appl. Polym. Sci. 2013, 127, 1885–1894. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, X.; Li, H.; Wu, W.; Sun, S. Effect of graphene oxide on tensile and flexural properties of carbon/glass hybrid fiber-reinforced polymer composite. Polym. Compos. 2021, 42, 5348–5360. [Google Scholar] [CrossRef]
- Coleman, J.N.; Cadek, M.; Ryan, K.P.; Fonseca, A.; Nagy, J.B.; Blau, W.J.; Ferreira, M.S. Reinforcement of polymers with carbon nanotubes. The role of an ordered polymer interfacial region. Experiment and modeling. Polymer 2006, 47, 8556–8561. [Google Scholar] [CrossRef]
- Eqra, R.; Moghim, M.H.; Eqra, N. A study on the mechanical properties of graphene oxide/epoxy nanocomposites. Polym. Polym. Compos. 2021, 29, S556–S564. [Google Scholar] [CrossRef]
- Chang, Y.; Yang, S.-T.; Liu, J.-H.; Dong, E.; Wang, Y.; Cao, A.; Liu, Y.; Wang, H. In vitro toxicity evaluation of graphene oxide on A549 cells. Toxicol. Lett. 2011, 200, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Na, H.-K.; Kim, M.-H.; Lee, J.; Kim, Y.-K.; Jang, H.; Lee, K.E.; Park, H.; Do Heo, W.; Jeon, H.; Choi, I.S.; et al. Cytoprotective effects of graphene oxide for mammalian cells against internalization of exogenous materials. Nanoscale 2013, 5, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Sawy, A.M.; Barhoum, A.; Abdel Gaber, S.A.; El-Hallouty, S.M.; Shousha, W.G.; Maarouf, A.A.; Khalil, A.S.G. Insights of Doxorubicin Loaded Graphene Quantum Dots: Synthesis, DFT Drug Interactions, and Cytotoxicity. Mater. Sci. Eng. C 2021, 122, 111921. [Google Scholar] [CrossRef]
- Yan, L.; Wang, Y.; Xu, X.; Zeng, C.; Hou, J.; Lin, M.; Xu, J.; Sun, F.; Huang, X.; Dai, L.; et al. Can Graphene Oxide Cause Damage to Eyesight? Chem. Res. Toxicol. 2012, 25, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Bernabò, N.; Fontana, A.; Sanchez, M.R.; Valbonetti, L.; Capacchietti, G.; Zappacosta, R.; Greco, L.; Marchisio, M.; Lanuti, P.; Ercolino, E.; et al. Graphene oxide affects in vitro fertilization outcome by interacting with sperm membrane in an animal model. Carbon 2018, 129, 428–437. [Google Scholar] [CrossRef]
- Duan, G.; Zhang, Y.; Luan, B.; Weber, J.K.; Zhou, R.W.; Yang, Z.; Zhao, L.; Xu, J.; Luo, J.; Zhou, R. Graphene-Induced Pore Formation on Cell Membranes. Sci. Rep. 2017, 7, 42767. [Google Scholar] [CrossRef]
- Li, R.; Guiney, L.M.; Chang, C.H.; Mansukhani, N.D.; Ji, Z.; Wang, X.; Liao, Y.-P.; Jiang, W.; Sun, B.; Hersam, M.C.; et al. Surface Oxidation of Graphene Oxide Determines Membrane Damage, Lipid Peroxidation, and Cytotoxicity in Macrophages in a Pulmonary Toxicity Model. ACS Nano 2018, 12, 1390–1402. [Google Scholar] [CrossRef]
- Liu, J.-H.; Wang, T.; Wang, H.; Gu, Y.; Xu, Y.; Tang, H.; Jia, G.; Liu, Y. Biocompatibility of graphene oxide intravenously administrated in mice—Effects of dose, size and exposure protocols. Toxicol. Res. 2015, 4, 83–91. [Google Scholar] [CrossRef]
- Pelin, M.; Fusco, L.; Martín, C.; Sosa, S.; Frontiñán-Rubio, J.; González-Domínguez, J.M.; Durán-Prado, M.; Vázquez, E.; Prato, M.; Tubaro, A. Graphene and graphene oxide induce ROS production in human HaCaT skin keratinocytes: The role of xanthine oxidase and NADH dehydrogenase. Nanoscale 2018, 10, 11820–11830. [Google Scholar] [CrossRef] [PubMed]
- Ali-Boucetta, H.; Bitounis, D.; Raveendran-Nair, R.; Servant, A.; Van den Bossche, J.; Kostarelos, K. Purified Graphene Oxide Dispersions Lack In Vitro Cytotoxicity and In Vivo Pathogenicity. Adv. Healthc. Mater. 2013, 2, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Li, Y.; Tjong, S.C. Graphene Nanomaterials: Synthesis, Biocompatibility, and Cytotoxicity. Int. J. Mol. Sci. 2018, 19, 3564. [Google Scholar] [CrossRef]
- Malhotra, R.; Halbig, C.E.; Sim, Y.F.; Lim, C.T.; Leong, D.T.; Neto, A.H.C.; Garaj, S.; Rosa, V. Cytotoxicity survey of commercial graphene materials from worldwide. npj 2D Mater. Appl. 2022, 6, 65. [Google Scholar] [CrossRef]
- Tupone, M.G.; Panella, G.; D’Angelo, M.; Castelli, V.; Caioni, G.; Catanesi, M.; Benedetti, E.; Cimini, A. An Update on Graphene-Based Nanomaterials for Neural Growth and Central Nervous System Regeneration. Int. J. Mol. Sci. 2021, 22, 13047. [Google Scholar] [CrossRef]
- Gurunathan, S.; Arsalan Iqbal, M.; Qasim, M.; Park, C.H.; Yoo, H.; Hwang, J.H.; Uhm, S.J.; Song, H.; Park, C.; Do, J.T.; et al. Evaluation of Graphene Oxide Induced Cellular Toxicity and Transcriptome Analysis in Human Embryonic Kidney Cells. Nanomaterials 2019, 9, 969. [Google Scholar] [CrossRef]
- Lammel, T.; Boisseaux, P.; Fernández-Cruz, M.-L.; Navas, J.M. Internalization and cytotoxicity of graphene oxide and carboxyl graphene nanoplatelets in the human hepatocellular carcinoma cell line Hep G2. Part. Fibre Toxicol. 2013, 10, 27. [Google Scholar] [CrossRef]
- Zhang, H.; Peng, C.; Yang, J.; Lv, M.; Liu, R.; He, D.; Fan, C.; Huang, Q. Uniform Ultrasmall Graphene Oxide Nanosheets with Low Cytotoxicity and High Cellular Uptake. ACS Appl. Mater. Interfaces 2013, 5, 1761–1767. [Google Scholar] [CrossRef]
- Lv, M.; Zhang, Y.; Liang, L.; Wei, M.; Hu, W.; Li, X.; Huang, Q. Effect of graphene oxide on undifferentiated and retinoic acid-differentiated SH-SY5Y cells line. Nanoscale 2012, 4, 3861–3866. [Google Scholar] [CrossRef]
- Hu, X.; Wei, Z.; Mu, L. Graphene oxide nanosheets at trace concentrations elicit neurotoxicity in the offspring of zebrafish. Carbon 2017, 117, 182–191. [Google Scholar] [CrossRef]
- Morotomi-Yano, K.; Hayami, S.; Yano, K.-I. Adhesion States Greatly Affect Cellular Susceptibility to Graphene Oxide: Therapeutic Implications for Cancer Metastasis. Int. J. Mol. Sci. 2024, 25, 1927. [Google Scholar] [CrossRef]
- Brisebois, P.P.; Siaj, M. Harvesting graphene oxide—Years 1859 to 2019: A review of its structure, synthesis, properties and exfoliation. J. Mater. Chem. C 2020, 8, 1517–1547. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.S.; Alemany, L.B.; Lu, W.; Tour, J.M. Correction to Improved Synthesis of Graphene Oxide. ACS Nano 2018, 12, 2078. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.J.; Xiao, P.; Lu, H. Pyrenebutyric acid-assisted room-temperature synthesis of large-size monolayer graphene oxide with high mechanical strength. Carbon 2021, 185, 224–233. [Google Scholar] [CrossRef]
- Sim, H.J.; Li, Z.; Xiao, P.; Lu, H. The Influence of Lateral Size and Oxidation of Graphene Oxide on Its Chemical Reduction and Electrical Conductivity of Reduced Graphene Oxide. Molecules 2022, 27, 7840. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Soussan, L.; Bechelany, M.; Teyssier, C.; Cavaillès, V.; Pochat-Bohatier, C.; Miele, P.; Kalkura, N.; Janot, J.-M.; Balme, S. Novel biocompatible electrospun gelatin fiber mats with antibiotic drug delivery properties. J. Mater. Chem. B 2016, 4, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Layek, R.K.; Parihar, V.S.; Skrifvars, M.; Javanshour, F.; Kroon, M.; Kanerva, M.; Vuorinen, J.; Kellomäki, M.; Sarlin, E. Tailoring of the physical and mechanical properties of biocompatible graphene oxide/gelatin composite nanolaminates via altering the crystal structure and morphology. Mater. Adv. 2021, 2, 4781–4791. [Google Scholar] [CrossRef]
- Kashyap, S.; Pratihar, S.K.; Behera, S.K. Strong and ductile graphene oxide reinforced PVA nanocomposites. J. Alloys Compd. 2016, 684, 254–260. [Google Scholar] [CrossRef]
- Sharma, H.; Kumar, A.; Rana, S.; Guadagno, L. An Overview on Carbon Fiber-Reinforced Epoxy Composites: Effect of Graphene Oxide Incorporation on Composites Performance. Polymers 2022, 14, 1548. [Google Scholar] [CrossRef] [PubMed]
- Vallés, C.; Kinloch, I.A.; Young, R.J.; Wilson, N.R.; Rourke, J.P. Graphene oxide and base-washed graphene oxide as reinforcements in PMMA nanocomposites. Compos. Sci. Technol. 2013, 88, 158–164. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Galarza Torre, A.; Shaw, J.E.; Wood, A.; Gilbert, H.T.J.; Dobre, O.; Genever, P.; Brennan, K.; Richardson, S.M.; Swift, J. An immortalised mesenchymal stem cell line maintains mechano-responsive behaviour and can be used as a reporter of substrate stiffness. Sci. Rep. 2018, 8, 8981. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Kupcho, K.; Shultz, J.; Hurst, R.; Hartnett, J.; Zhou, W.; Machleidt, T.; Grailer, J.; Worzella, T.; Riss, T.; Lazar, D.; et al. A real-time, bioluminescent annexin V assay for the assessment of apoptosis. Apoptosis 2019, 24, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Mposula, S.; Amoako, D.G.; Somboro, A.M.; Arhin, I.; Kumalo, H.M.; Khan, R.B. Apoptosis-inducing effects of Terminalia phanerophlebia leaf extracts on human renal cells. S. Afr. J. Bot. 2021, 139, 273–280. [Google Scholar] [CrossRef]
- Tsotetsi, N.; Amoako, D.G.; Somboro, A.M.; Khumalo, H.M.; Khan, R.B. Molecular mechanisms underlying the renoprotective effects of 1,4,7-triazacyclononane: A βeta-lactamase inhibitor. Cytotechnology 2020, 72, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.H.; Bhang, S.H.; Kim, T.; Yu, T.; Hyeon, T.; Kim, B.S. Dual Roles of Graphene Oxide in Chondrogenic Differentiation of Adult Stem Cells: Cell-Adhesion Substrate and Growth Factor-Delivery Carrier. Adv. Funct. Mater. 2014, 24, 6455–6464. [Google Scholar] [CrossRef]
- Gopal, R.; Park, J.S.; Seo, C.H.; Park, Y. Applications of Circular Dichroism for Structural Analysis of Gelatin and Antimicrobial Peptides. Int. J. Mol. Sci. 2012, 13, 3229–3244. [Google Scholar] [CrossRef]
- Díaz-Calderón, P.; Flores, E.; González-Muñoz, A.; Pepczynska, M.; Quero, F.; Enrione, J. Influence of extraction variables on the structure and physical properties of salmon gelatin. Food Hydrocoll. 2017, 71, 118–128. [Google Scholar] [CrossRef]
- An, J.; Gou, Y.; Yang, C.; Hu, F.; Wang, C. Synthesis of a biocompatible gelatin functionalized graphene nanosheets and its application for drug delivery. Mater. Sci. Eng. C 2013, 33, 2827–2837. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Chen, B. One-pot synthesis and characterization of reduced graphene oxide–gelatin nanocomposite hydrogels. RSC Adv. 2016, 6, 6171–6181. [Google Scholar] [CrossRef]
- Stankovich, S.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Synthesis and exfoliation of isocyanate-treated graphene oxide nanoplatelets. Carbon 2006, 44, 3342–3347. [Google Scholar] [CrossRef]
- Kim, S.; Nimni, M.E.; Yang, Z.; Han, B. Chitosan/gelatin–based films crosslinked by proanthocyanidin. J. Biomed. Mater. Res. Part B Appl. Biomater. 2005, 75B, 442–450. [Google Scholar] [CrossRef]
- Li, C.; Luo, J.; Qin, Z.; Chen, H.; Gao, Q.; Li, J. Mechanical and thermal properties of microcrystalline cellulose-reinforced soy protein isolate–gelatin eco-friendly films. RSC Adv. 2015, 5, 56518–56525. [Google Scholar] [CrossRef]
- Piao, Y.; Chen, B. Self-assembled graphene oxide–gelatin nanocomposite hydrogels: Characterization, formation mechanisms, and pH-sensitive drug release behavior. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 356–367. [Google Scholar] [CrossRef]
- Park, S.; Dikin, D.A.; Nguyen, S.T.; Ruoff, R.S. Graphene Oxide Sheets Chemically Cross-Linked by Polyallylamine. J. Phys. Chem. C 2009, 113, 15801–15804. [Google Scholar] [CrossRef]
- Lavogina, D.; Lust, H.; Tahk, M.-J.; Laasfeld, T.; Vellama, H.; Nasirova, N.; Vardja, M.; Eskla, K.-L.; Salumets, A.; Rinken, A.; et al. Revisiting the Resazurin-Based Sensing of Cellular Viability: Widening the Application Horizon. Biosensors 2022, 12, 196. [Google Scholar] [CrossRef]
- Rychlewski, J. On Hooke’s law. J. Appl. Math. Mech. 1984, 48, 303–314. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Property | Value |
|---|---|---|
| AFM | Lateral Size | 1.0 ± 0.4 µm |
| Flake Thickness | 1–2 nm | |
| Monolayer | 91% | |
| Raman | ID/IG ratio | 0.93 ± 0.1 |
| XPS | Purity (%) | 99 |
| C/O ratio | 2.2 ± 0.1 | |
| XPS C1s Deconvolution | C=C/C–C | 33.8% |
| C–O | 42.9% | |
| O–C=O | 21.4% | |
| π-π* | 1.9% |
| C/N Ratio | C=C/C–C (%) | C=N (%) | C–N (%) | O=C–N (%) | |
|---|---|---|---|---|---|
| Gelatine | 2.02 ± 0.06 | 32 ± 3 | 26 ± 3 | 38 ± 2 | 4 ± 1 |
| 0.25% GO–gelatine | 2.20 ± 0.05 | 34 ± 1 | 26 ± 2 | 37 ± 3 | 3 ± 1 |
| 0.5% GO–gelatine | 2.24 ± 0.03 | 35 ± 2 | 26 ± 2 | 38 ± 1 | 2 ± 1 |
| 1% GO–gelatine | 2.32 ± 0.01 | 36 ± 1 | 24 ± 1 | 38 ± 3 | 2 ± 2 |
| 2% GO–gelatine | 2.49 ± 0.03 | 38 ± 2 | 23 ± 2 | 37 ± 4 | 2 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, H.J.; Marinkovic, K.; Xiao, P.; Lu, H. Graphene Oxide Strengthens Gelatine through Non-Covalent Interactions with Its Amorphous Region. Molecules 2024, 29, 2700. https://doi.org/10.3390/molecules29112700
Sim HJ, Marinkovic K, Xiao P, Lu H. Graphene Oxide Strengthens Gelatine through Non-Covalent Interactions with Its Amorphous Region. Molecules. 2024; 29(11):2700. https://doi.org/10.3390/molecules29112700
Chicago/Turabian StyleSim, Hak Jin, Katarina Marinkovic, Ping Xiao, and Hui Lu. 2024. "Graphene Oxide Strengthens Gelatine through Non-Covalent Interactions with Its Amorphous Region" Molecules 29, no. 11: 2700. https://doi.org/10.3390/molecules29112700
APA StyleSim, H. J., Marinkovic, K., Xiao, P., & Lu, H. (2024). Graphene Oxide Strengthens Gelatine through Non-Covalent Interactions with Its Amorphous Region. Molecules, 29(11), 2700. https://doi.org/10.3390/molecules29112700