
Connecting GSK-3β Inhibitory Activity with IKK-β or ROCK-1 Inhibition to Target Tau Aggregation and Neuroinflammation in Alzheimer’s Disease—Discovery, In Vitro and In Cellulo Activity of Thiazole-Based Inhibitors

 , , , , , , , , and
, , , , , , , , and
Abstract

1. Introduction
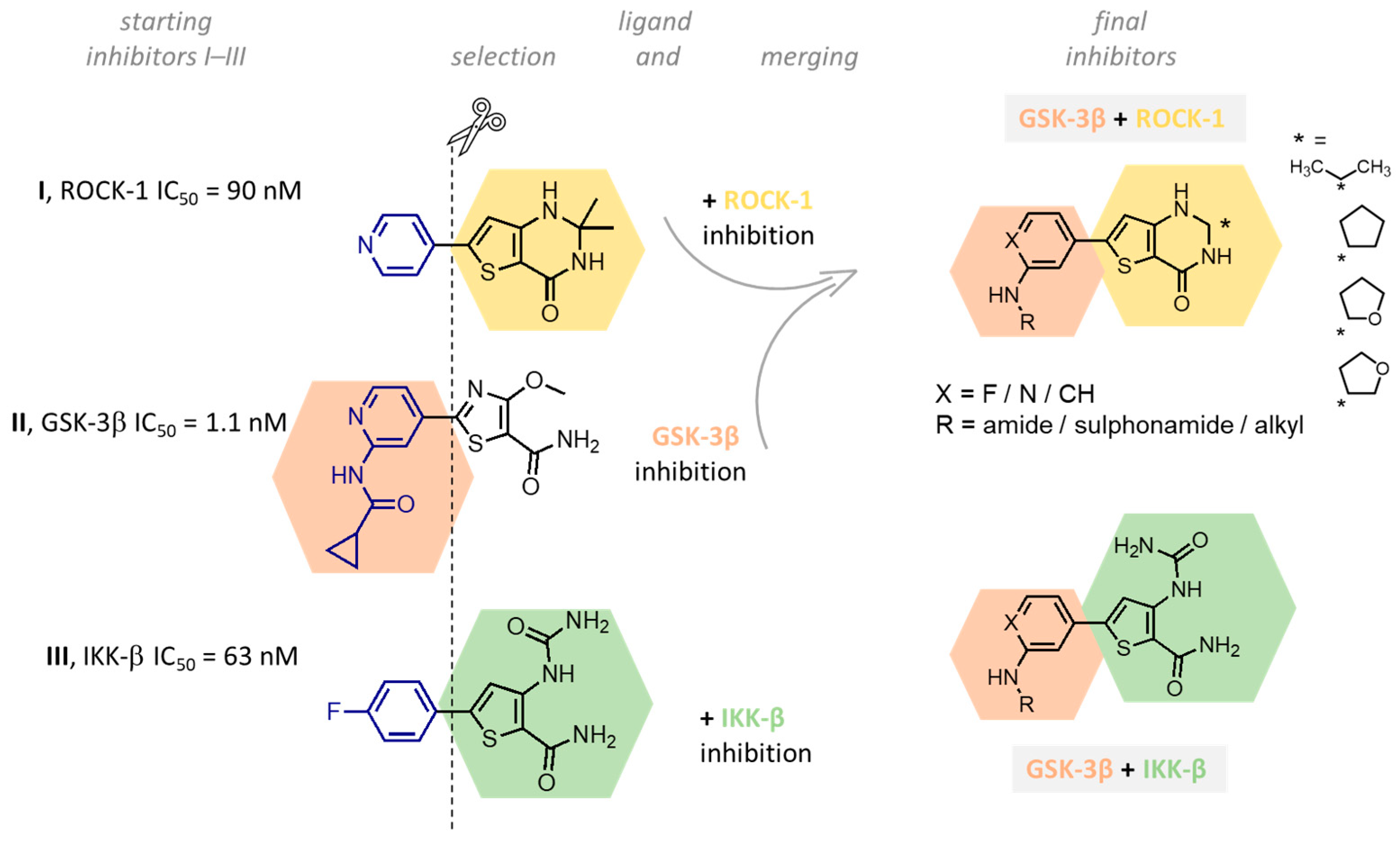
2. Design
3. Chemistry
4. Biological Evaluation
4.1. Inhibitory Activity against GSK-3β, IKK-β, and ROCK-1 Kinases
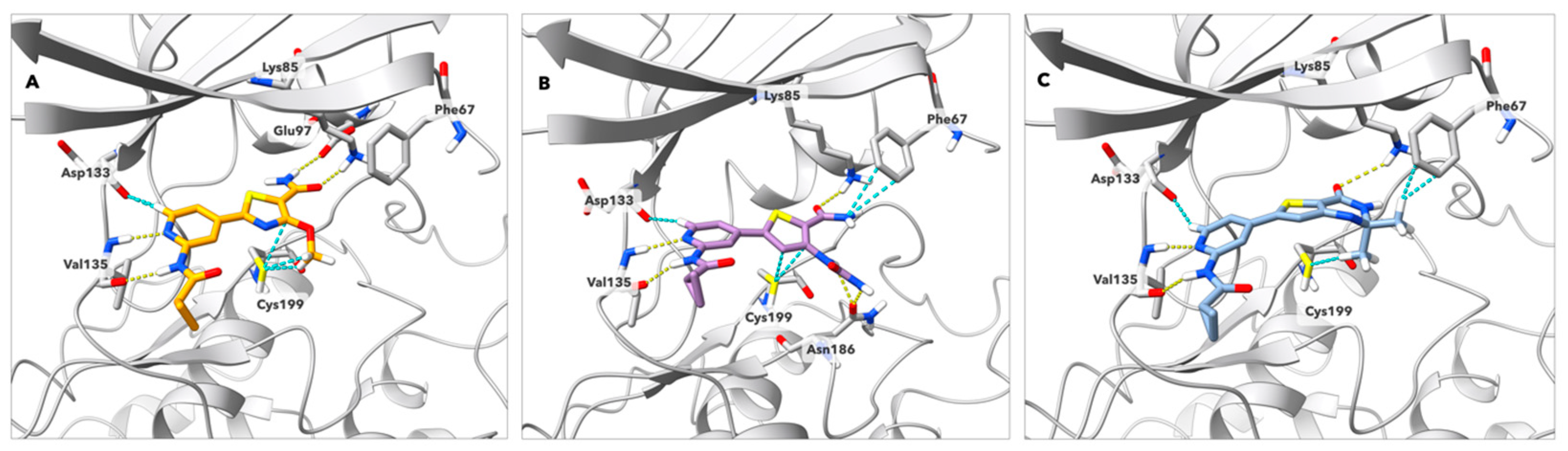
4.2. Kinetic Studies of GSK-3β Inhibition by Compound 62
4.3. Kinase Selectivity
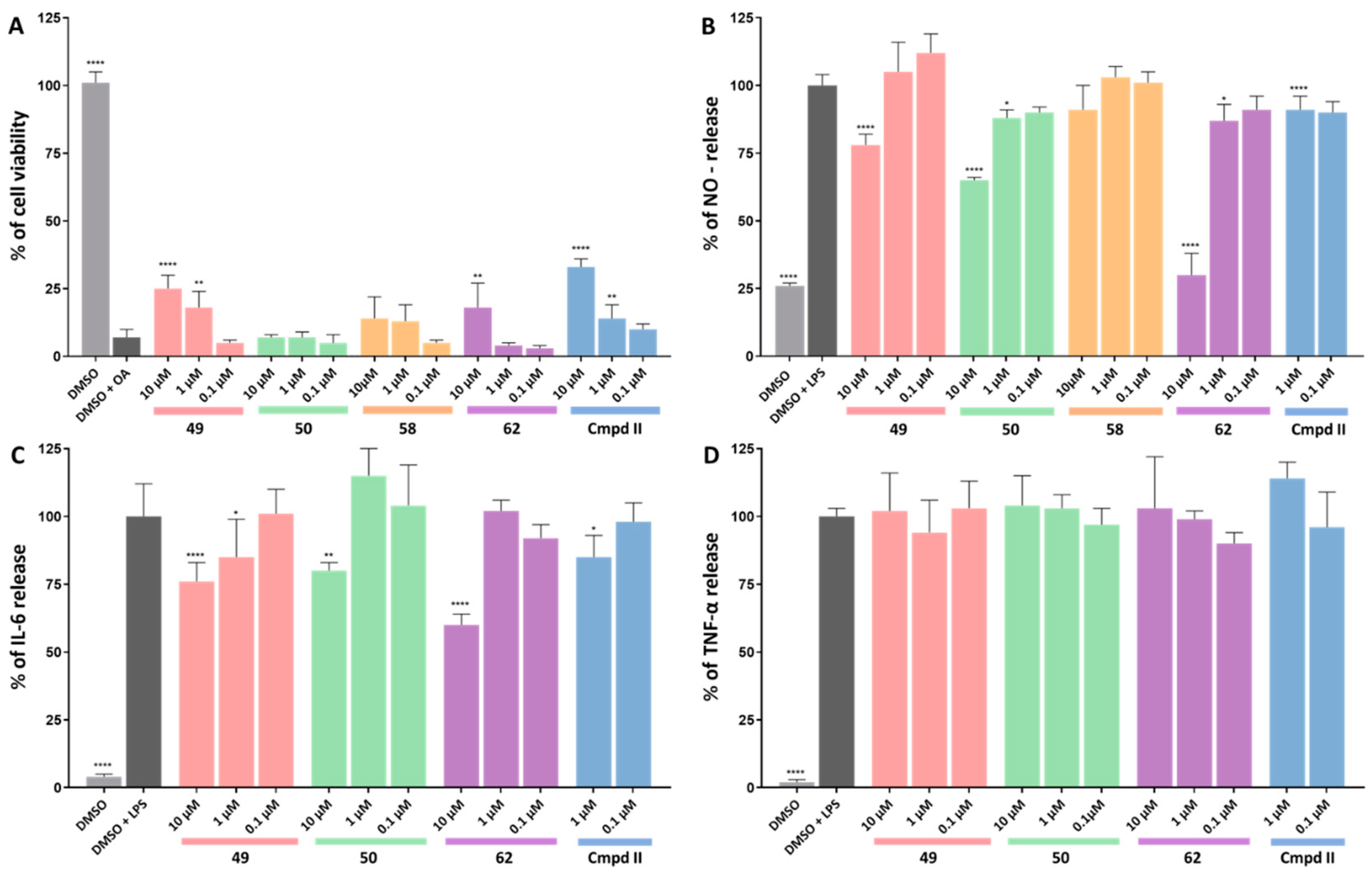
4.4. Cytotoxicity in HT-22 and BV-2 Cells
4.5. Evaluation of Inhibitory Activity towards Okadaic-Acid-Induced Hyperphosphorylation
4.6. Evaluation of Anti-Inflammatory Activity in BV-2 Microglial Cells
5. Preliminary In Vitro ADME
5.1. Thermodynamic Solubility
5.2. Metabolic Stability in Mouse Liver Microsomes
6. Experimental Section
6.1. General Chemistry Information
6.2. Previously Reported Compounds
6.3. Chemical Synthesis
6.3.1. Methyl 5-Bromo-3-((tert-butoxycarbonyl)amino)thiophene-2-carboxylate (1)
- At rt, 25 mL of anhydrous pyridine DMAP (104 mg, 0.85 mmol, 0.1 equiv.) was added to a stirred solution of methyl 3-amino-5-bromothiophene-2-carboxylate (2.00 g, 8.47 mmol, 1.0 equiv.). Then, the mixture was cooled to 0 °C and, under Ar, di-tert-butyl dicarbonate (2.03 g, 9.32 mmol, 1.1 equiv.) was added portion-wise over 20 min. The reaction mixture was warmed up to rt and stirred overnight. After that time, pyridine was evaporated under reduced pressure. The residue was then purified via flash chromatography (PE/EtOAc 95:5). Yield: 2.29 g (80%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.52 (s, 9H), 3.86 (s, 3H), 7.97 (br s, 1H), and 9.33 (br s, 1H). Formula: C10H12BrNO4S.
6.3.2. 5-Bromo-3-((tert-butoxycarbonyl)amino)thiophene-2-carboxylic Acid (2)
- To a stirred solution of 1 (785 mg, 2.33 mmol), 18 mL of MeOH 10% KOH solution (7 mL) was added and the mixture was heated under reflux for 1 h. Then, the reaction mixture was cooled to rt, the pH was adjusted to 2–3 by adding 6 M of HCl, and the mixture concentrated under vacuum to remove MeOH. The residue was extracted using EtOAc. Then, the combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. Yield: 726 mg (97%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.44 (s, 6H), 7.78 (s, 1H), 9.37 (s, 1H), and 13.72 (br s, 1H). Formula: C10H12BrNO4S.
6.3.3. Tert-Butyl (5-Bromo-2-carbamoylthiophen-3-yl)carbamate (3)
- To a solution of 2 (726 mg, 2.25 mmol, 1.0 equiv.) in 18 mL of DMF HATU (1.11 g, 2.93 mmol, 1.3 equiv.), DIPEA (1.18 mL, 6.75 mmol, 3.0 equiv.) and NH4HCO3 (543 mg, 6.75 mmol, 3.0 equiv.) were added and the mixture was stirred at rt overnight. After that time, DMF was evaporated under reduced pressure and the residue was extracted using DCM. The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was then purified via flash chromatography (DCM/PE/EtOAc 6:3:1). Yield: 621 mg (86%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.46 (s, 9H), 7.68 (br s, 2H), 7.81 (s, 1H), and 10.43 (s, 1H). Formula: C10H13BrN2O3S.
6.3.4. 3-Amino-5-bromothiophene-2-carboxamide (4)
- To a solution of 3 (620 mg, 1.93 mmol) in 25 mL DCM TFA (4.5 mL) was added and the mixture was stirred at rt for 1 h. When all of the starting material was consumed, the pH was adjusted to 8 by the addition of saturated aqueous NaHCO3 solution and extracted first to DCM and then to EtOAc. The residue was purified by flash chromatography (DCM/PE/EtOAc 5:2:3). Yield: 407 mg (95%). 1H NMR (500 MHz, DMSO-d6) δ ppm 6.56 (s, 2H), 6.70 (s, 1H), 6.92 (br s, 2H). Formula: C5H5BrN2OS.
6.3.5. 5-Bromo-3-ureidothiophene-2-carboxamide (5)
- A stirred solution of 4 (315 mg, 1.42 mmol) in 13 mL anhydrous THF was cooled to 0 °C on an ice bath and then trichloroacetyl isocyanate (186 µL) was added dropwise. The reaction mixture was warmed up to rt and stirred for 1.5 h. After that time, 4 M of ammonia solution in methanol (20 mL) was added and stirred for another 1.5 h. Then, the solvents were evaporated under reduced pressure and the solid residue was washed with Et2O. Yield: 129 mg (34%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 6.64 (br s, 2H), 7.43 (br s, 2H), 7.96 (s, 1H), and 10.01 (s, 1H). Formula: C6H6BrN3O2S.
6.3.6. 6-Bromo-2,2-dimethyl-2,3-dihydrothieno [3,2-d]pyrimidin-4(1H)-one (6)
- To a stirred solution of 4 (200 mg, 0.91 mmol, 1.0 equiv.) in 6 mL acetone, CH3COOH (3 mL) and p-TSA (17 mg) were added; then, the mixture was heated to 80 °C overnight. After that time, the reaction mixture was cooled to rt, solvents were evaporated under reduced pressure, and the residue was extracted using DCM. The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. Yield: 217 mg (92%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.47 (s, 6H), 6.48 (s, 1H), 6.57 (s, 1H), and 7.24 (s, 1H). Formula: C8H9BrN2OS.
6.3.7. 6′-Bromo-1′H-spiro[cyclopentane-1,2′-thieno [3,2-d]pyrimidin]-4′(3′H)-one (7)
- A stirred solution of cyclopentanone (3.3 mL) and p-TSA (16 mg) in 6 mL of anhydrous toluene was heated to 80 °C. After 5 min, 4 (50 mg, 0.23 mmol) was added, and the mixture was heated to 110 °C for 5 h. Then, the reaction mixture was cooled to rt, the solvent was evaporated under reduced pressure, and the solid residue was washed with MeCN/MeOH 2:1. Yield: 24 mg (37%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.58–1.70 (m, 4H), 1.77–1.87 (m, 4H), 6.70 (s, 1H), 7.30 (br s, 1H), and 7.71 (br s, 1H). Formula: C10H11BrN2OS.
6.3.8. 6′-Bromo-4,5-dihydro-1′H,3H-spiro[furan-2,2′-thieno [3,2-d]pyrimidin]-4′(3′H)-one (8)
- A stirred solution of γ-butyrolactone (6.6 mL) and p-TSA (40 mg) in 8 mL of anhydrous toluene was heated to 80 °C. After 5 min, 4 (150 mg, 0.68 mmol) was added, and the mixture was heated to 110 °C overnight. Then, the reaction mixture was cooled to rt and the solvent was evaporated under reduced pressure. The residue was purified via flash chromatography (DCM/MeOH 91:9). Yield: 104 mg (53%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.83 (quin, J = 6.9 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H), 3.44 (t, J = 6.3 Hz, 2H), 4.57 (br s, 1H), 7.56 (s, 1H), and 12.26 (br s, 1H). Formula: C9H9BrN2O2S.
6.3.9. 6′-Bromo-4,5-dihydro-1′H,2H-spiro[furan-3,2′-thieno [3,2-d]pyrimidin]-4′(3′H)-one (9)
- To the tube filled with the solution of 4 (25 mg, 0.11 mmol) in 400 µL anhydrous toluene dihydro-3(2H)-furanone (100 µL) was added, sealed and placed in a microwave reactor for 2 h in 120 °C. Then reaction mixture was cooled to rt, solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (DCM/MeOH 93:7). Yield: 30 mg (92%). 1H NMR (500 MHz, DMSO-d6) δ ppm 2.10–2.18 (m, 2H), 3.56 (d, J = 8.9 Hz, 1H), 3.73 (d, J = 8.9 Hz, 1H), 3.80–3.89 (m, 2H), 6.75 (s, 1H), 7.67 (br s, 1H), 7.97 (br s, 1H). Formula: C9H9BrN2O2S.
6.4. General Procedure for the Synthesis of Compounds 10–14, and 17 (GP1)
- An appropriate aromatic amine (1.0 equiv.) was dissolved in DCM, the solution was cooled to 0 °C on an ice bath, and then a base (2.0 equiv.) and an appropriate acid chloride (1.2 equiv.) were added dropwise. The reaction mixture was then warmed up to rt and stirred overnight. After that time, the mixture was extracted using DCM, and the combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The product did not require further purification.
6.4.1. N-(4-Bromopyridin-2-yl)acetamide (10)
- Following GP1, compound 10 was prepared using 2-amino-4-bromopyridine (750 mg, 4.33 mmol), acetyl chloride (354 µL, 4.98 mmol), and pyridine (698 µL, 8.66 mmol) in 15 mL of DCM. Yield: 744 mg (79%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 2.19 (s, 3H), 6.77–6.82 (m, 1H), 7.19 (dd, J = 5.4, 1.7 Hz, 1H), 8.05 (d, J = 5.4 Hz, 1H), and 8.45 (s, 1H). Formula: C7H7BrN2O.
6.4.2. N-(4-Bromopyridin-2-yl)isobutyramide (11)
- Following GP1, compound 11 was prepared using 2-amino-4-bromopyridine (500 mg, 2.89 mmol), isobutyryl chloride (350 µL, 3.32 mmol), and pyridine (466 µL, 5.78 mmol) in 10 mL of DCM. Yield: 430 mg (61%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.24 (d, J = 6.9 Hz, 6H), 2.54 (dt, J = 13.7, 6.9 Hz, 1H), 7.18 (dd, J = 5.2, 1.7 Hz, 1H), 8.05 (d, J = 5.4 Hz, 1H), 8.13 (br s, 1H), and 8.50 (d, J = 1.4 Hz, 1H). Formula: C9H11BrN2O.
6.4.3. N-(4-Bromopyridin-2-yl)cyclopropanecarboxamide (12)
- Following GP1, compound 12 was prepared using 2-amino-4-bromopyridine (2.00 g, 11.56 mmol), cyclopropanecarbonyl chloride (1.21 mL, 13.29 mmol), and pyridine (1.87 mL, 23.12 mmol) in 40 mL of DCM. Yield: 2.54 g (91%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 0.91–0.96 (m, 2H), 1.08–1.14 (m, 2H), 1.63–1.70 (m, 1H), 7.23 (dd, J = 5.7, 1.7 Hz, 1H), 8.02 (d, J = 5.7 Hz, 1H), 8.58 (d, J = 1.7 Hz, 1H), and 9.61 (br s, 1H). Formula: C9H9BrN2O.
6.4.4. N-(3-Bromophenyl)cyclopropanecarboxamide (13)
- Following GP1, compound 13 was prepared using 3-bromoaniline (544 µL, 5.00 mmol), cyclopropanecarbonyl chloride (544 µL, 6.00 mmol), and triethylamine (832 µL, 6.00 mmol) in 5 mL of DCM. Yield: 1100 mg (92%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.80 (br s, 1H), 7.54 (br s, 1H), 7.38–7.43 (m, 1H), 7.20–7.24 (m, 1H), 7.14–7.19 (m, 1H), 1.47–1.54 (m, 1H), 1.06–1.12 (m, 2H), and 0.84–0.90 (m, 2H). Formula: C10H10BrNO.
6.4.5. N-(5-Bromo-2-fluorophenyl)cyclopropanecarboxamide (14)
- Following GP1, compound 14 was prepared using 5-bromo-2-fluoroaniline (1.00 g, 5.26 mmol), cyclopropanecarbonyl chloride (549 µL, 6.05 mmol), and pyridine (847 µL, 10.52 mmol) in 10 mL of DCM. Yield: 1.32 g (97%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 0.86–0.91 (m, 2H), 1.07–1.12 (m, 2H), 1.55 (tt, J = 7.8, 4.5 Hz, 1H), 6.95 (dd, J = 10.7, 8.7 Hz, 1H), 7.12 (ddd, J = 8.7, 4.6, 2.4 Hz, 1H), 7.56 (br s, 1H), and 8.55 (br d, J = 5.7 Hz, 1H). Formula: C10H9BrFNO.
6.4.6. N-(4-Bromopyridin-2-yl)benzamide (17)
- Following GP1, compound 17 was prepared using 2-amino-4-bromopyridine (500 mg, 2.89 mmol), benzoyl chloride (383 µL, 3.32 mmol), and pyridine (466 µL, 5.78 mmol) in 10 mL of DCM. Yield: 272 mg (34%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 7.41 (dd, J = 5.4, 1.7 Hz, 1H), 7.45–7.51 (m, 2H), 7.54–7.60 (m, 1H), 7.94–8.01 (m, 2H), 8.27 (d, J = 5.4 Hz, 1H), 8.43 (d, J = 1.7 Hz, 1H), and 11.01 (s, 1H). Formula: C12H9BrN2O.
6.5. General Procedure for the Synthesis of Compounds 15, 16 (GP2)
- An appropriate cycloalkyl acid (1.0 equiv.) was dissolved in anhydrous DCM, and then 2-amino-4-bromopyridine (1.1 equiv.), anhydrous pyridine (2.0 equiv.), and 50% T3P sol. in ethyl acetate (2.2 equiv.) were added under Ar. The reaction mixture was stirred for 1 h at rt. After that time, the residue was extracted using DCM. The crude product was purified via flash chromatography.
6.5.1. N-(4-Bromopyridin-2-yl)cyclobutanecarboxamide (15)
- Following GP2, compound 15 was prepared using cyclobutanecarboxylic acid (101 µL, 1.06 mmol), 2-amino-4-bromopyridine (200 mg, 1.16 mmol), pyridine (171 µL, 2.12 mmol), and T3P (1.37 mL, 2.33 mmol) in 12 mL of DCM. Purification: flash chromatography (DCM/MeOH 98:2). Yield: 268 mg (91%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.86–1.95 (m, 1H), 1.97–2.07 (m, 1H), 2.18–2.29 (m, 2H), 2.31–2.43 (m, 2H), 3.19–3.29 (m, 1H), 7.22 (dd, J = 5.4, 1.7 Hz, 1H), 8.04 (d, J = 5.4 Hz, 1H), 8.61 (d, J = 1.4 Hz, 1H), and 8.82 (br s, 1H). Formula: C10H11BrN2O.
6.5.2. N-(4-Bromopyridin-2-yl)cyclohexanecarboxamide (16)
- Following GP2, compound 16 was prepared using cyclohexanecarboxylic acid (506 mg, 3.95 mmol), 2-amino-4-bromopyridine (750 mg, 4.34 mmol), pyridine (636 µL, 7.90 mmol), and T3P (5.12 mL, 8.69 mmol) in 30 mL of DCM. Purification: flash chromatography (DCM/MeOH 98:2). Yield: 833 mg (68%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.22–1.37 (m, 3H), 1.53 (qd, J = 12.2, 3.2 Hz, 2H), 1.67–1.74 (m, 1H), 1.79–1.87 (m, 2H), 1.96 (br dd, J = 13.5, 2.0 Hz, 2H), 2.27 (tt, J = 11.7, 3.7 Hz, 1H), 7.20 (dd, J = 5.3, 1.9 Hz, 1H), 8.06 (d, J = 5.4 Hz, 1H), 8.31 (br s, 1H), and 8.54 (d, J = 1.4 Hz, 1H). Formula: C12H15BrN2O.
6.6. General Procedure for the Synthesis of Compounds 18–23 (GP3)
- An appropriate amine (1.0–1.2 equiv.) was dissolved in anhydrous pyridine or DCM, the solution was cooled to 0 °C on an ice bath, and then sulfonyl chloride (1.0–2.0 equiv.) and base (optional; 1.2–1.5 equiv.) were added dropwise. The reaction mixture was stirred at rt overnight. After that time, pyridine was evaporated under reduced pressure. The crude product was purified using different methods, as described below.
6.6.1. N-(4-Bromopyridin-2-yl)methanesulfonamide (18)
- Following GP3, compound 18 was prepared using 2-amino-4-bromopyridine (600 mg, 3.47 mmol) and mesyl chloride (537 µL, 6.94 mmol) in 11 mL of pyridine. Water was added directly to the residue and the solid was filtered. Then, the solid product was washed with Et2O. Yield: 530 mg (61%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 3.25 (s, 3H), 7.12 (s, 1H), 7.23 (br d, J = 4.6 Hz, 1H), 8.11 (d, J = 5.4 Hz, 1H), and 10.91 (br s, 1H). Formula: C6H7BrN2O2S.
6.6.2. N-(4-Bromopyridin-2-yl)ethanesulfonamide (19)
- Following GP3, compound 19 was prepared using 2-amino-4-bromopyridine (500 mg, 2.89 mmol) and ethanesulfonyl chloride (546 µL, 5.78 mmol) in 9 mL of pyridine. The residue was extracted using DCM. The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude product was then purified via flash chromatography (DCM/MeOH 98:2). Yield: 216 mg (28%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.34–1.40 (m, 3H), 3.21–3.28 (m, 2H), 7.11–7.17 (m, 1H), 7.66 (d, J = 1.7 Hz, 1H), and 8.17 (d, J = 5.7 Hz, 1H). The proton of the -NH- group was not detected. Formula: C7H9BrN2O2S.
6.6.3. N-(3-Bromophenyl)ethanesulfonamide (20)
- Following GP3, compound 20 was prepared using 3-bromoaniline (435 µL, 4.00 mmol), ethanesulfonyl chloride (492 µL, 5.20 mmol), and pyridine (484 µL, 6.00 mmol) in 20 mL of DCM. The mixture was transferred to a separatory funnel and washed sequentially with saturated NH4Cl(aq.) and saturated NaCl(aq.). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified using flash chromatography (DCM/MeOH 98:2 then 96:4). Yield: 940 mg (89%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.41–7.43 (m, 1H), 7.32 (s, 1H), 7.27–7.30 (m, 1H), 7.18–7.22 (m, 2H), 3.18 (q, J = 7.3 Hz, 2H), and 1.39 (t, J = 7.4 Hz, 3H). Formula: C8H10BrNO2S.
6.6.4. 4-Bromo-N-(cyclopropylmethyl)pyridin-2-amine (21)
- To a suspension of NaH (60% suspension in mineral oil, 240 mg, 6.00 mmol, 2.0 equiv.) in DMF (8 mL), 4-bromopyridin-2-amine (519 mg, 3.00 mmol, 1.0 equiv.) was added, the mixture was stirred for 1 h at 50 ºC, and then cyclopropylmethyl bromide (405 mg, 3.00 mmol, 1.0 equiv.) was added. The reaction mixture was stirred overnight at 100 ºC. After that time, the mixture was concentrated under vacuum, diluted with water, and extracted using DCM. The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified via flash chromatography (PE/EtOAc 8:2). Yield: 192 mg (28%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.88 (d, J = 5.4 Hz, 1 H), 6.71 (dd, J = 5.4, 1.7 Hz, 1 H), 6.56 (d, J = 1.4 Hz, 1 H), 4.89 (br s, 1 H), 3.10 (dd, J = 7.0, 5.3 Hz, 2 H), 1.02–1.14 (m, 1 H), 0.51–0.60 (m, 2 H), and 0.21–0.29 (m, 2 H). Formula: C9H11BrN2.
6.6.5. N-(3-Bromophenyl)cyclohexanesulfonamide (22)
- Following GP3, compound 22 was prepared using 3-bromoaniline (163 mL, 1.50 mmol), cyclohexylsulfonyl chloride (283 mL, 1.95 mmol), and pyridine (182 mL, 2.25 mmol) in 7.5 mL of DCM. The mixture was transferred to a separatory funnel and washed sequentially with saturated NH4Cl(aq.) and saturated NaCl(aq.). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was purified using flash chromatography (0–4% MeOH in DCM). Yield: 425 mg (98%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.38 (t, J = 2.0 Hz, 1H), 7.23–7.27 (m, 1H), 7.12–7.20 (m, 2H), 6.78 (s, 1H), 3.01 (tt, J = 12.2, 3.4 Hz, 1H), 2.10–2.19 (m, 2H), 1.82–1.92 (m, 2H), 1.63–1.72 (m, 1H), 1.46–1.62 (m, 2H), and 1.10–1.30 (m, 3H). Formula: C12H16BrNO2S.
6.6.6. 1-((3-Bromophenyl)sulfonyl)piperidine (23)
- Following GP3, compound 23 was prepared using piperidine (475 µL, 4.80 mmol), 3-bromobenzene-1-sulfonyl chloride (577 µL, 4.00 mmol), and triethylamine (665 µL, 4.80 mmol) in 4.5 mL of DCM. The mixture was transferred to a separatory funnel and washed sequentially with saturated NH4Cl(aq.) and saturated NaCl(aq.). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. Yield: 1.08 g (88%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.91 (t, J = 1.9 Hz, 1H), 7.67–7.75 (m, 2H), 7.42 (t, J = 7.9 Hz, 1H), 2.96–3.06 (m, 4H), 1.63–1.71 (m, 4H), and 1.41–1.49 (m, 2H). Formula: C11H14BrNO2S.
6.7. General Procedure for the Synthesis of Compounds 24–37 (GP4)
- An appropriate aryl bromide (1.0 equiv.) and bis(pinacolato)diboron (1.0–1.5 equiv.) were dissolved in anhydrous dioxane. Then, potassium acetate (2.0 equiv.) was added and, under Ar, Pd(dppf)Cl2 (0.1–0.4 equiv.) was added. The reaction was stirred under reflux overnight. After that time, the reaction mixture was diluted with DCM, filtered through Celite, and evaporated under reduced pressure. The residue was dissolved in EtOAc. Then, activated charcoal (5–10 g) was added and the mixture was stirred under reflux for 1 h. The mixture was then filtered again through Celite and evaporated under reduced pressure. The crude product was purified using different methods, as described below.
6.7.1. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide (24)
- Following GP4, compound 24 was prepared using N-(4-bromopyridin-2-yl)acetamide (10) (744 mg, 3.46 mmol), bis(pinacolato)diboron (879 mg, 3.46 mmol), potassium acetate (679 mg, 6.92 mmol), Pd(dppf)Cl2 (255 mg, 0.35 mmol) in 10 mL dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 520 mg (57%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm 1.35 (s, 12H), 2.22 (s, 3H), 7.40 (d, J = 4.6 Hz, 1H), 8.26 (br d, J = 4.6 Hz, 1H), 8.55 (br s, 1H), 8.79 (br s, 1H). Formula: C13H19BN2O3.
6.7.2. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)isobutyramide (25)
- Following GP4, compound 25 was prepared using N-(4-bromopyridin-2-yl)isobutyramide (11) (430 mg, 1.77 mmol), bis(pinacolato)diboron (450 mg, 1.77 mmol), potassium acetate (348 mg, 3.54 mmol), and Pd(dppf)Cl2 (130 mg, 0.18 mmol) in 5 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 200 mg (39%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.24 (d, J = 6.9 Hz, 6H), 1.31 (s, 12H), 2.47–2.58 (m, 1H), 7.34 (d, J = 4.6 Hz, 1H), 8.19 (br s, 1H), 8.25 (br d, J = 4.6 Hz, 1H), and 8.57 (s, 1H). Formula: C15H23BN2O3.
6.7.3. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclopropanecarboxamide (26)
- Following GP4, compound 26 was prepared using N-(4-bromopyridin-2-yl)cyclopropanecarboxamide (12) (2.54 g, 8.82 mmol), bis(pinacolato)diboron (2.54 g, 8.82 mmol), potassium acetate (1.73 g, 17.64 mmol), and Pd(dppf)Cl2 (650 mg, 0.89 mmol) in 21 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 1.03 g (40%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 0.83–0.90 (m, 2H), 1.07–1.12 (m, 2H), 1.30 (s, 12H), 1.55–1.64 (m, 1H), 7.34 (d, J = 4.6 Hz, 1H), 8.24 (d, J = 4.6 Hz, 1H), 8.55 (s, 1H), and 8.95 (br s, 1H). Formula: C15H21BN2O3.
6.7.4. N-(3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclopropanecarboxamide (27)
- Following GP4, compound 27 was prepared using N-(3-bromophenyl)cyclopropanecarboxamide (13) (600 mg, 2.50 mmol), bis(pinacolato)diboron (635 mg, 2.50 mmol), potassium acetate (491 mg, 5.00 mmol), and Pd(dppf)Cl2 (180 mg, 0.25 mmol) in 7 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 475 mg (66%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 0.82 (dq, J = 7.6, 3.7 Hz, 2H), 1.03–1.10 (m, 2H), 1.32 (s, 12H), 1.44 (m, J = 2.3 Hz, 1H), 7.28–7.34 (m, 1H), 7.37 (br s, 1H), 7.51 (br d, J = 7.2 Hz, 1H), 7.69 (s, 1H), and 7.83 (br d, J = 7.4 Hz, 1H). Formula: C16H22BNO3.
6.7.5. N-(2-Fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclopropanecarboxamide (28)
- Following GP4, compound 28 was prepared using N-(5-bromo-2-fluorophenyl)cyclopropanecarboxamide (14) (660 mg, 2.56 mmol), bis(pinacolato)diboron (975 mg, 3.84 mmol), potassium acetate (754 mg, 7.68 mmol), and Pd(dppf)Cl2 (180 mg, 0.25 mmol) in 7 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 291 mg (37%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 0.83–0.88 (m, 2H), 1.06–1.13 (m, 2H), 1.29 (s, 12H), 1.50–1.58 (m, 1H), 7.06 (dd, J = 11.2, 8.3 Hz, 1H), 7.47 (br t, J = 6.4 Hz, 1H), 7.52 (br s, 1H), and 8.67 (br d, J = 7.7 Hz, 1H). Formula: C16H21BFNO3.
6.7.6. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclobutanecarboxamide (29)
- Following GP4, compound 29 was prepared using N-(4-bromopyridin-2-yl)cyclobutanecarboxamide (15) (405 mg, 1.59 mmol), bis(pinacolato)diboron (404 mg, 1.59 mmol), potassium acetate (312 mg, 3.18 mmol), and Pd(dppf)Cl2 (120 mg, 0.16 mmol) in 5 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 120 mg (25%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm 1.31 (s, 12H), 1.87–1.96 (m, 1H), 1.97–2.04 (m, 1H), 2.19–2.28 (m, 2H), 2.34–2.44 (m, 2H), 3.21 (quin, J = 8.4 Hz, 1H), 7.37 (d, J = 4.9 Hz, 1H), 8.22 (d, J = 4.9 Hz, 1H), 8.38 (br s, 1H), and 8.63 (s, 1H). Formula: C16H23BN2O3.
6.7.7. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclohexanecarboxamide (30)
- Following GP4, compound 30 was prepared using N-(4-bromopyridin-2-yl)cyclohexanecarboxamide (16) (831 mg, 2.93 mmol), bis(pinacolato)diboron (744 mg, 2.93 mmol), potassium acetate (575 mg, 5.86 mmol), and Pd(dppf)Cl2 (215 mg, 0.29 mmol) in 8.5 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 602 mg (62%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.22–1.29 (m, 2H), 1.32 (s, 12H), 1.34 (s, 1H), 1.48–1.61 (m, 2H), 1.69 (br d, J = 12.0 Hz, 1H), 1.77–1.87 (m, 2H), 1.97 (br d, J = 12.6 Hz, 2H), 2.32 (br t, J = 11.0 Hz, 1H), 7.39 (d, J = 4.6 Hz, 1H), 8.21 (d, J = 4.9 Hz, 1H), 8.67 (s, 1H), and 8.78 (br s, 1H). Formula: C18H27BN2O3.
6.7.8. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)benzamide (31)
- Following GP4, compound 31 was prepared using N-(4-bromopyridin-2-yl)benzamide (17) (398 mg, 1.44 mmol), bis(pinacolato)diboron (366 mg, 1.44 mmol), potassium acetate (283 mg, 2.88 mmol), and Pd(dppf)Cl2 (110 mg, 0.15 mmol) in 4 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 195 mg (42%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.35 (s, 12H), 7.44 (d, J = 4.0 Hz, 1H), 7.47–7.53 (m, 2H), 7.55–7.60 (m, 1H), 7.99 (d, J = 7.4 Hz, 2H), 8.27 (br d, J = 4.3 Hz, 1H), 8.81 (s, 1H), and 9.22 (br s, 1H). Formula: C18H21BN2O3.
6.7.9. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)methanesulfonamide (32)
- Following GP4, compound 32 was prepared using N-(4-bromopyridin-2-yl)methanesulfonamide (18) (518 mg, 2.06 mmol), bis(pinacolato)diboron (680 mg, 2.68 mmol), potassium acetate (404 mg, 4.12 mmol), and Pd(dppf)Cl2 (240 mg, 0.33 mmol) in 6 mL of dioxane. The solid product was used further without purification. Formula: C12H19BN2O4S. MW: 298.16. LC-MS: m/z 217 [M + H]+ of (2-(methylsulfonamido)pyridin-4-yl)boronic acid, MW: 216.02.
6.7.10. N-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)ethanesulfonamide (33)
- Following GP4, compound 33 was prepared using N-(4-bromopyridin-2-yl)ethanesulfonamide (19) (360 mg, 1.36 mmol), bis(pinacolato)diboron (518 mg, 2.04 mmol), potassium acetate (267 mg, 2.72 mmol), and Pd(dppf)Cl2 (200 mg, 0.27 mmol) in 5 mL of dioxane. Purification: crystallization from EtOAc/PE 1:2. Yield: 241 mg (57%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 1.32 (s, 12H), 1.34–1.38 (m, 3H), 3.26 (q, J = 7.4 Hz, 2H), 7.25 (s, 1H), 7.69 (s, 1H), and 8.25 (d, J = 5.2 Hz, 1H). The proton of the -NH- group was not detected. Formula: C13H21BN2O4S.
6.7.11. N-(3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)ethanesulfonamide (34)
- Following GP4, compound 34 was prepared using N-(3-bromophenyl)ethanesulfonamide (20) (528 mg, 2.00 mmol), bis(pinacolato)diboron (762 mg, 3.00 mmol), potassium acetate (393 mg, 4.00 mmol), and Pd(dppf)Cl2 (294 mg, 0.40 mmol) in 7.5 mL of dioxane. Purification: crystallization from the mixture of Et2O and EtOAc. Yield: 433 mg (70%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.62 (dt, J = 7.3, 1.1 Hz, 1H), 7.49–7.51 (m, 1H), 7.43–7.47 (m, 1H), 7.34–7.39 (m, 1H), 6.41 (s, 1H), 3.13 (q, J = 7.4 Hz, 2H), 1.36–1.40 (m, 3H), and 1.35 (s, 12H). Formula: C14H22BNO4S.
6.7.12. N-(Cyclopropylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine (35)
- Following GP4, compound 35 was prepared using 4-bromo-N-(cyclopropylmethyl)pyridin-2-amine (21) (242 mg, 1.07 mmol), bis(pinacolato)diboron (259 mg, 1.07 mmol), potassium acetate (210 mg, 2.14 mmol), and Pd(dppf)Cl2 (79 mg, 0.11 mmol) in 4 mL of dioxane. Purification: crystallization from the mixture of PE and EtOAc. Yield: 260 mg (89%). Formula: C15H23BN2O2. MW: 274.17.
6.7.13. N-(3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclohexanesulfonamide (36)
- Following GP4, compound 36 was prepared using N-(3-bromophenyl)cyclohexanesulfonamide (22) (416 mg, 1.31 mmol), bis(pinacolato)diboron (500 mg, 1.97 mmol), potassium acetate (257 mg, 2.62 mmol), and Pd(dppf)Cl2 (200 mg, 0.27 mmol) in 5 mL of dioxane. Purification: flash chromatography (0–3% MeOH gradient in DCM). Yield: 415 mg (87%). 1H NMR (500 MHz, CHLOROFORM-d) δ ppm: 7.59 (d, J = 7.2 Hz, 1 H), 7.42–7.52 (m, 2 H), 7.32–7.39 (m, 1H), 6.25 (s, 1H), 3.00 (tt, J = 12.0, 3.4 Hz, 1H), 2.17 (dd, J = 13.2, 1.4 Hz, 2H), 1.83–1.91 (m, 2H), 1.51–1.72 (m, 4H), 1.35 (s, 12H), and 1.18–1.22 (m, 2H). Formula: C18H28BNO4S.
6.7.14. 1-((3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)sulfonyl)piperidine (37)
- Following GP4, compound 27 was prepared using 1-((3-bromophenyl)sulfonyl)piperidine (23) (650 mg, 2.14 mmol), bis(pinacolato)diboron (760 mg, 2.99 mmol), potassium acetate (420 mg, 4.28 mmol), and Pd(dppf)Cl2 (260 mg, 0.36 mmol) in 7 mL of dioxane. Purification: preparative HPLC (20–100% MeCN gradient). Yield: 306 mg (41%). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.28 (s, 12H), 1.30–1.34 (m, 2H), 1.50 (quin, J = 5.6 Hz, 4H), 7.63 (t, J = 7.6 Hz, 1H), 7.81 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.86–7.89 (m, 1H), and 7.92 (dt, J = 7.2, 1.1 Hz, 1H). Formula: C17H26BNO4S.
6.8. General Procedure for the Synthesis of Compounds 38–62 (GP5)
- An appropriate pinacol ester of 4-arylboronic acid (1.0 equiv.) and thiophene-based building block with urea 5 or cyclic moiety 6–9 (1.0 equiv.) were dissolved in anhydrous DMF. Then, K2CO3 (3.00 equiv.) was added and, under Ar, [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (Pd(dppf)Cl2) (0.4–0.5 equiv.) was added. The reaction was stirred overnight at 80 °C. After that time, the reaction mixture was diluted with DCM, filtered through Celite®, and evaporated under reduced pressure. The crude product was purified using different methods, as described below.
6.8.1. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)acetamide (38)
- Following GP5, compound 38 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide (24) (100 mg, 0.38 mmol), 6 (99 mg, 0.38 mmol), K2CO3 (158 mg, 1.14 mmol), and Pd(dppf)Cl2 (125 mg, 0.17 mmol) in 4 mL of DMF. Purification: column chromatography (DCM/MeOH/NH3(aq) 95:5:0.05), and then the solid residue was washed with MeCN. Yield: 39 mg (33%), yellow solid, mp 284–285 °C. Purity: 97% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.42 (s, 6H), 2.11 (s, 3H), 7.05 (d, J = 2.6 Hz, 1H), 7.13 (s, 1H), 7.35 (dd, J = 5.2, 2.0 Hz, 1H), 7.66 (s, 1H), 8.30–8.37 (m, 2H), and 10.62 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 23.99, 28.23 (2C), 69.10, 105.91, 108.61, 115.30, 116.00, 141.82, 145.13, 148.92, 151.70, 153.05, 160.73, and 169.67. Formula: C15H16N4O2S. MW: 316.38. MS: m/z 317 (M + H+).
6.8.2. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)isobutyramide (39)
- Following GP5, compound 39 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)isobutyramide (25) (100 mg, 0.34 mmol), 6 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), Pd(dppf)Cl2 (112 mg, 0.15 mmol), and DMF (3.5 mL). Purification: column chromatography (5% MeOH in DCM). Yield: 44 mg (38%), yellow solid, mp 226–227 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.53 (s, 1 H), 8.34 (d, J = 1.1 Hz, 1 H), 8.29 (d, J = 5.7 Hz, 1 H), 7.63 (s, 1 H), 7.32 (dd, J = 5.3, 1.9 Hz, 1 H), 7.08 (d, J = 0.9 Hz, 1 H), 7.02 (s, 1 H), 2.73 (quin, J = 6.8 Hz, 1 H), and 1.38 (s, 6 H), 1.06 (d, J = 6.9 Hz, 6 H). 13C NMR (DMSO-d6, 126 MHz) δ ppm: 176.6, 160.7, 153.2, 151.7, 148.9, 145.2, 141.8, 116.0, 115.3, 108.7, 105.9, 69.1, 34.5, 28.2 (2C), and 19.4 (2C). Formula: C17H20N4O2S. MW: 344.43. MS: m/z 344 (M + H+).
6.8.3. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)-cyclopropanecarboxamide (40)
- Following GP5, compound 40 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclopropanecarboxamide (26) (110 mg, 0.38 mmol), 6 (100 mg, 0.38 mmol), K2CO3 (158 mg, 1.14 mmol), and Pd(dppf)Cl2 (125 mg, 0.17 mmol) in 4 mL of DMF. Purification: flash chromatography (DCM/MeOH/NH3(aq) 9:1:0.1). Yield: 54 mg (41%), brown solid, mp 257–258 °C. Purity: 95% (UPLC/MS). 1H NMR (500 MHz, METHANOL-d4) δ ppm: 0.89–0.94 (m, 2H), 0.99–1.03 (m, 2H), 1.54 (s, 6H), 1.86–1.96 (m, 1H), 3.35 (s, 1H), 7.04 (s, 1H), 7.31 (dd, J = 5.2, 1.1 Hz, 1H), 7.59 (br s, 2H), 8.30 (d, J = 5.2 Hz, 1H), and 8.38 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.85 (2C), 14.27, 28.21 (2C), 69.14, 105.96, 108.66, 115.25, 116.07, 141.90, 145.20, 148.97, 151.74, 153.05, 160.77, and 173.05. Formula: C17H18N4O2S. MW: 342.42. MS: m/z 343 (M + H+).
6.8.4. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)-cyclobutanecarboxamide (41)
- Following GP5, compound 41 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclobutanecarboxamid (29) (62 mg, 0.21 mmol), 6 (55 mg, 0.21 mmol), K2CO3 (87 mg, 0.63 mmol), and Pd(dppf)Cl2 (65 mg, 0.09 mmol) in 2.5 mL of DMF. Purification: preparative HPLC (10–60% MeCN gradient). Yield: 16 mg (22%), yellow solid, mp 252–253 °C. Purity: 98% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.42 (s, 6H), 1.76–1.85 (m, 1H), 1.87–1.98 (m, 1H), 2.06–2.15 (m, 2H), 2.19–2.29 (m, 2H), 7.07 (s, 1H), 7.13 (s, 1H), 7.35 (dd, J = 5.4, 1.7 Hz, 1H), 7.67 (s, 1H), 8.30–8.34 (m, 1H), 8.39 (s, 1H), and 10.45 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 17.70, 24.46 (2C), 28.24 (2C), 40.10, 69.12, 105.95, 108.75, 115.26, 116.06, 141.84, 145.18, 148.92, 151.72, 153.14, 160.75, and 174.12. Formula: C18H20N4O2S. MW: 356.44. MS: m/z 257 (M + H+).
6.8.5. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)-cyclohexanecarboxamide (42)
- Following GP5, compound 42 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclohexanecarboxamide (30) (112 mg, 0.34 mmol), 6 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), and Pd(dppf)Cl2 (110 mg, 0.15 mmol) in 3.5 mL of DMF. Purification: column chromatography (DCM/MeOH 97:3), and then the solid residue was washed with MeCN. Yield: 19 mg (15%), yellow solid, mp 268–269 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.12–1.32 (m, 4H), 1.39 (br s, 1H), 1.40–1.44 (m, 7H), 1.64 (br d, J = 11.5 Hz, 1H), 1.74 (br d, J = 12.3 Hz, 2H), 1.80 (br d, J = 12.3 Hz, 2H), 7.06 (s, 1H), 7.11 (s, 1H), 7.35 (dd, J = 5.2, 1.7 Hz, 1H), 7.66 (s, 1H), 8.32 (d, J = 5.2 Hz, 1H), 8.36 (s, 1H), and 10.50 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 25.17 (2C), 25.39, 28.24 (2C), 29.02 (2C), 44.29, 69.10, 105.91, 108.72, 115.22, 116.03, 141.79, 145.17, 148.90, 151.70, 153.21, 160.72, and 175.64. Formula: C20H24N4O2S. MW: 384.50. MS: m/z 385 (M + H+).
6.8.6. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)benzamide (43)
- Following GP5, compound 43 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)benzamide (31) (94 mg, 0.29 mmol), 6 (75 mg, 0.29 mmol), K2CO3 (120 mg, 0.87 mmol), and Pd(dppf)Cl2 (95 mg, 0.13 mmol) in 3 mL of DMF. Purification: column chromatography (DCM/MeOH 98:2), and then the solid residue was washed with MeCN. Yield: 30 mg (28%), yellow solid, mp 242–243 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.43 (s, 6H), 7.12 (s, 1H), 7.15 (s, 1H), 7.45 (dd, J = 5.3, 1.6 Hz, 1H), 7.51–7.55 (m, 2H), 7.59–7.63 (m, 1H), 7.68 (s, 1H), 8.04 (d, J = 1.4 Hz, 1H), 8.05 (s, 1H), 8.43 (d, J = 5.2 Hz, 1H), 8.47 (d, J = 1.1 Hz, 1H), and 10.95 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 28.25 (2C), 69.13, 106.04, 110.03, 115.91, 116.19, 128.08 (2C), 128.41 (2C), 132.08, 133.96, 141.90, 145.06, 148.96, 151.75, 153.17, 160.74, and 166.33. Formula: C20H18N4O2S. MW: 378.45. MS: m/z 379 (M + H+).
6.8.7. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)-methanesulfonamide (44)
- Following GP5, compound 44 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)methanesulfonamide (32) (146 mg, 0.49 mmol), 6 (127 mg, 0.49 mmol), K2CO3 (203 mg, 1.47 mmol), and Pd(dppf)Cl2 (160 mg, 0.22 mmol) in 5 mL of DMF. Purification: column chromatography (DCM/MeOH/NH3(aq) 9:1:0.01), and then the solid residue was washed with MeCN. Yield: 12 mg (7%), dark yellow solid, mp 303–304 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.42 (s, 6H), 3.30 (s, 3H), 7.05 (s, 1H), 7.14 (br s, 1H), 7.16 (s, 1H), 7.27 (br s, 1H), 7.50 (br s, 1H), 7.69 (s, 1H), and 8.26 (br s, 1H). Due to the low solubility of the compound, no signals were detected in the 13C NMR. Formula: C14H16N4O3S2. MW: 352.43. MS: m/z 353 (M + H+).
6.8.8. N-(4-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)pyridin-2-yl)-ethanesulfonamide (45)
- Following GP5, compound 45 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)ethanesulfonamide (33) (106 mg, 0.34 mmol), 6 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), and Pd(dppf)Cl2 (115 mg, 0.16 mmol) in 3.5 mL of DMF. Purification: preparative HPLC (10–60% MeCN gradient). Yield: 14 mg (11%), yellow solid, mp 284–285 °C. Purity: 96% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.23 (t, J = 7.3 Hz, 3H), 1.42 (s, 6H), 3.45 (br d, J = 7.4 Hz, 2H), 7.05 (s, 1H), 7.16 (s, 2H), 7.26 (br d, J = 4.0 Hz, 1H), 7.68 (s, 1H), 8.24 (br d, J = 2.3 Hz, 1H), and 10.79 (br s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 8.06, 28.23 (2C), 47.33, 69.12, 106.22, 107.56, 107.59, 116.42, 142.52, 144.34, 151.68, 153.33, 153.41, and 160.69. Formula: C15H18N4O3S2. MW: 366.45. MS: m/z 367 (M + H+).
6.8.9. 6-(2-((Cyclopropylmethyl)amino)pyridin-4-yl)-2,2-dimethyl-2,3-dihydrothieno [3,2-d]pyrimidin-4(1H)-one (46)
- Following GP5, compound 46 was prepared using N-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine (35) (93 mg, 0.34 mmol), 6 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), and Pd(dppf)Cl2 (110 mg, 0.15 mmol) in 3 mL of DMF. Purification: flash chromatography (DCM/MeOH 95:5). Yield: 45 mg (40%), yellow solid, mp 232–233 °C. Purity: 95% (UPLC/MS). 1H NMR (DMSO-d6, 500 MHz) δ ppm: 7.97 (d, 1H, J = 5.4 Hz), 7.59 (s, 1H), 7.08 (s, 1H), 6.93 (s, 1H), 6.77 (t, 1H, J = 5.6 Hz), 6.6–6.7 (m, 2H), 3.14 (t, 2H, J = 6.2 Hz), 1.41 (s, 6H), 1.0–1.1 (m, 1H), 0.4–0.5 (m, 2H), and 0.2–0.2 (m, 2H). 13C NMR (DMSO-d6, 126 MHz) δ ppm: 160.8, 159.5, 151.6, 148.6, 146.4, 140.4, 115.1, 107.8, 104.9, 103.6, 69.0, 45.2, 28.2 (2C), 10.9, and 3.4 (2C). Formula: C17H20N24OS. MW: 328.43. MS: m/z 329 (M + H+).
6.8.10. 5-(2-Acetamidopyridin-4-yl)-3-ureidothiophene-2-carboxamide (47)
- Following GP5, compound 47 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide (24) (81 mg, 0.31 mmol), 5 (82 mg, 0.31 mmol), K2CO3 (129 mg, 0.93 mmol), and Pd(dppf)Cl2 (102 mg, 0.14 mmol) in 3 mL of DMF. Purification: column chromatography (10% MeOH in DCM). Yield: 8 mg (8%), brownish solid, mp degradation over 365 °C. 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.61 (s, 1H), 10.00 (s, 1H), 8.38 (s, 1H), 8.27–8.36 (m, 2H), 7.41–7.70 (m, 2H), 7.28 (dd, J = 5.2, 1.4 Hz, 1H), 6.65 (br s, 2H), and 2.08 (s, 3H). 13C NMR (DMSO-d6, 126 MHz) δ ppm: 170.2, 165.7, 155.4, 153.6, 149.6, 145.7, 142.2, 141.1, 120.7, 115.7, 110.8, 109.1, and 24.5. Formula: C13H13N5O3S. MW: 319.34. MS: m/z 320 (M + H+).
6.8.11. 5-(2-Isobutyramidopyridin-4-yl)-3-ureidothiophene-2-carboxamide (48)
- Following GP5, compound 48 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)isobutyramide (25) (92 mg, 0.32 mmol), 5 (85 mg, 0.32 mmol), K2CO3 (133 mg, 0.96 mmol), and Pd(dppf)Cl2 (100 mg, 0.14 mmol) in 3 mL of DMF. Purification: preparative HPLC (5–50% MeCN gradient). Yield: 7 mg (6%), yellow solid, mp degradation over 308 °C. Purity: 96% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.10 (d, J = 6.9 Hz, 6H), 2.72–2.83 (m, 1H), 6.69 (br s, 1H), 7.33 (dd, J = 5.2, 1.7 Hz, 1H), 7.56 (br s, 2H), 8.35 (d, J = 5.4 Hz, 1H), 8.42 (s, 1H), 8.45 (d, J = 1.1 Hz, 1H), 10.04 (s, 1H), and 10.60 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 19.36 (2C), 34.50, 108.79, 110.26, 115.10, 120.13, 140.62, 141.67, 145.25, 149.06, 153.27, 154.90, 165.22, and 176.60. Formula: C15H17N5O3S. MW: 347.39. MS: m/z 348 (M + H+).
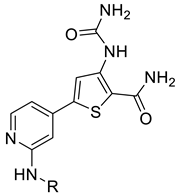
6.8.12. 5-(2-(Cyclopropanecarboxamido)pyridin-4-yl)-3-ureidothiophene-2-carboxamide (49)
- Following GP5, compound 49 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclopropanecarboxamide (26) (98 mg, 0.34 mmol), 5 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), and Pd(dppf)Cl2 (120 mg, 0.16 mmol) in 3.5 mL of DMF. Purification: column chromatography (DCM/MeOH/TEA 90:10:0.1). Yield: 48 mg (41%), brown solid, mp degradation over 290 °C. Purity: 100% (UPLC/MS). HRMS (ESI-QTOF): m/z calcd. for 346.0929; found 346.0958. 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.76–0.94 (m, 4H), 1.97–2.08 (m, 1H), 6.69 (br s, 1H), 7.32 (s, 1H), 7.57 (br s, 3H), 8.37 (s, 1H), 8.42 (s, 2H), 10.04 (br s, 1H), and 10.97 (br s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.89 (2C), 14.23, 99.49, 108.69, 110.24, 120.12, 140.60, 141.71, 145.25, 149.10, 153.11, 154.89, 165.22, and 173.03. Formula: C15H15N5O3S. MW: 345.38. MS: m/z 346 (M + H+).
6.8.13. 5-(2-(Cyclobutanecarboxamido)pyridin-4-yl)-3-ureidothiophene-2-carboxamide (50)
- Following GP5, compound 50 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclobutanecarboxamide (29) (60 mg, 0.20 mmol), 5 (53 mg, 0.20 mmol), K2CO3 (101 mg, 0.60 mmol), and Pd(dppf)Cl2 (65 mg, 0.09 mmol) in 2 mL of DMF. Purification: preparative HPLC (10–60% MeCN gradient). Yield: 16 mg (22%), cream solid, mp degradation over 346 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.77–1.86 (m, 1H), 1.88–1.99 (m, 1H), 2.07–2.15 (m, 2H), 2.17–2.29 (m, 2H), 2.51–2.52 (m, 1H), 6.69 (br s, 1H), 7.32 (dd, J = 5.2, 1.7 Hz, 1H), 7.59 (br s, 1H), 8.34 (dd, J = 5.4, 0.6 Hz, 1H), 8.43 (s, 1H), 8.46 (s, 1H), 10.04 (s, 1H), and 10.49 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 17.73, 24.45 (2C), 40.10, 108.74, 110.26, 115.07, 120.14, 140.61, 141.67, 145.24, 149.06, 153.22, 154.89, 165.22, and 174.16. Formula: C16H17N5O3S. MW: 359.40. MS: m/z 360 (M + H+).
6.8.14. 5-(2-(Cyclohexanecarboxamido)pyridin-4-yl)-3-ureidothiophene-2-carboxamide (51)
- Following GP5, compound 51 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclohexanecarboxamide (30) (112 mg, 0.34 mmol), 5 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), Pd(dppf)Cl2 (112 mg, 0.15 mmol), and DMF (3.5 mL). Purification: column chromatography (5% MeOH in DCM). Yield: 15 mg (11%), greyish solid, mp degradation over 315 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.54 (s, 1 H), 10.04 (s, 1 H), 8.40–8.45 (m, 2 H), 8.35 (d, J = 5.4 Hz, 1 H), 7.39–7.78 (m, 2 H), 7.32 (dd, J = 5.2, 1.7 Hz, 1 H), 6.69 (br s, 2 H), 2.08 (s, 1 H), 1.70–1.86 (m, 4 H), 1.60–1.68 (m, 1 H), 1.34–1.47 (m, 2 H), and 1.16–1.32 (m, 3 H). 13C NMR (DMSO-d6, 126 MHz) δ ppm: 175.7, 165.2, 154.9, 153.3, 149.1, 145.2, 141.6, 140.6, 120.1, 115.1, 110.2, 108.8, 44.2, 29.0 (2C), 25.4, and 25.2 (2C). Formula: C18H21N5O3S. MW: 387.46. MS: m/z 388 (M + H+).
6.8.15. 5-(2-Benzamidopyridin-4-yl)-3-ureidothiophene-2-carboxamide (52)
- Following GP5, compound 52 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)benzamide (31) (100 mg, 0.31 mmol), 5 (82 mg, 0.31 mmol), K2CO3 (129 mg, 0.93 mmol), Pd(dppf)Cl2 (102 mg, 0.14 mmol), and DMF (3 mL). Purification: column chromatography (5% MeOH in DCM). Yield: 13 mg (11%), brown solid, mp degradation over 300 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.97 (s, 1 H), 10.06 (s, 1 H), 8.55 (d, J = 1.1 Hz, 1 H), 8.42–8.51 (m, 2 H), 8.02–8.10 (m, 2 H), 7.58–7.65 (m, 2 H), 7.49–7.57 (m, 3 H), 7.42 (dd, J = 5.3, 1.6 Hz, 1 H), and 6.64–6.80 (m, 1 H). 13C NMR (DMSO-d6, 126 MHz) δ ppm:166.8, 165.7, 155.4, 153.8, 149.6, 145.8, 142.3, 141.0, 134.4, 132.6, 128.9 (2C), 128.6 (2C), 120.8, 116.3, 110.9, and 110.4. Formula: C18H15N5O3S. MW: 381.41. MS: m/z 382 (M + H+).
6.8.16. N-(3-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)phenyl)cyclopropanecarboxamide (53)
- Following GP5, compound 53 was prepared using N-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclopropanecarboxamide (27) (98 mg, 0.34 mmol), 6 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), Pd(dppf)Cl2 (112 mg, 0.15 mmol), and DMF (3.5 mL). Purification: column chromatography (5–10% MeOH in DCM). Yield: 38 mg (33%), brown–green solid, mp 157–158 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.30 (s, 1 H), 7.88 (t, J = 1.6 Hz, 1 H), 7.50–7.56 (m, 1 H), 7.48 (s, 1 H), 7.24–7.34 (m, 2 H), 7.01 (d, J = 0.9 Hz, 1 H), 6.78 (s, 1 H), 1.70–1.79 (m, 1 H), 1.38 (s, 6 H), AND 0.74–0.81 (m, 4 H). 13C NMR (DMSO-d6, 126 MHz) δ ppm: 172.4, 161.5, 152.4, 148.8, 140.6, 134.1, 130.2, 120.6, 119.7, 116.2, 114.3, 104.6, 69.5, 28.7 (2C), 15.2, and 7.9 (2C). Formula: C18H19N3O2S. MW: 341.43. MS: m/z 342 (M + H+).
6.8.17. N-(5-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)-2-fluorophenyl)- cyclopropanecarboxamide (54)
- Following GP5, compound 54 was prepared using N-(2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclopropanecarboxamide (28) (70 mg, 0.23 mmol), 6 (60 mg, 0.23 mmol), K2CO3 (95 mg, 0.69 mmol), and Pd(dppf)Cl2 (80 mg, 0.11 mmol) in 3 mL of DMF. Purification: flash chromatography (DCM/MeOH/NH3(aq) 9.5:0.5:0.05), and then the solid residue was crystallized from MeCN. Yield: 31 mg (37%), beige solid, mp 199–200 °C. Purity: 95% (UPLC/MS). 1H NMR (500 MHz, ACETONE-d6) δ ppm: 0.81–0.86 (m, 2H), 0.92–0.97 (m, 2H), 1.57 (s, 6H), 2.00–2.04 (m, 1H), 6.29 (br s, 1H), 6.66 (br s, 1H), 6.85 (s, 1H), 7.22 (dd, J = 10.9, 8.6 Hz, 1H), 7.35 (ddd, J = 8.5, 4.7, 2.3 Hz, 1H), 8.66 (dd, J = 7.3, 1.9 Hz, 1H), and 9.32 (br s, 1H). 13C NMR (126 MHz, ACETONE-d6) δ ppm: 7.66 (2C), 14.74, 28.28 (2C), 69.81, 106.20, 114.47, 115.96 (d, J = 20.5 Hz), 120.03, 121.56 (br d, J = 7.2 Hz) 128.17 (d, J = 11.5 Hz) 130.66 (d, J = 3.6 Hz) 148.57, 153.10 (d, J = 252.3 Hz) 152.22, 161.30, and 172.76. Formula: C18H18FN3O2S. MW: 359.42. MS: m/z 360 (M + H+).
6.8.18. N-(3-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)phenyl)ethanesulfonamide (55)
- Following GP5, compound 55 was prepared using N-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)ethanesulfonamide (34) (106 mg, 0.34 mmol), 6 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), and Pd(dppf)Cl2 (115 mg, 0.16 mmol) in 3.5 mL of DMF. Purification: preparative HPLC (10–60% MeCN gradient). Yield: 78 mg (62%), brown solid, mp 119–200 °C. Purity: 96% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.20 (t, J = 7.3 Hz, 3H), 1.41 (s, 6H), 3.12–3.17 (m, 2H), 6.83 (s, 1H), 7.07 (s, 1H), 7.22 (dt, J = 7.7, 1.8 Hz, 1H), 7.34–7.40 (m, 2H), 7.44–7.46 (m, 1H), 7.54 (s, 1H), and 9.96 (br s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 8.05, 28.20 (2C), 45.27, 69.00, 104.29, 114.13, 115.77, 119.28, 120.73, 130.29, 134.20, 139.26, 147.68, 151.87, and 160.94. Formula: C16H19N3O3S2. MW: 365.47. MS: m/z 366 (M + H+).
6.8.19. N-(3-(2,2-Dimethyl-4-oxo-1,2,3,4-tetrahydrothieno [3,2-d]pyrimidin-6-yl)phenyl)cyclohexanesulfonamide (56)
- Following GP5, compound 56 was prepared using N-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclohexanesulfonamide (36) (84 mg, 0.23 mmol), 6 (60 mg, 0.23 mmol), K2CO3 (95 mg, 0.69 mmol), Pd(dppf)Cl2 (76 mg, 0.10 mmol), and DMF (2 mL). Purification: column chromatography (2% MeOH in DCM). Yield: 28 mg (29%), yellow solid, mp 275–276 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 9.90 (br s, 1 H), 7.50 (s, 1 H), 7.41–7.44 (m, 1 H), 7.27–7.35 (m, 2 H), 7.16–7.20 (m, 1 H), 7.04 (d, J = 0.9 Hz, 1 H), 6.79 (s, 1 H), 2.99 (tt, J = 11.8, 3.4 Hz, 1 H), 1.95–2.03 (m, 2 H), 1.65–1.77 (m, 2 H), 1.47–1.57 (m, 1 H), 1.33–1.42 (m, 8 H), 1.12–1.23 (m, 2 H), and 1.03–1.11 (m, 1 H). 13C NMR (DMSO-d6, 126 MHz) δ ppm: 160.9, 151.9, 147.7, 139.5, 134.1, 130.2, 120.5, 119.1, 115.5, 114.1, 104.2, 69.0, 59.3, 28.2 (2C), 26.0, 24.7, and 24.3 (2C). Formula: C20H25N3O3S2. MW: 419.56. MS: m/z 420 (M + H+).
6.8.20. 2,2-Dimethyl-6-(3-(piperidin-1-ylsulfonyl)phenyl)-2,3-dihydrothieno [3,2-d]pyrimidin-4(1H)-one (57)
- Following GP5, compound 57 was prepared using 1-((3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)sulfonyl)piperidine (37) (97 mg, 0.28 mmol), 6 (73 mg, 0.28 mmol), K2CO3 (116 mg, 0.84 mmol), and Pd(dppf)Cl2 (95 mg, 0.13 mmol) in 3 mL of DMF. Purification: column chromatography (DCM/MeOH 97:3), and then the solid residue was washed with MeCN. Yield: 24 mg (21%), beige solid, mp 202–203 °C. Purity: 97% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 1.34–1.39 (m, 2H), 1.42 (s, 6H), 1.51–1.57 (m, 4H), 2.92 (br t, J = 5.3 Hz, 4H), 7.04 (s, 1H), 7.10 (s, 1H), 7.61 (s, 1H), 7.68–7.73 (m, 2H), 7.83 (d, J = 0.9 Hz, 1H), and 7.98 (ddd, J = 5.4, 3.4, 2.0 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 22.79, 24.70 (2C), 28.20 (2C), 46.62 (2C), 69.08, 105.17, 115.47, 123.54, 127.34, 129.95, 130.53, 134.27, 136.62, 145.91, 151.92, and 160.83. Formula: C19H23N3O3S2. MW: 405.53. MS: m/z 406 (M + H+).
6.8.21. 5-(3-(Cyclopropanecarboxamido)phenyl)-3-ureidothiophene-2-carboxamide (58)
- Following GP5, compound 58 was prepared using N-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclopropanecarboxamide (27) (98 mg, 0.34 mmol), 5 (90 mg, 0.34 mmol), K2CO3 (141 mg, 1.02 mmol), and Pd(dppf)Cl2 (110 mg, 0.15 mmol) in 3.5 mL of DMF. Purification: column chromatography (DCM/MeOH 9:1), and then the solid residue was washed with MeCN. Yield: 24 mg (21%), beige solid, mp 240–241 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.79–0.84 (m, 4H), 1.75–1.82 (m, 1H), 6.65 (br s, 2H), 7.26–7.29 (m, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.45 (br d, J = 3.4 Hz, 1H), 7.56 (dd, J = 8.2, 1.0 Hz, 1H), 8.05 (s, 1H), 8.23 (s, 1H), 10.07 (s, 1H), and 10.39 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.36 (2C), 14.64, 108.40, 115.60, 118.03, 119.09, 119.87, 129.81, 133.37, 140.22, 143.64, 145.37, 154.93, 165.53, and 171.96. Formula: C16H16N4O3S. MW: 344.39. MS: m/z 345 (M + H+).
6.8.22. 5-(3-(Cyclopropanecarboxamido)-4-fluorophenyl)-3-ureidothiophene-2-carboxamide (59)
- Following GP5, compound 59 was prepared using N-(2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclopropanecarboxamide (28) (80 mg, 0.26 mmol), 5 (68 mg, 0.26 mmol), K2CO3 (108 mg, 0.78 mmol), and Pd(dppf)Cl2 (95 mg, 0.13 mmol) in 3 mL of DMF. Purification: column chromatography (DCM/MeOH/NH3(aq) 9.5:0.5:0.05), and then the solid residue was washed with MeCN. Yield: 50 mg (53%), brown solid, mp degradation over 305 °C. Purity: 95% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.72–0.87 (m, 4H), 1.95–2.08 (m, 1H), 6.61 (br s, 1H), 7.31 (br s, 2H), 7.47 (br s, 3H), 8.13 (s, 1H), 8.32 (br d, J = 6.3 Hz, 1H), 10.02 (s, 1H), and 10.11 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.69 (2C), 14.08, 108.46, 116.40 (d, J = 20.5 Hz), 118.18, 120.19, 121.54 (d, J = 6.6 Hz), 127.24 (d, J = 12.1 Hz), 129.39 (d, J = 6.0 Hz), 142.65, 145.39, 153.03 (d, J = 238.4 Hz), 154.90, 165.46, and 172.52. Formula: C16H15FN4O3S. MW: 362.38 MS: m/z 363 (M + H+).
6.8.23. N-(4-(4’-Oxo-3’,4’-dihydro-1’H-spiro[cyclopentane-1,2’-thieno [3,2-d]pyrimidin]-6’-yl)pyridin-2-yl)cyclopropanecarboxamide (60)
- Following GP5, compound 60 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclopropanecarboxamide (26) (92 mg, 0.32 mmol), 7 (92 mg, 0.32 mmol), K2CO3 (133 mg, 0.96 mmol), and Pd(dppf)Cl2 (55 mg, 0.08 mmol) in 3 mL of DMF. Purification: preparative HPLC (10–60% MeCN gradient). Yield: 60 mg (51%), yellow solid, mp 225–226 °C. Purity: 99% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.79–0.89 (m, 4H), 1.60–1.73 (m, 4H), 1.78–1.91 (m, 4H), 1.98–2.06 (m, 1H), 7.07 (s, 1H), 7.22 (s, 1H), 7.34 (dd, J = 5.3, 1.6 Hz, 1H), 7.82 (s, 1H), 8.31–8.37 (m, 2H), and 10.93 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.83 (2C), 14.27, 21.87 (2C), 38.65 (2C), 79.01, 106.99, 108.73, 115.20, 116.11, 141.86, 145.11, 148.94, 152.23, 153.02, 161.14, and 173.01. Formula: C19H20N4O2S. MW: 368.46. MS: m/z 369 (M + H+).
6.8.24. N-(4-(4′-Oxo-3′,4,4′,5-tetrahydro-1′H,3H-spiro[furan-2,2′-thieno [3,2-d]pyrimidin]-6′-yl)pyridin-2-yl)cyclopropanecarboxamide (61)
- Following GP5, compound 61 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclopropanecarboxamide (26) (104 mg, 0.36 mmol), 8 (102 mg, 0.36 mmol), K2CO3 (149 mg, 1.08 mmol), and Pd(dppf)Cl2 (60 mg, 0.08 mmol) in 3.5 mL of DMF. Purification: flash chromatography (DCM/MeOH/NH3(aq) 9:1:0.01). Yield: 12 mg (9%), white solid, mp 314–315 °C. Purity: 100% (UPLC/MS). 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.81–0.91 (m, 4H), 1.83–1.91 (m, 2H), 2.01–2.07 (m, 1H), 2.69 (t, J = 7.4 Hz, 2H), 3.43–3.50 (m, 2H), 4.56 (t, J = 4.9 Hz, 1H), 7.59 (dd, J = 5.2, 1.7 Hz, 1H), 7.95 (s, 1H), 8.41 (dd, J = 5.3, 0.7 Hz, 1H), 8.47 (d, J = 1.1 Hz, 1H), 11.01 (s, 1H), and 12.51 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.87 (2C), 14.28, 29.97, 30.91, 59.97, 109.32, 115.75, 121.08, 123.60, 141.38, 147.46, 149.07, 153.14, 157.87, 158.35, 160.11, and 173.10. Formula: C18H18N4O3S. MW: 370.43. MS: m/z 371 (M + H+).
6.8.25. N-(4-(4′-Oxo-3′,4,4′,5-tetrahydro-1′H,2H-spiro[furan-3,2′-thieno [3,2-d]pyrimidin]-6′-yl)pyridin-2-yl)cyclopropanecarboxamide (62)
- Following GP5, compound 62 was prepared using N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)cyclopropanecarboxamide (26) (81 mg, 0.28 mmol), 9 (81 mg, 0.28 mmol), K2CO3 (116 mg, 0.84 mmol), and Pd(dppf)Cl2 (50 mg, 0.07 mmol) in 2.5 mL of DMF. Purification: preparative HPLC (5–50% MeCN gradient). Yield: 18 mg (18%), yellow solid, mp 249–250 °C. Purity: 98% (UPLC/MS). HRMS (ESI-QTOF): m/z calcd. for 371.1133; found 371.1161. 1H NMR (500 MHz, DMSO-d6) δ ppm: 0.78–0.89 (m, 4H), 1.99–2.07 (m, 1H), 2.10–2.24 (m, 2H), 3.60 (d, J = 8.9 Hz, 1H), 3.76 (d, J = 8.9 Hz, 1H), 3.80–3.95 (m, 2H), 7.11 (s, 1H), 7.36 (dd, J = 5.2, 1.7 Hz, 1H), 7.59 (s, 1H), 8.07 (s, 1H), 8.31–8.39 (m, 2H), and 10.95 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ ppm: 7.85 (2C), 14.28, 38.77, 65.98, 76.48, 77.68, 107.07, 108.79, 115.28, 116.06, 141.73, 145.71, 148.99, 152.08, 153.03, 160.94, and 173.04. Formula: C18H18N4O3S. MW: 370.43. MS: m/z 371 (M + H+).
6.9. Evaluation of In Vitro Inhibitory Potencies towards Kinases GSK-3β, IKK-β, and ROCK-1
6.10. Computational Analysis and Visualizations
6.11. Kinetics Studies of GSK-3β Inhibition by 62
7. In Cellulo Studies
7.1. Cells Preparation
7.2. Preparation of Test Compound Solutions
7.3. Cell Viability Assay
7.4. Okadaic Acid Treated HT-22 Cells
7.5. LPS-Treated BV-2 Cells
7.6. NO Release Measurement
7.7. Measurement of Cytokine Levels
7.8. Statistical Analysis
7.9. Thermodynamic Solubility Assay
7.10. Chemical Stability Assay
7.11. Metabolic Stability Assay
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association 2022 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2022, 18, 700–789. [CrossRef] [PubMed]
- Wong, W. Economic Burden of Alzheimer Disease and Managed Care Considerations. Am. J. Manag. Care 2020, 26, 177–183. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau Pathology Is an Initiating Factor in Sporadic Alzheimer’s Disease. Alzheimer’s Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The Revitalized Tau Hypothesis on Alzheimer’s Disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Song, L.; Oseid, D.E.; Wells, E.A.; Robinson, A.S. The Interplay between GSK3β and Tau Ser262 Phosphorylation during the Progression of Tau Pathology. Int. J. Mol. Sci. 2022, 23, 11610. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Onishi, T.; Iwashita, H.; Uno, Y.; Kunitomo, J.; Saitoh, M.; Kimura, E.; Fujita, H.; Uchiyama, N.; Kori, M.; Takizawa, M. A Novel Glycogen Synthase Kinase-3 Inhibitor 2-methyl-5-(3-{4-[(S)-}-1--5-)-1,3,4- Decreases Tau Phosphorylation and Ameliorates Cognitive Deficits in a Transgenic Model of Alzheimer’s Disease. J. Neurochem. 2011, 119, 1330–1340. [Google Scholar] [CrossRef]
- Bao, X.Q.; Li, N.; Wang, T.; Kong, X.C.; Tai, W.J.; Sun, H.; Zhang, D. FLZ Alleviates the Memory Deficits in Transgenic Mouse Model of Alzheimer’s Disease via Decreasing Beta-Amyloid Production and Tau Hyperphosphorylation. PLoS ONE 2013, 8, e78033. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased Signalling up-Regulates BACE1 Expression and Its Therapeutic Potential in Alzheimer’s Disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-Mediated BACE1 Expression Reduces Alzheimer-Associated Phenotypes. J. Clin. Invest. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of Glycogen Synthase Kinase-3 Ameliorates β-Amyloid Pathology and Restores Lysosomal Acidification and Mammalian Target of Rapamycin Activity in the Alzheimer Disease Mouse Model: In Vivo and In Vitro Studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef]
- Ding, Y.; Qiao, A.; Fan, G.H. Indirubin-3’-monoxime Rescues Spatial Memory Deficits and Attenuates β-Amyloid-Associated Neuropathology in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2010, 39, 156–168. [Google Scholar] [CrossRef]
- Gianferrara, T.; Cescon, E.; Grieco, I.; Spalluto, G.; Federico, S. Glycogen Synthase Kinase 3β Involvement in Neuroinflammation and Neurodegenerative Diseases. Curr. Med. Chem. 2022, 29, 4631–4697. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.L.; Zhu, L.; Wu, P.Y.; Dai, W.B.; Rakesh, K.P. Structure-Activity Relationship (SAR) Studies of Synthetic Glycogen Synthase Kinase-3β Inhibitors: A Critical Review. Eur. J. Med. Chem. 2019, 164, 448–470. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, J.; Zhang, J.; Yang, J.; Wang, G.; Wang, Y. Development of Inhibitors Targeting Glycogen Synthase Kinase-3β for Human Diseases: Strategies to Improve Selectivity. Eur. J. Med. Chem. 2022, 236, 114301. [Google Scholar] [CrossRef] [PubMed]
- Shri, S.R.; Manandhar, S.; Nayak, Y.; Pai, K.S.R. Role of GSK-3β Inhibitors: New Promises and Opportunities for Alzheimer’s Disease. Adv. Pharm. Bull. 2023, 13, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Balboni, B.; Masi, M.; Rocchia, W.; Girotto, S.; Cavalli, A. GSK-3β Allosteric Inhibition: A Dead End or a New Pharmacological Frontier? Int. J. Mol. Sci. 2023, 24, 7541. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Han, T.; Yao, J.; Wang, K.; Dong, X.; Yu, F.; Huang, H.; Han, M.; Liao, Q.; He, S.; et al. Targeting Glycogen Synthase Kinase-3β for Alzheimer’s Disease: Recent Advances and Future Prospects. Eur. J. Med. Chem. 2024, 265, 116065. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas Ruiz, S.M.; Eldar-Finkelman, H. Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward. Front. Mol. Neurosci. 2022, 14, 792364. [Google Scholar] [CrossRef] [PubMed]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.B.; Gich, I.; Agulló, J.M.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A Novel GSK-3β Inhibitor Reduces Alzheimer’s Pathology and Rescues Neuronal Loss In Vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef] [PubMed]
- DaRocha-Souto, B.; Coma, M.; Pérez-Nievas, B.G.; Scotton, T.C.; Siao, M.; Sánchez-Ferrer, P.; Hashimoto, T.; Fan, Z.; Hudry, E.; Barroeta, I.; et al. Activation of Glycogen Synthase Kinase-3 Beta Mediates β-Amyloid Induced Neuritic Damage in Alzheimer’s Disease. Neurobiol. Dis. 2013, 45, 425–437. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A Phase II Trial of Tideglusib in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-Associated Protein Kinase 1 (ROCK1) Is Increased in Alzheimer’s Disease and ROCK1 Depletion Reduces Amyloid-β Levels in Brain. J. Neurochem. 2016, 138, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Ren, R.J.; Zhang, Y.F.; Huang, Y.; Cui, H.L.; Ma, C.; Qiu, W.Y.; Wang, H.; Cui, P.J.; Chen, H.Z.; et al. Rho-Associated Coiled-Coil Kinase 1 Activation Mediates Amyloid Precursor Protein Site-Specific Ser655 Phosphorylation and Triggers Amyloid Pathology. Aging Cell 2019, 18, e13001. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, J.H.; Feng, Y.; Mattheyses, A.L.; Hales, C.M.; Higginbotham, L.A.; Duong, D.M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Seyfried, N.T.; et al. Pharmacologic Inhibition of ROCK2 Suppresses Amyloid-β Production in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2013, 33, 19086–19098. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Shirafuji, N.; Yen, S.H.; Yoshida, H.; Kanaan, N.M.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Fujita, Y.; Kuriyama, M.; et al. Rho-Kinase ROCK Inhibitors Reduce Oligomeric Tau Protein. Neurobiol. Aging 2020, 89, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.; Ohtomo, E. Effects of Fasudil in Acute Ischemic Stroke: Results of a Prospective Placebo-Controlled Double-Blind Trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wong-Guerra, M.; Calfio, C.; Maccioni, R.B.; Rojo, L.E. Revisiting the Neuroinflammation Hypothesis in Alzheimer’s Disease: A Focus on the Druggability of Current Targets. Front. Pharmacol. 2023, 14, 1161850. [Google Scholar] [CrossRef]
- Sun, E.; Motolani, A.; Campos, L.; Lu, T. The Pivotal Role of NF-κB in the Pathogenesis and Therapeutics of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 38, 8972. [Google Scholar] [CrossRef] [PubMed]
- Sivamaruthi, B.S.; Raghani, N.; Chorawala, M.; Bhattacharya, S.; Prajapati, B.G.; Elossaily, G.M.; Chaiyasut, C. NF-κB Pathway and Its Inhibitors: A Promising Frontier in the Management of Alzheimer’ s Disease. Biomedicines 2023, 11, 2587. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, H.; Davies, A.M. Regulation of Neural Process Growth, Elaboration and Structural Plasticity by NF-κB. Trends Neurosci. 2011, 34, 316–325. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, R.; Li, W.; Ma, X.C.; Qiu, F.; Sun, C.P. IκB Kinase β (IKKβ): Structure, Transduction Mechanism, Biological Function, and Discovery of Its Inhibitors. Int. J. Biol. Sci. 2023, 19, 4181–4203. [Google Scholar] [CrossRef] [PubMed]
- Awasthee, N.; Rai, V.; Chava, S.; Nallasamy, P.; Kunnumakkara, A.B.; Bishayee, A.; Chauhan, S.C.; Challagundla, K.B.; Gupta, S.C. Targeting IκappaB Kinases for Cancer Therapy. Semin. Cancer Biol. 2019, 56, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-Molecule Inhibitors of IκB Kinase (IKK) and IKK-Related Kinases. Pharm. Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Hao, W.; Decker, Y.; Schomburg, R.; Fülöp, L.; Pasparakis, M.; Menger, M.D.; Fassbender, K. IKKβ Deficiency in Myeloid Cells Ameliorates Alzheimer’s Disease-Related Symptoms and Pathology. J. Neurosci. 2014, 34, 12982–12999. [Google Scholar] [CrossRef] [PubMed]
- Schnöder, L.; Quan, W.; Yu, Y.; Tomic, I.; Luo, Q.; Hao, W.; Peng, G.; Li, D.; Fassbender, K.; Liu, Y. Deficiency of IKKβ in Neurons Ameliorates Alzheimer’s Disease Pathology in APP- and Tau-Transgenic Mice. FASEB J. 2023, 37, e22778. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Zhang, Y.H.; Zhang, W.; Zhao, P. Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 472. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kurasawa, O.; Oguro, Y.; Miyazaki, T.; Homma, M.; Mori, K.; Iwai, K.; Hara, H.; Skene, R.; Hoffman, I.; Ohashi, A.; et al. Identification of a New Class of Potent Cdc7 Inhibitors Designed by Putative Pharmacophore Model: Synthesis and Biological Evaluation of 2,3-dihydrothieno[3,2-d]pyrimidin-4(1H)-ones. Bioorganic Med. Chem. 2017, 25, 2133–2147. [Google Scholar] [CrossRef]
- Sivaprakasam, P.; Han, X.; Civiello, R.L.; Jacutin-Porte, S.; Kish, K.; Pokross, M.; Lewis, H.A.; Ahmed, N.; Szapiel, N.; Newitt, J.A.; et al. Discovery of New Acylaminopyridines as GSK-3 Inhibitors by a Structure Guided in-Depth Exploration of Chemical Space around a Pyrrolopyridinone Core. Bioorganic Med. Chem. Lett. 2015, 25, 1856–1863. [Google Scholar] [CrossRef]
- Baxter, A.; Brough, S.; Cooper, A.; Floettmann, E.; Foster, S.; Harding, C.; Kettle, J.; McInally, T.; Martin, C.; Mobbs, M.; et al. Hit-to-Lead Studies: The Discovery of Potent, Orally Active, Thiophenecarboxamide IKK-2 Inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 2817–2822. [Google Scholar] [CrossRef]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and Homogeneous ADP Monitoring Assay for Kinases. Assay Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Sippl, W. Activation, Regulation, and Inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Park, S.Y.; Jung, M.S.; Yoon, S.H.; Kwen, M.Y.; Lee, S.Y.; Choi, S.H.; Radnaabazar, C.; Kim, M.K.; Kim, H.; et al. Dyrk1A-Mediated Phosphorylation of Presenilin 1: A Functional Link between Down Syndrome and Alzheimer’s Disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, B.; Xu, W.F.; Liu, R.F.; Yang, J.; Yu, C.X. Effects of PTEN Inhibition on Regulation of Tau Phosphorylation in an Okadaic Acid-Induced Neurodegeneration Model. Int. J. Dev. Neurosci. 2012, 30, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tung, Y.C.; Wang, Y.; Li, X.T.; Iqbal, K.; Grundke-Iqbal, I. Hyperphosphorylation and Accumulation of Neurofilament Proteins in Alzheimer Disease Brain and in Okadaic Acid-Treated SY5Y Cells. FEBS Lett. 2001, 507, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, L.; Li, D.; Huang, Y.; Liu, M.; Liu, W.; Tian, C.; Liu, X.; Jiang, X.; Hu, X.; et al. Discovery of Novel Tacrine Derivatives as Potent Antiproliferative Agents with CDKs Inhibitory Property. Bioorg. Chem. 2022, 126, 105875. [Google Scholar] [CrossRef]
- Sudau, A.; Es-Sayed, M.; Braun, C.A.; Meissner, R.; Sirven, C.; Benting, J.; Dahmen, P.; Portz, D.; Wachendorff-Neumann, U.; Desbordes, P.; et al. Phenylpyri(mi)dinylazoles. CA2773205A1, 14 April 2011. [Google Scholar]
- Sudau, A.; Es-Sayed, M.; Braun, C.A.; Meissner, R.; Sirven, C.; Benting, J.; Dahmen, P.; Portz, D.; Wachendorff-Neumann, U.; Desbordes, P.; et al. Phenylpyri(mi)dinylazoles. U.S. Patent US20110183978A1, 28 July 2011. [Google Scholar]
- Hou, S.; Yang, X.; Yang, Y.; Tong, Y.; Chen, Q.; Wan, B.; Wei, R.; Lu, T.; Chen, Y.; Hu, Q. Design, Synthesis and Biological Evaluation of 1H-Indazole Derivatives as Novel ASK1 Inhibitors. Eur. J. Med. Chem. 2021, 220, 113482. [Google Scholar] [CrossRef]
- Houze, J.B.; Dransfield, P.; Pattaropong, V.; Du, X.; Fu, Z.; Lai, S.; Park, J.; Jiao, X.; Kohn, T.J.; Aicher, T.D.; et al. Urea Compounds as GKA Activators. WO 2013/086397, 13 June 2013. [Google Scholar]
- Sutherland, M.; Li, A.; Kaghad, A.; Panagopoulos, D.; Li, F.; Szewczyk, M.; Smil, D.; Scholten, C.; Bouché, L.; Stellfeld, T.; et al. Rational Design and Synthesis of Selective PRMT4 Inhibitors: A New Chemotype for Development of Cancer Therapeutics. ChemMedChem 2021, 16, 1116–1125. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, Y.; Liu, J.; Jiang, Q.; Yang, S.; Guo, L.; He, G. Design, Synthesis, and Biological Evaluation of Polo-like Kinase 1/Eukaryotic Elongation Factor 2 Kinase (PLK1/EEF2K) Dual Inhibitors for Regulating Breast Cancer Cells Apoptosis and Autophagy. Eur. J. Med. Chem. 2018, 144, 517–528. [Google Scholar] [CrossRef]
- Castro, A.C.; Chan, K.; Evans, C.A.; Janardanannair, S.; Lescarbeau, A.; Li, L.; Liu, T.; Liu, Y.; Ren, P.; Snyder, D.A.; et al. Heterocyclic Compounds and Uses Thereof. U.S. Patent US20130267521A1, 10 October 2013. [Google Scholar]
- Castro, A.C.; Evans, C.A.; Janardanannair, S.; Lescarbeau, A.; Liu, T.; Snyder, D.A.; Tremblay, M.R.; Ren, P.; Liu, Y.; Li, L.; et al. Heterocyclic Compounds and Uses Thereof. U.S. Patent US20130053362A1, 28 February 2013. [Google Scholar]
- Qu, B.; Xu, Y.; Lu, Y.; Zhuang, W.; Jin, X.; Shi, Q.; Yan, S.; Guo, Y.; Shen, Z.; Che, J.; et al. Design, Synthesis and Biological Evaluation of Sulfonamides Inhibitors of XPO1 Displaying Activity against Multiple Myeloma Cells. Eur. J. Med. Chem. 2022, 235, 114257. [Google Scholar] [CrossRef] [PubMed]
- Boys, M.L.; Schretzman, L.A.; Tollefson, M.B.; Chandrakumar, N.S.; Khanna, I.K.; Nguyen, M.; Downs, V.; Mohler, S.B.; Gesicki, G.J.; Penning, T.D.; et al. Heteroarylalkanoic Acids as Integrin Receptor Antagonists. WO 2004/058254 2004, 15 July 2004. [Google Scholar]
- Oguro, Y.; Kurasawa, O. Thienopyrimidine as CDC7 Kinase Inhibitors. WO 2010/101302, 10 September 2010. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Tools Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Tools Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- PrestoBlue™ Cell Viability Reagent. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0018370-PrestoBlueCellViabilityReagent-PI.pdf (accessed on 18 February 2022).
- Nussler, A.K.; Glanemann, M.; Schirmeier, A.; Liu, L.; Nüssler, N.C. Fluorometric Measurement of Nitrite/Nitrate by 2,3-Diaminonaphthalene. Nat. Protoc. 2006, 1, 2223–2226. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Series I | Series II | ||||||||
|---|---|---|---|---|---|---|---|---|---|
 |  | ||||||||
| Cmpd. | IC50 [nM] a or % inh. at 10 µM b | R | Cmpd. | IC50 [nM] a or % inh. at 10 µM b | |||||
| GSK-3β | IKK-β | ROCK-1 | GSK-3β | IKK-β | ROCK-1 | ||||
| 38 | 1130 ± 43 | 6524 ± 281 | 50% ± 0 | 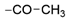 | 47 | 34% ± 9 | 17% ± 8 | nd | |
| 39 | 12 ± 0 | 33% ± 6 | 32% ± 3 | 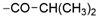 | 48 | 22 ± 0 | <10% | <10% | |
| 40 | 10 ± 0 | 4380 ± 544 | 1760 ± 109 | 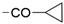 | 49 | 20 ± 0 | 16% ± 3 | 14% d ± 3 | |
| 41 | 32 ± 1 | 32% ± 4 | 48% ± 3 | 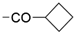 | 50 | 34 ± 0 | <10% | <10% d | |
| 42 | 830 ± 30 | 18% ± 3 | 30% ± 2 | 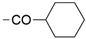 | 51 | 614 ± 26 | <10% | 11% d ± 0 | |
| 43 | 1314 ± 42 | 38% ± 5 | 3742 ± 217 | 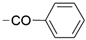 | 52 | 398 ± 12 | 30% ± 2 | 15% ± 7 | |
| 44 | 32% ± 1 | <10% | <10% |  | - | ||||
| 45 | 40% ± 9 | <10% | <10% | 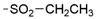 | - | ||||
| 46 | 39% ± 2 | 41% ± 2 | 1729 ± 78 | 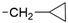 | - | ||||
| I | 161 ± 2 | 46% | 119 ± 6 | 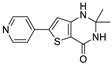 | |||||
| II | 5 ± 0 | 22% ± 7 | nd | 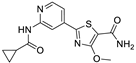 | |||||
| III | 47% ± 9 | 405 ± 9 | nd | 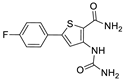 | |||||
| IV c | 80 ± 2 | 894 ± 42 | nd | Staurosporine | |||||
| Series III | Series IV | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
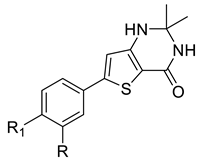 |  | |||||||||
| Cmpd. | IC50 [nM] a or % inh. at 10 µM b | R | R1 | Cmpd. | IC50 [nM] a or % inh. at 10 µM b | |||||
| GSK-3β | IKK-β | ROCK-1 | GSK-3β | IKK-β | ROCK-1 | |||||
| 53 | 47% ± 4 | <10% | <10% |  | - | 58 | 36% ± 8 | 422 ± 12 | 16% c ± 2 | |
| 54 | 453 ± 13 | 10% ± 7 | nd | 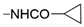 | F | 59 | 1847 ± 38 | 7621 ± 695 | <10% | |
| 55 | 11% ± 3 | 16% ± 0 | <10% | 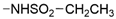 | - | - | ||||
| 56 | <10% | <10% | 33% ± 2 | 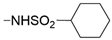 | - | - | ||||
| 57 | <10% | 17% ± 7 | <10% |  | - | - | ||||
| Series V | ||||
|---|---|---|---|---|
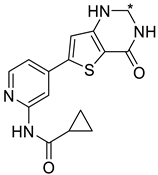 | ||||
| Cmpd. | * | IC50 [nM] a or % inh. at 10 µM b | ||
| GSK-3β | IKK-β | ROCK-1 | ||
| 40 |  | 10 ± 0 | 4380 ± 544 | 1760 ± 109 |
| 60 | 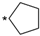 | 10 ± 0 | 7351 | 1361 ± 79 |
| 61 | 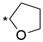 | 12 ± 0 | <10% | 40% ± 3 |
| 62 | 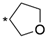 | 8 ± 0 | 39% ± 9 | 2310 ± 131 |
| Compound | Cytotoxicity in HT-22 Cells ± SEM µM) | Cytotoxicity in BV-2 Cells ( ± SEM µM) |
|---|---|---|
| 39 | 8.4 ± 5.3 | 4.5 ± 1.9 |
| 40 | 19.9 ± 4.1 | 3.6 ± 0.7 |
| 41 | >100 | 6.1 ± 3.1 |
| 48 | 36.6 ± 4.3 | 21.8 ± 3.8 |
| 49 | >100 | >100 |
| 50 | >100 | >100 |
| 58 | 67.1 ± 14.5 | 51.4 ± 0.8 |
| 60 | 9.7 ± 5.8 | 3.0 ± 0.0 |
| 62 | 58.3 ± 10.0 | 54.8 ± 1.9 |
| II | >100 | 15.4 ± 0.6 |
| Compd. | Solubility (µg/mL) a ± SD | MLM Stability (%) b ± SD |
|---|---|---|
| 62 | 31 ± 1 | 100 ± 1 |
| II | 5 ± 1 | 100 ± 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Góral, I.; Wichur, T.; Sługocka, E.; Godyń, J.; Szałaj, N.; Zaręba, P.; Głuch-Lutwin, M.; Mordyl, B.; Panek, D.; Więckowska, A. Connecting GSK-3β Inhibitory Activity with IKK-β or ROCK-1 Inhibition to Target Tau Aggregation and Neuroinflammation in Alzheimer’s Disease—Discovery, In Vitro and In Cellulo Activity of Thiazole-Based Inhibitors. Molecules 2024, 29, 2616. https://doi.org/10.3390/molecules29112616
Góral I, Wichur T, Sługocka E, Godyń J, Szałaj N, Zaręba P, Głuch-Lutwin M, Mordyl B, Panek D, Więckowska A. Connecting GSK-3β Inhibitory Activity with IKK-β or ROCK-1 Inhibition to Target Tau Aggregation and Neuroinflammation in Alzheimer’s Disease—Discovery, In Vitro and In Cellulo Activity of Thiazole-Based Inhibitors. Molecules. 2024; 29(11):2616. https://doi.org/10.3390/molecules29112616
Chicago/Turabian StyleGóral, Izabella, Tomasz Wichur, Emilia Sługocka, Justyna Godyń, Natalia Szałaj, Paula Zaręba, Monika Głuch-Lutwin, Barbara Mordyl, Dawid Panek, and Anna Więckowska. 2024. "Connecting GSK-3β Inhibitory Activity with IKK-β or ROCK-1 Inhibition to Target Tau Aggregation and Neuroinflammation in Alzheimer’s Disease—Discovery, In Vitro and In Cellulo Activity of Thiazole-Based Inhibitors" Molecules 29, no. 11: 2616. https://doi.org/10.3390/molecules29112616
APA StyleGóral, I., Wichur, T., Sługocka, E., Godyń, J., Szałaj, N., Zaręba, P., Głuch-Lutwin, M., Mordyl, B., Panek, D., & Więckowska, A. (2024). Connecting GSK-3β Inhibitory Activity with IKK-β or ROCK-1 Inhibition to Target Tau Aggregation and Neuroinflammation in Alzheimer’s Disease—Discovery, In Vitro and In Cellulo Activity of Thiazole-Based Inhibitors. Molecules, 29(11), 2616. https://doi.org/10.3390/molecules29112616