
Expedient Synthesis of Alkyl and Aryl Thioethers Using Xanthates as Thiol-Free Reagents


Abstract

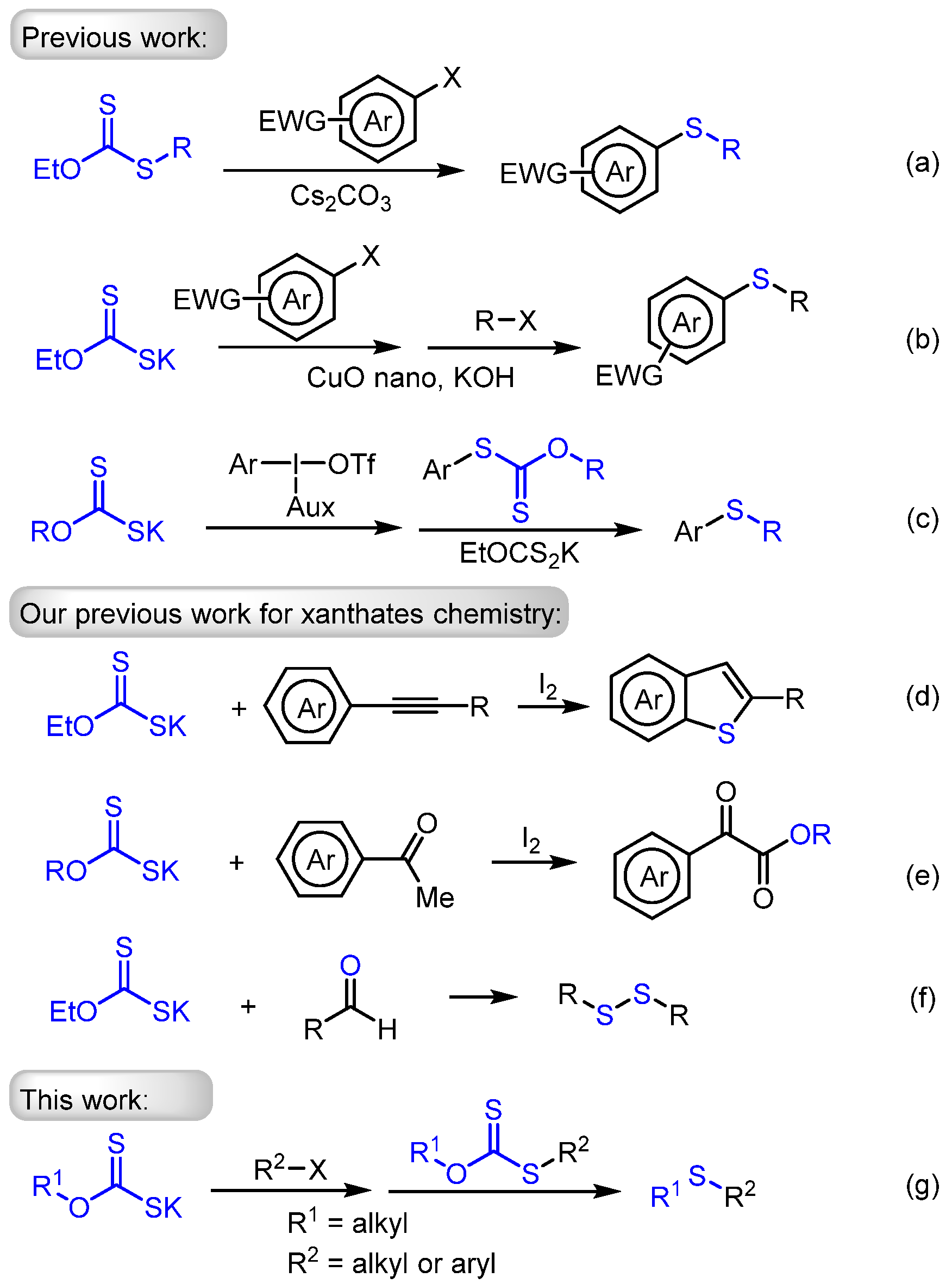
1. Introduction
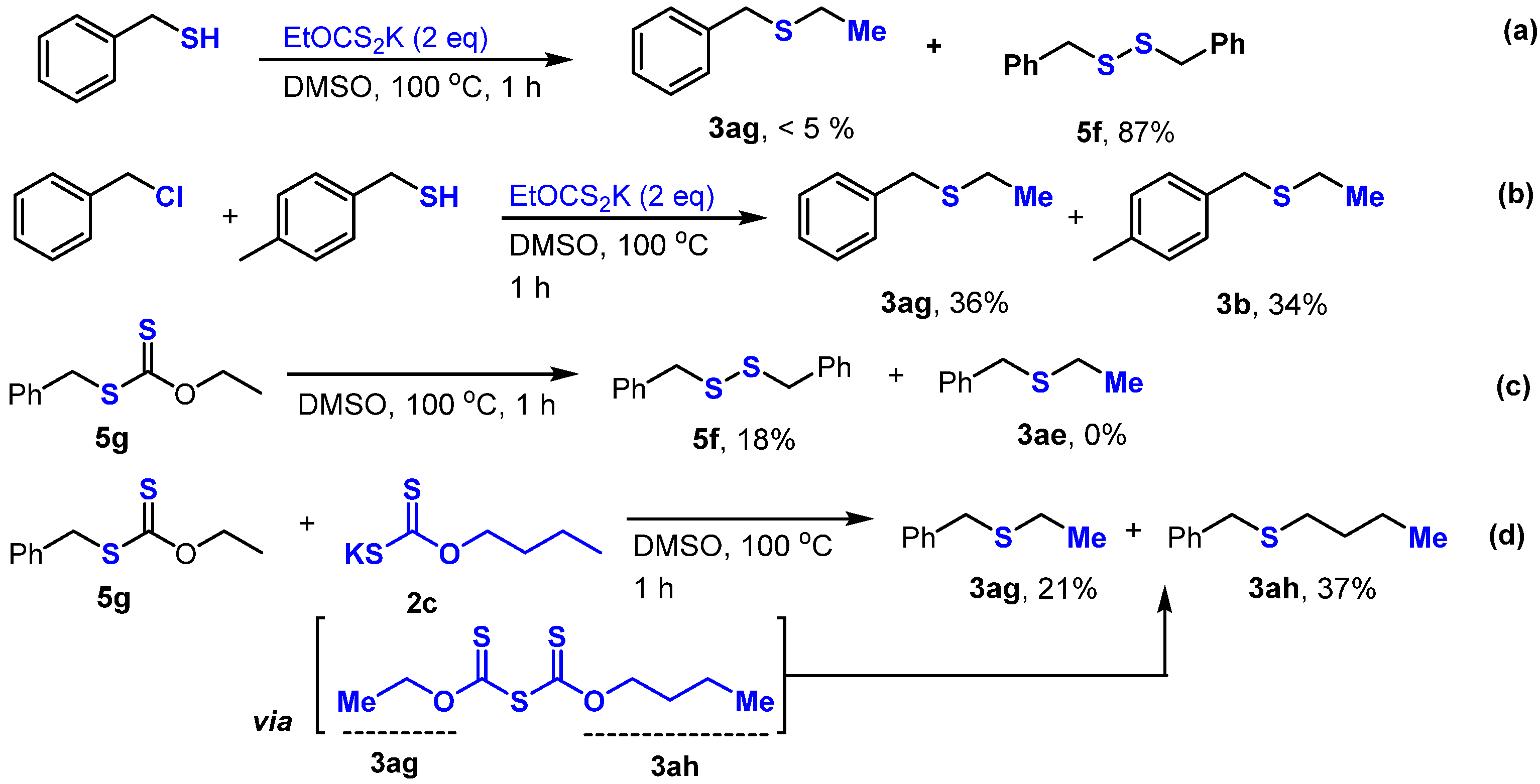
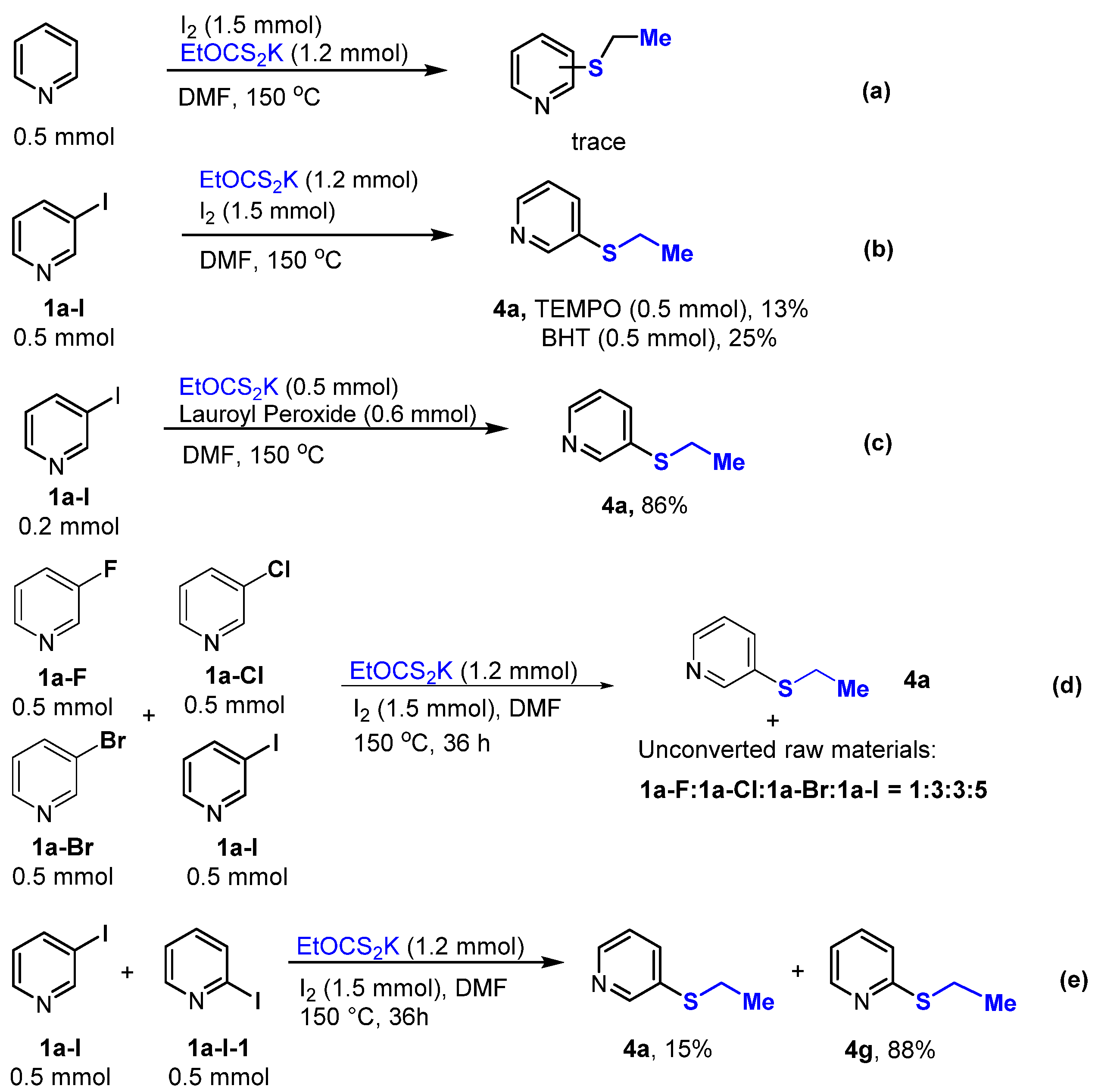
2. Results and Discussion
3. Materials and Methods
3.1. General Methods (Chemistry)
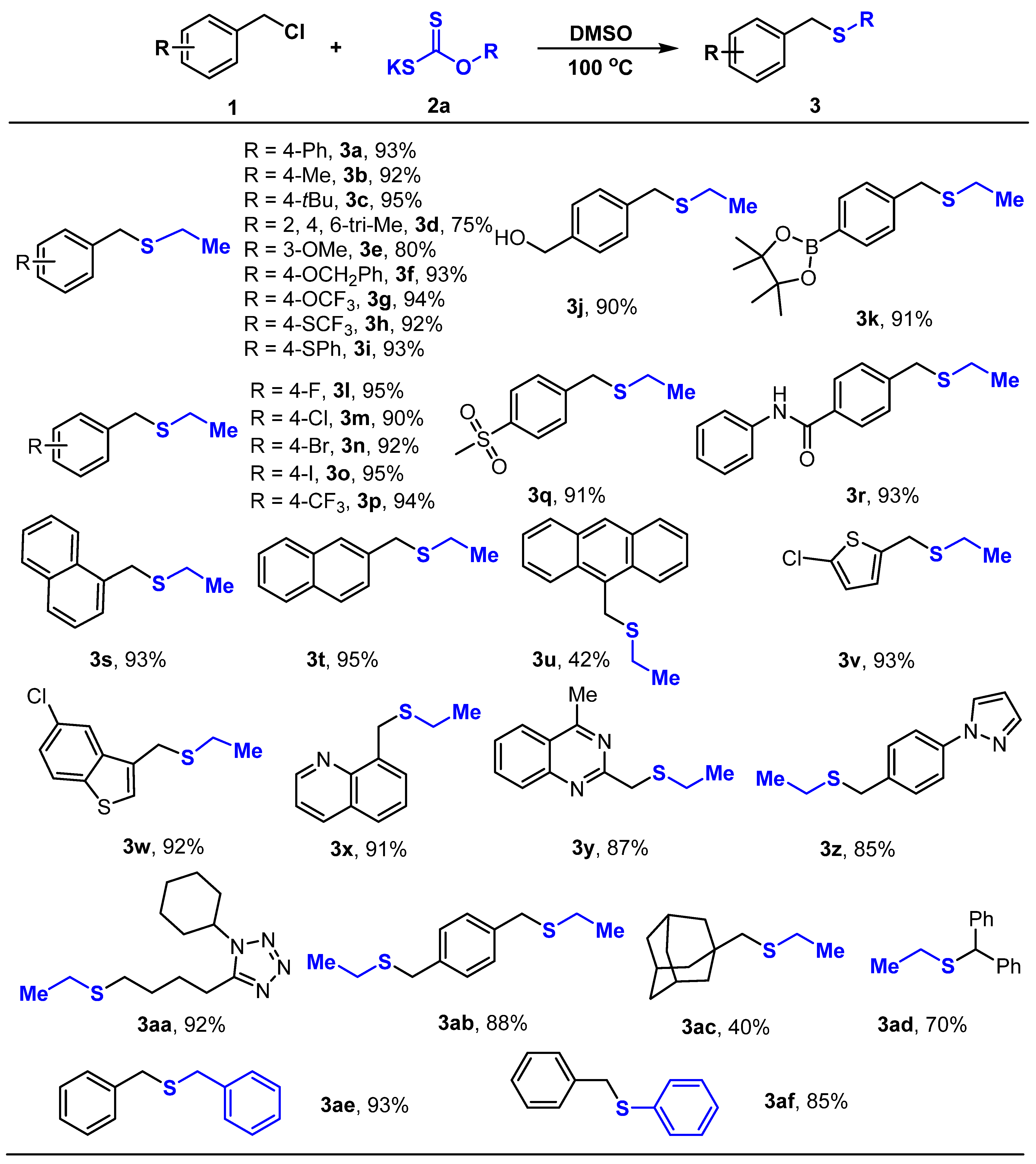
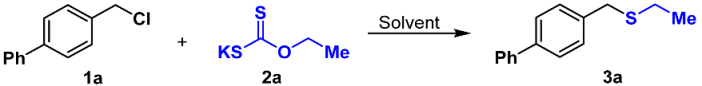
3.2. General Procedures for the Preparation of Compounds 3a–3af
- ([1,1′-biphenyl]-4-ylmethyl)(ethyl)sulfane (3a)
- Yellow liquid (106 mg, 93% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.63–7.55 (m, 4H), 7.49–7.39 (m, 4H), 7.39–7.33 (m, 1H), 3.79 (s, 2H), 2.51 (d, J = 7.4 Hz, 2H), 1.29 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 140.8, 139.8, 137.7, 129.2 (2C), 128.7 (2C), 127.2, 127.2 (2C), 127.0 (2C), 35.5, 25.2, 14.4; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C15H17S+, 229.1045; found, 229.1042.
- Ethyl(4-methylbenzyl)sulfane (3b)
- Yellow liquid (76 mg, 92% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.25–7.19 (m, 2H), 7.15–7.10 (m, 2H), 3.87 (s, 2H), 2.48 (q, J = 7.3 Hz, 2H), 2.34 (s, 3H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.1, 134.4, 129.2 (2C), 129.1 (2C), 43.5, 32.4, 21.1, 14.3; HRMS (ESI-TOF) (m/z): [M + K]+ calcd for C10H14KS+, 205.0448; found, 205.0445.
- (4-(tert-butyl)benzyl)(ethyl)sulfane (3c)
- Yellow liquid (99 mg, 95% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.37–7.32 (m, 2H), 7.28–7.23 (m, 2H), 3.71 (s, 2H), 2.47 (q, J = 7.4 Hz, 2H), 1.33 (s, 9H), 1.26 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.7, 135.5, 128.4 (2C), 125.3 (2C), 35.4, 34.4, 31.3, 25.3, 14.3; HRMS (ESI-TOF) (m/z): [M + K]+ calcd for C13H20KS+, 247.0917; found, 247.0914.
- Ethyl(2,4,6-trimethylbenzyl)sulfane (3d)
- Yellow liquid (73 mg, 75% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 6.85 (s, 2H), 3.78 (s, 2H), 2.61 (q, J = 7.4 Hz, 2H), 2.40 (s, 6H), 2.27 (s, 3H), 1.33 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 136.8 (2C), 136.3, 131.4, 129.0 (2C), 30.5, 26.8, 20.9, 19.6 (2C), 14.8; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C12H19S+, 195.1202; found, 195.1207.
- Ethyl(3-methoxybenzyl)sulfane (3e)
- Yellow liquid (73 mg, 80% yield); Rf = 0.4 (Hexane/EtOAc = 10:1); 1H NMR (400 MHz, CDCl3) δ 7.22 (t, J = 7.8 Hz, 1H), 6.94–6.85 (m, 2H), 6.82–6.76 (m, 1H), 3.81 (s, 3H), 3.70 (s, 2H), 2.45 (q, J = 7.4 Hz, 2H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.7, 140.2, 129.4, 121.2, 114.3, 112.4, 55.2, 35.9, 25.3, 14.3; HRMS (ESI-TOF) (m/z): [M + K]+ calcd for C10H14KOS+, 221.0397; found, 221.0395.
- (4-(benzyloxy)benzyl)(ethyl)sulfane (3f)
- Yellow solid (120 mg, 93% yield), MP: 61–62 °C; Rf = 0.4 (Hexane/EtOAc = 20:1); 1H NMR (400 MHz, CDCl3) δ 7.46–7.37 (m, 4H), 7.36–7.30 (m, 1H), 7.26–7.21 (m, 2H), 6.95–6.90 (m, 2H), 5.06 (s, 2H), 3.69 (s, 2H), 2.44 (q, J = 7.2 Hz, 2H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 157.7, 137.0, 130.9, 129.8 (2C), 128.6 (2C), 127.9, 127.4 (2C), 114.8 (2C), 79.7–74.5 (m), 70.0, 35.2, 25.1, 14.4; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C16H19OS+, 259.1151; found, 259.1149.
- Ethyl(4-(trifluoromethoxy)benzyl)sulfane (3g)
- Yellow liquid (111 mg, 94% yield); Rf = 0.5 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.3 Hz, 2H), 7.15 (d, J = 8.2 Hz, 2H), 3.71 (s, 2H), 2.44 (q, J = 7.4 Hz, 2H), 1.23 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 148.1 (q, J = 1.8 Hz), 137.4, 130.1 (2C), 121.0 (2C), 120.5 (q, J = 204.2 Hz), 35.1, 25.3, 14.3; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C10H11F3NaOS+, 259.0375; found, 259.0371.
- Ethyl(4-((trifluoromethyl)thio)benzyl)sulfane (3h)
- Yellow liquid (166 mg, 92% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.63–7.56 (m, 2H), 7.40–7.34 (m, 2H), 3.73 (s, 2H), 2.44 (q, J = 7.4 Hz, 2H), 1.23 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.0, 136.4 (2C), 129.9 (2C), 129.5 (q, J = 306.1 Hz), 122.6 (q, J = 2.3 Hz), 35.4, 25.4, 14.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C10H12F3S2+, 253.0327; found, 253.0320.
- Ethyl(4-(phenylthio)benzyl)sulfane (3i)
- Yellow liquid (121 mg, 93% yield); Rf = 0.5 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.37–7.21 (m, 9H), 3.70 (s, 2H), 2.45 (q, J = 7.4 Hz, 2H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.7, 135.8, 134.0, 131.2 (4C), 130.8 (4C), 129.6 (4C), 129.1 (4C), 126.9, 35.4, 25.3, 14.3; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C15H16NaS2+, 283.0586; found, 283.0593.
- (4-((ethylthio)methyl)phenyl)methanol (3j)
- Yellow liquid (82 mg, 90% yield); Rf = 0.5 (Hexane/EtOAc = 2:1); 1H NMR (400 MHz, CDCl3) δ 7.33–7.27 (m, 4H), 4.65 (s, 2H), 3.71 (s, 2H), 2.42 (q, J = 7.3 Hz, 2H), 1.91 (s, 1H), 1.22 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 139.5, 138.0, 128.9 (2C), 127.1 (2C), 65.0, 35.5, 25.2, 14.3; HRMS (ESI-TOF) (m/z): [M + K]+ calcd for C10H14KOS+, 221.0397; found, 221.0395.
- 2-(4-((ethylthio)methyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3k)
- Yellow liquid (127 mg, 91% yield); Rf = 0.5 (Hexane/EtOAc = 20:1); 1H NMR (400 MHz, CDCl3) δ 7.78–7.73 (m, 2H), 7.36–7.30 (m, 2H), 3.72 (s, 2H), 2.41 (q, J = 7.4 Hz, 2H), 1.34 (s, 12H), 1.21 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.9, 141.8, 134.9 (2C), 128.2 (2C), 83.7 (2C), 35.9, 25.1, 24.8 (4C), 14.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C15H24BO2S+, 278.1621; found, 278.1624.
- Ethyl(4-fluorobenzyl)sulfane (3l)
- Yellow liquid (81 mg, 95% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.31–7.25 (m, 2H), 7.03–6.95 (m, 2H), 3.69 (s, 2H), 2.43 (q, J = 7.3 Hz, 2H), 1.23 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 161.8 (d, J = 245.2 Hz), 134.3 (d, J = 3.0 Hz), 130.3 (d, J = 8.0 Hz, 2C), 115.3 (d, J = 21.4 Hz, 2C), 35.1, 25.2, 14.3; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C9H11FNaS+, 193.0458; found, 193.0466.
- (4-chlorobenzyl)(ethyl)sulfane (3m)
- Yellow liquid (84 mg, 90% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.30–7.26 (m, 2H), 7.24 (d, J = 8.7 Hz, 2H), 3.68 (s, 2H), 2.42 (q, J = 7.4 Hz, 2H), 1.22 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.1, 132.6, 130.1 (2C), 128.6 (2C), 35.2, 25.2, 14.3; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C9H11ClNaS+, 209.0162; found, 209.0152.
- (4-bromobenzyl)(ethyl)sulfane (3n)
- Yellow liquid (106 mg, 92% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.47–7.39 (m, 2H), 7.22–7.16 (m, 2H), 3.66 (s, 2H), 2.42 (q, J = 7.4 Hz, 2H), 1.22 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.7, 131.5 (2C), 130.5 (2C), 120.6, 35.2, 25.2, 14.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C9H12BrS+, 230.9838; found, 230.9828.
- Ethyl(4-Iodobenzyl)sulfane (3o)
- Yellow liquid (132 mg, 95% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.67–7.58 (m, 2H), 7.10–7.03 (m, 2H), 3.65 (s, 2H), 2.41 (q, J = 7.4 Hz, 2H), 1.22 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 138.3, 137.5 (2C), 130.8 (2C), 92.1, 35.3, 25.2, 14.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C9H12IS+, 278.9699; found, 278.9693.
- Ethyl(4-(trifluoromethyl)benzyl)sulfane (3p)
- Yellow liquid (103 mg, 94% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 3.75 (s, 2H), 2.43 (q, J = 7.4 Hz, 2H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.9, 129.1 (2C), 129.1 (q, J = 31.7 Hz), 125.4 (q, J = 3.8 Hz, 2C), 124.2 (q, J = 269.9 Hz), 35.4, 25.3, 14.2; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C10H11F3NaOS+, 259.0375; found, 259.0371.
- Ethyl(4-(methylsulfonyl)benzyl)sulfane (3q)
- Yellow liquid (105 mg, 91% yield); Rf = 0.4 (Hexane/EtOAc = 1:1); 1H NMR (400 MHz, CDCl3) δ 7.91–7.82 (m, 2H), 7.54–7.46 (m, 2H), 3.75 (s, 2H), 3.03 (s, 3H), 2.42 (q, J = 7.4 Hz, 2H), 1.21 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 145.3, 138.9, 129.6 (2C), 127.5 (2C), 44.4, 35.4, 25.4, 14.2; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C10H15O2S2+, 231.0508; found, 231.0503.
- 4-((ethylthio)methyl)-N-phenylbenzamide (3r)
- White solid (126 mg, 93% yield), MP: 118–120 °C; Rf = 0.4 (Hexane/EtOAc = 3:1); 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.83–7.76 (m, 2H), 7.66–7.61 (m, 2H), 7.40–7.30 (m, 4H), 7.16–7.10 (m, 1H), 3.74 (s, 2H), 2.50–2.36 (m, 2H), 1.23 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.6, 142.8, 137.9, 133.5, 129.1 (2C), 129.0 (2C), 127.3 (2C), 124.5, 120.3 (2C), 35.5, 25.3, 14.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C16H18NOS+, 272.1104; found, 272.1098.
- Ethyl(naphthalen-1-ylmethyl)sulfane (3s)
- Yellow liquid (94 mg, 93% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.3 Hz, 1H), 7.89–7.84 (m, 1H), 7.81–7.73 (m, 1H), 7.59–7.53 (m, 1H), 7.52–7.47 (m, 1H), 7.44–7.35 (m, 2H), 4.19 (s, 2H), 2.51 (q, J = 7.3 Hz, 2H), 1.28 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 134.1, 133.9, 131.4, 128.8, 128.0, 126.9, 126.1, 125.8, 125.1, 124.1, 33.7, 26.0, 14.4; HRMS (ESI-TOF) (m/z): [M]+ calcd for C13H14S+, 202.0811; found, 202.0816.
- Ethyl(naphthalen-2-ylmethyl)sulfane (3t)
- Yellow liquid (96 mg, 95% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.87–7.79 (m, 3H), 7.72 (s, 1H), 7.55–7.44 (m, 3H), 3.90 (s, 2H), 2.46 (q, J = 7.4 Hz, 2H), 1.26 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.2, 132.5, 128.3, 127.6, 127.6, 127.1, 126.1, 125.6, 36.1, 25.1, 14.3; HRMS (ESI-TOF) (m/z): [M]+ calcd for C13H14S+, 202.0811; found, 202.0818.
- (Anthracen-9-ylmethyl)(ethyl)sulfane (3u)
- Yellow solid (53 mg, 42% yield), MP: 68–70 °C; Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H), 8.35 (d, J = 8.9 Hz, 2H), 8.01 (d, J = 8.4 Hz, 2H), 7.61–7.53 (m, 2H), 7.52–7.44 (m, 2H), 4.75 (s, 2H), 2.70 (q, J = 7.4 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 131.5 (2C), 129.9 (2C), 129.5, 129.2 (2C), 127.2, 126.0 (2C), 125.0 (2C), 124.2 (2C), 28.7, 27.1, 14.8; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C17H17S+, 253.1045; found, 253.1045.
- 2-chloro-5-((ethylthio)methyl)thiophene (3v)
- Yellow liquid (89 mg, 93% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 6.73–6.66 (m, 2H), 3.82 (s, 2H), 2.51 (q, J = 7.4 Hz, 2H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.4, 128.8, 125.5, 125.0, 30.4, 25.4, 14.2; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C7H10ClS2+, 192.9907; found, 192.9902.
- 5-chloro-3-((ethylthio)methyl)benzo[b]thiophene (3w)
- Yellow liquid (111 mg, 92% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.88–7.84 (m, 1H), 7.74 (d, J = 8.6 Hz, 1H), 7.35–7.29 (m, 2H), 3.92 (s, 3H), 2.48 (q, J = 7.4 Hz, 2H), 1.26 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 139.1, 138.7, 131.9, 130.4, 125.4, 124.9, 123.8, 121.9, 28.9, 25.7, 14.2; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C11H11ClNaS2+, 264.9883; found, 264.9884.
- 8-((ethylthio)methyl)quinoline (3x)
- Yellow liquid (92 mg, 91% yield); Rf = 0.4 (Hexane/EtOAc = 10:1); 1H NMR (400 MHz, CDCl3) δ 8.97–8.93 (m, 1H), 8.16–8.09 (m, 1H), 7.71 (d, J = 7.7 Hz, 2H), 7.52–7.45 (m, 1H), 7.42–7.37 (m, 1H), 4.44 (s, 2H), 2.56 (q, J = 7.4 Hz, 2H), 1.28 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 149.6, 146.3, 137.3, 136.3, 129.3, 128.5, 127.0, 126.1, 121.1, 31.1, 26.1, 14.5; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C12H14NS+, 204.0842; found, 204.0843.
- 2-((ethylthio)methyl)-4-methylquinazoline (3y)
- Yellow solid (95 mg, 87% yield), MP: 52–54 °C; Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 8.03–7.98 (m, 1H), 7.95–7.89 (m, 1H), 7.82–7.76 (m, 1H), 7.56–7.50 (m, 1H), 4.01 (s, 2H), 2.89 (s, 3H), 2.62 (q, J = 7.4 Hz, 2H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 163.6, 149.6, 133.5, 128.5, 126.9, 124.8, 122.5, 39.1, 25.9, 21.7, 14.4; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C12H15N2S+, 219.0950; found, 219.0942.
- 1-(4-((ethylthio)methyl)phenyl)-1H-pyrazole (3z)
- Yellow liquid (93 mg, 85% yield); Rf = 0.5 (Hexane/EtOAc = 10:1); 1H NMR (400 MHz, CDCl3) δ 7.95–7.88 (m, 1H), 7.74–7.70 (m, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.45–7.37 (m, 2H), 3.75 (s, 2H), 2.45 (q, J = 7.3, 6.8 Hz, 2H), 1.24 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.0, 139.0, 136.9, 129.8 (2C), 126.7, 119.3 (2C), 107.6, 35.3, 25.2, 14.4; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C12H15N2S+, 219.0950; found, 219.0946.
- 1-cyclohexyl-5-(4-(ethylthio)butyl)-1H-tetrazole (3aa)
- Yellow liquid (123 mg, 92% yield); Rf = 0.4 (Hexane/EtOAc = 2:1); 1H NMR (400 MHz, CDCl3) δ 4.21–4.04 (m, 1H), 2.85 (t, 2H), 2.61–2.48 (m, 3H), 2.09–1.87 (m, 8H), 1.82–1.66 (m, 4H), 1.48–1.30 (m, 3H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 153.5, 57.6, 32.9 (2C), 30.9, 28.6, 26.2, 25.9, 25.3 (2C), 24.8, 22.9, 14.8; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H25N4S+, 269.1794; found, 269.1790.
- 1,4-bis((ethylthio)methyl)benzene (3ab)
- Yellow liquid (99 mg, 88% yield); Rf = 0.5 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.25 (s, 4H), 3.70 (s, 4H), 2.43 (q, J = 7.4 Hz, 4H), 1.23 (t, J = 7.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 137.2 (2C), 128.9 (4C), 35.5, 25.2, 14.4; HRMS (ESI-TOF) (m/z): [M]+ calcd for C12H18S2+, 226.0845; found, 226.0836.
- (((1s,3s)-adamantan-1-yl)methyl)(ethyl)sulfane (3ac)
- Yellow liquid (42 mg, 40% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 2.51 (q, J = 7.4 Hz, 2H), 2.32 (s, 2H), 1.97 (s, 3H), 1.73–1.66 (m, 3H), 1.65–1.59 (m, 3H), 1.58–1.54 (m, 6H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 47.3, 41.9 (3C), 36.9 (3C), 33.9, 28.6 (3C), 28.2, 15.0; HRMS (ESI-TOF) (m/z): [M + K]+ calcd for C13H22KS+, 249.1074; found, 249.1075.
- Benzhydryl(ethyl)sulfane (3ad)
- Yellow liquid (80 mg, 70% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.50–7.44 (m, 4H), 7.38–7.31 (m, 4H), 7.30–7.21 (m, 2H), 5.22 (s, 1H), 2.44 (q, J = 7.4 Hz, 2H), 1.25 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.5 (2C), 128.5 (4C), 128.2 (4C), 127.0 (2C), 53.7, 26.2, 14.2; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C15H17S+, 229.1045; found, 229.1046.
- Dibenzylsulfane (3ae) [52]
- White solid (100 mg, 93% yield), MP: 61–62 °C; Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.36–7.22 (m, 10H), 3.61 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 138.1 (2C), 129.0 (4C), 128.5 (4C), 127.0 (2C), 35.6 (2C).
- benzyl(phenyl)sulfane (3af) [53]
- White solid (90 mg, 90% yield), MP: 39–40 °C; Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.34–7.22 (m, 9H), 7.21–7.15 (m, 1H), 4.13 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 137.4, 136.3, 129.8 (2C), 128.8 (2C), 128.8 (2C), 128.5 (2C), 127.2, 126.3, 39.0.
- Benzyl(ethyl)sulfane (3ag)
- Yellow liquid (27 mg, 36% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (500 MHz, CDCl3) δ 7.35–7.28 (m, 4H), 7.26–7.21 (m, 1H), 3.73 (s, 2H), 2.44 (q, J = 7.4 Hz, 2H), 1.23 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 138.6, 128.8 (2C), 128.4 (2C), 126.8, 35.8, 25.2, 14.3; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C9H13S+, 153.0732; found, 153.0735.
- Benzyl(butyl)sulfane (3ah)
- Yellow liquid (33 mg, 37% yield); Rf = 0.6 (Hexane/EtOAc = 50:1); 1H NMR (400 MHz, CDCl3) δ 7.35–7.25 (m, 4H), 7.28–7.19 (m, 1H), 3.71 (s, 2H), 2.49–2.35 (m, 2H), 1.60–1.49 (m, 2H), 1.38 (dt, J = 8.1, 7.0 Hz, 2H), 0.89 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) 138.7, 128.8 (2C), 128.4 (2C), 126.8, 36.2, 31.3, 31.0, 22.0, 13.7; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C11H17S+, 181.1045; found, 181.1050.
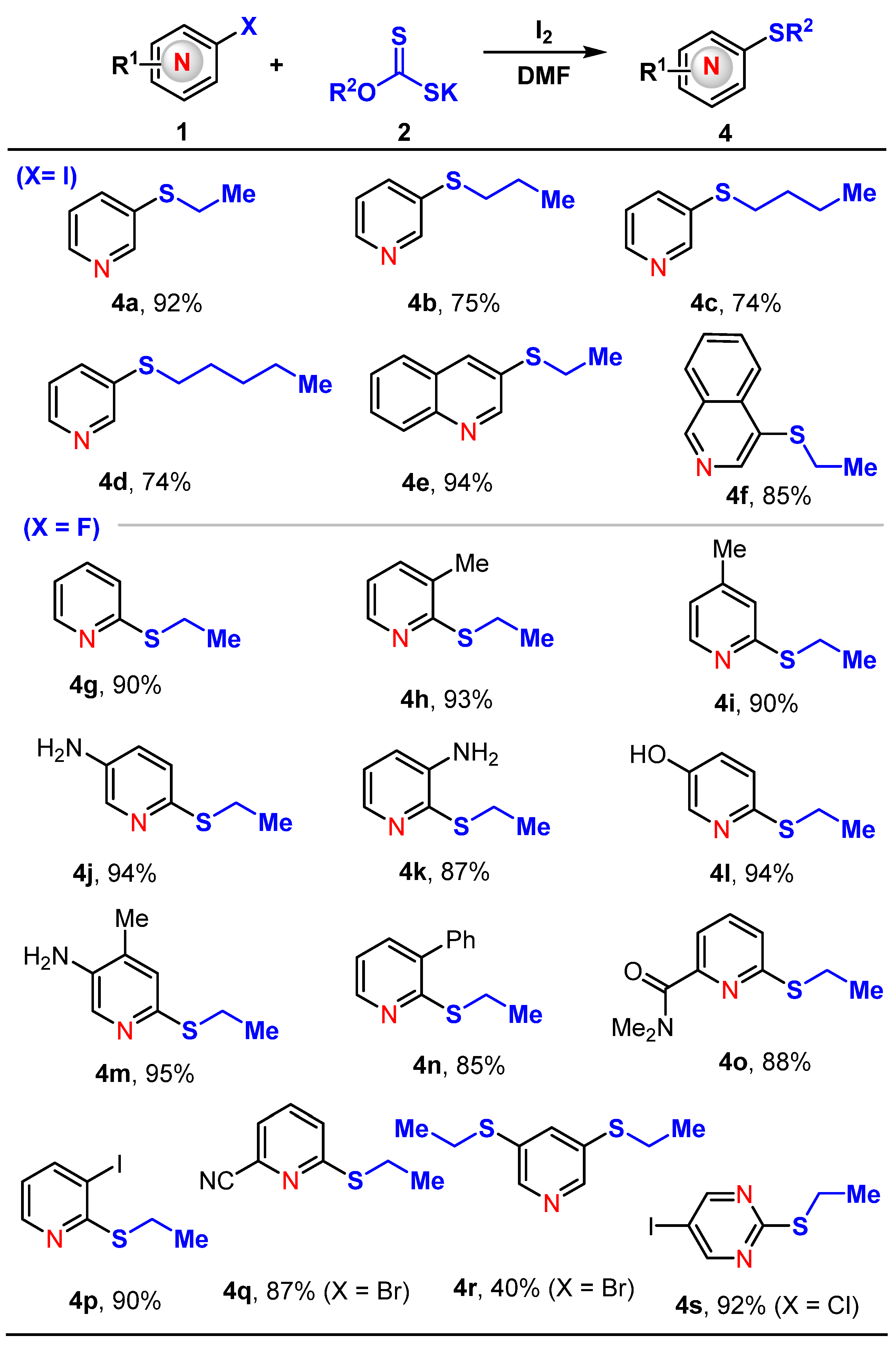
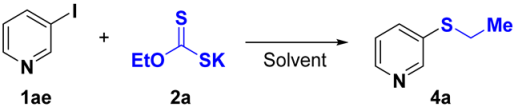
3.3. General Procedures for the Preparation of Compounds 4a–4s
- 3-(Ethylthio)pyridine (4a) [54]
- Yellow liquid (64 mg, 92% yield); Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.56 (s, 1H), 8.41 (d, J = 4.8 Hz, 1H), 7.66 (ddd, J = 8.0, 2.4, 1.5 Hz, 1H), 7.23 (dd, J = 8.0, 4.8 Hz, 1H), 2.96 (q, J = 7.4 Hz, 2H), 1.32 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 149.6, 146.5, 137.1, 134.0, 123.7, 27.7, 14.3.
- 3-(Propylthio)pyridine (4b) [55]
- Yellow liquid (58 mg, 75%); Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.56 (d, J = 1.8 Hz, 1H), 8.48–8.35 (m, 1H), 7.69–7.60 (m, 1H), 7.21 (dd, J = 7.9, 4.8 Hz, 1H), 2.96–2.74 (m, 2H), 1.67 (h, J = 7.3 Hz, 2H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 149.9, 146.7, 136.8, 134.1, 123.6, 35.7, 22.4, 13.3.
- 3-(Butylthio)pyridine (4c) [56]
- Yellow liquid (62 mg, 74%); Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 8.41 (d, J = 4.2 Hz, 1H), 7.66 (dt, J = 8.0, 1.8 Hz, 1H), 7.24 (dd, J = 7.9, 4.8 Hz, 1H), 2.97–2.90 (m, 2H), 1.63 (p, J = 7.4 Hz, 2H), 1.45 (dq, J = 14.6, 7.3 Hz, 2H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 149.2, 146.2, 137.0, 134.6, 123.7, 33.3, 31.1, 21.8, 13.6.
- 3-(Pentylthio)pyridine (4d)
- Yellow liquid (67 mg, 74%); Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 8.41 (s, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.24–7.21 (m, 1H), 2.92 (t, J = 7.4 Hz, 2H), 1.64 (p, J = 7.4 Hz, 2H), 1.40 (dt, J = 14.3, 6.9 Hz, 2H), 1.32 (dq, J = 14.3, 6.9 Hz, 2H), 0.89 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 149.4, 146.4, 136.9, 134.4, 123.6, 33.6, 30.8, 28.7, 22.2, 13.9. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C10H16NS+, 182.0998; found, 182.0995.
- 3-(Ethylthio)quinoline (4e) [57]
- Yellow liquid (89 mg, 94% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.84 (d, J = 2.3 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.07 (d, J = 2.3 Hz, 1H), 7.75 (dd, J = 8.2, 1.4 Hz, 1H), 7.68 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.56 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 3.06 (q, J = 7.4 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.6, 146.2, 134.8, 130.6, 129.3, 129.0, 128.2, 127.2, 126.9, 27.9, 14.3.
- 4-(Ethylthio)isoquinoline (4f) [57]
- Yellow liquid (80 mg, 85% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 9.12 (s, 1H), 8.55 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.1 Hz, 1H), 7.77 (ddd, J = 8.3, 6.8, 1.3 Hz, 1H), 7.64 (ddd, J = 8.1, 6.8, 1.1 Hz, 1H), 3.01 (q, J = 7.3 Hz, 2H), 1.32 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.0, 143.7, 135.9, 130.9, 128.7, 128.4, 128.2, 127.67, 124.1, 28.3, 14.5.
- 2-(Ethylthio)pyridine (4g) [58]
- Yellow liquid (63 mg, 90% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.41 (ddd, J = 5.0, 1.9, 1.0 Hz, 1H), 7.50–7.41 (m, 1H), 7.15 (dt, J = 8.1, 1.1 Hz, 1H), 6.95 (ddd, J = 7.3, 4.9, 1.1 Hz, 1H), 3.16 (q, J = 7.4 Hz, 2H), 1.36 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 159.3, 149.3, 135.8, 122.1, 119.2, 24.4, 14.5.
- 2-(Ethylthio)-3-methylpyridine (4h)
- Yellow liquid (71 mg, 93% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.29 (dd, J = 4.9, 1.7 Hz, 1H), 7.30 (ddd, J = 7.4, 1.8, 0.9 Hz, 1H), 6.90 (dd, J = 7.4, 4.9 Hz, 1H), 3.22 (q, J = 7.4 Hz, 2H), 2.24 (s, 3H), 1.38 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 158.2, 146.4, 136.2, 130.9, 118.8, 24.0, 18.6, 14.6. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C8H12NS+, 154.0685; found, 154.0685.
- 4-2-(Ethylthio)-4-methylpyridine (4i)
- Yellow liquid (69 mg, 90% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.28 (d, J = 5.1 Hz, 1H), 6.99 (s, 1H), 6.79 (dd, J = 5.2, 1.5 Hz, 1H), 3.15 (q, J = 7.4 Hz, 2H), 2.26 (s, 3H), 1.36 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 158.9, 148.9, 147.2, 122.7, 120.7, 24.4, 20.8, 14.6. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C8H12NS+, 154.0685; found, 154.0685.
- 6-(Ethylthio)pyridin-3-amine (4j) [59]
- Brown liquid (72 mg, 94% yield); Rf = 0.4 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.02 (d, J = 2.9 Hz, 1H), 7.04 (dd, J = 8.4, 0.7 Hz, 1H), 6.91 (dd, J = 8.4, 2.9 Hz, 1H), 3.51 (s, 2H), 3.07 (q, J = 7.3 Hz, 2H), 1.31 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 139.9, 136.9, 123.9, 123.4, 25.9, 14.7.
- 2-(Ethylthio)pyridin-3-amine (4k) [60]
- Brown liquid (67 mg, 87% yield); Rf = 0.4 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 7.97 (dd, J = 4.2, 2.0 Hz, 1H), 6.93–6.83 (m, 2H), 3.23 (q, J = 7.4 Hz, 2H), 1.35 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 143.0, 140.9, 139.4, 120.6, 120.4, 25.2, 14.9.
- 6-(Ethylthio)pyridin-3-ol (4l)
- Pale-yellow solid (73 mg, 94% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.12 (dd, J = 2.8, 0.8 Hz, 1H), 7.22 (dd, J = 8.6, 2.8 Hz, 1H), 7.18 (dd, J = 8.7, 0.7 Hz, 1H), 3.02 (q, J = 7.3 Hz, 2H), 1.29 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 152.0, 148.1, 137.0, 125.8, 125.1, 26.9, 14.5. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C7H10NOS+, 156.0478; found, 156.0476.
- 6-(Ethylthio)-4-methylpyridin-3-amine (4m)
- Red liquid (80 mg, 95% yield); Rf = 0.5 (Hexane/EtOAc = 2:1); 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 1H), 6.95 (t, J = 0.7 Hz, 1H), 3.07 (q, J = 7.4 Hz, 2H), 2.13 (d, J = 0.8 Hz, 3H), 1.31 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 138.8, 136.5, 132.2, 124.9, 25.8, 16.8, 14.8. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C8H13N2S+, 169.0794; found, 169.0792.
- 2-(Ethylthio)-3-phenylpyridine (4n)
- Yellow liquid (91 mg, 85% yield); Rf = 0.5 (Hexane/EtOAc = 10:1); 1H NMR (500 MHz, CDCl3) δ 8.46 (dd, J = 4.9, 1.7 Hz, 1H), 7.50–7.42 (m, 5H), 7.41 (dd, J = 7.4, 1.8 Hz, 1H), 7.06 (dd, J = 7.5, 4.9 Hz, 1H), 3.19 (q, J = 7.3 Hz, 2H), 1.35 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 157.5, 147.9, 138.2, 136.3, 136.0, 129.1, 128.3, 128.0, 118.8, 24.6, 14.3. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C13H14NS+, 216.0841; found, 216.0839.
- 6-(Ethylthio)-N,N-dimethylpicolinamide (4o)
- Yellow liquid (92 mg, 88% yield); Rf = 0.4 (Hexane/EtOAc = 2:1); 1H NMR (500 MHz, CDCl3) δ 7.54 (t, J = 7.8 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 7.16 (d, J = 8.1 Hz, 1H), 3.15 (q, J = 7.4 Hz, 2H), 3.10 (d, J = 17.2 Hz, 6H), 1.34 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.5, 158.1, 154.0, 136.6, 122.7, 119.1, 39.0, 35.8, 24.2, 14.6. HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C10H14N2NaOS+, 233.0719; found, 233.0715.
- 2-(Ethylthio)-3-iodopyridine (4p)
- Brown liquid (119 mg, 90% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.39 (dd, J = 4.7, 1.6 Hz, 1H), 7.90 (dd, J = 7.7, 1.6 Hz, 1H), 6.70 (dd, J = 7.7, 4.7 Hz, 1H), 3.13 (q, J = 7.4 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 161.8, 148.1, 145.7, 119.9, 93.7, 26.9, 14.0. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C7H9INS+, 265.9500; found, 265.9504.
- 6-(Ethylthio)picolinonitrile (4q)
- Brown solid (71 mg, 87% yield); MP: 50–52 °C, Rf = 0.4 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.64 (d, J = 2.5 Hz), 7.63 (dd, J = 8.4, 2.2 Hz), 7.21 (dd, J = 8.4, 0.9 Hz), 3.19 (q, J = 7.4 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.6, 152.1, 137.6, 121.7, 117.1, 104.3, 24.5, 14.2. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C8H9N2S+, 165.0481; found, 165.0481.
- 3,5-Bis(ethylthio)pyridine (4r)
- Brown liquid (40 mg, 40% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.32 (s, 2H), 7.55 (s, 1H), 2.95 (q, J = 7.4 Hz, 4H), 1.31 (t, J = 7.4 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 146.5 (2C), 136.6 (2C), 134.0, 27.6 (2C), 14.2 (2C). HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C9H14NS2+, 200.0562; found, 200.0559.
- 2-(Ethylthio)-5-iodopyrimidine (4s)
- Brown solid (122 mg, 92% yield); MP: 64–65 °C, Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.64 (s, 2H), 3.09 (q, J = 7.4 Hz, 2H), 1.36 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.1, 162.2 (2C), 86.2, 25.4, 14.2. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C6H8IN2S+, 266.9447; found, 266.9450.
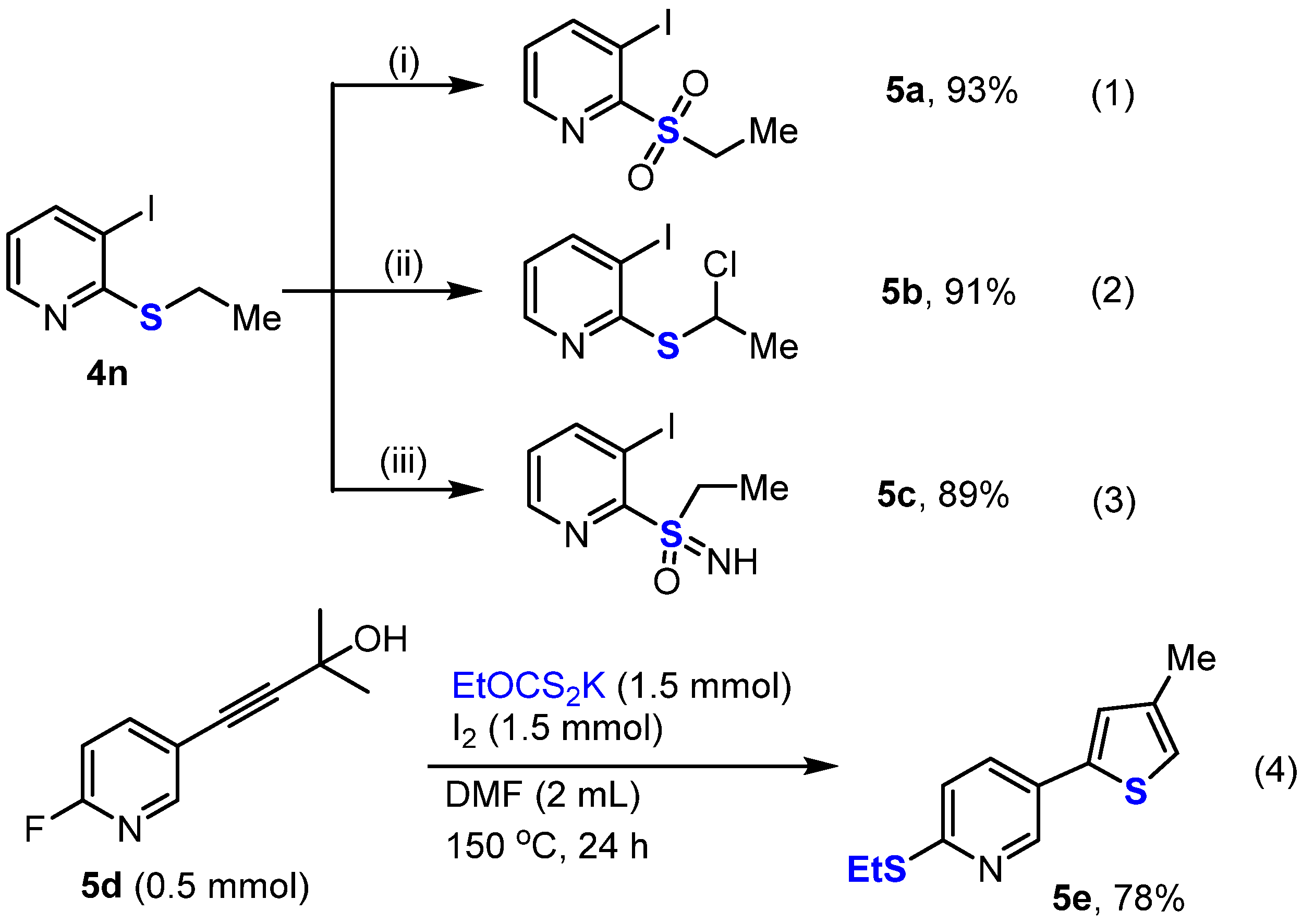
- 4-(Ethylsulfonyl)aniline (5a)
- Yellow liquid (138 mg, 93% yield); Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.58 (dd, J = 4.6, 1.5 Hz, 1H), 8.40 (dd, J = 8.0, 1.5 Hz, 1H), 7.20 (dd, J = 8.0, 4.5 Hz, 1H), 3.67 (q, J = 7.4 Hz, 2H), 1.46 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 157.3, 150.9, 147.2, 127.3, 86.1, 46.0, 7.2. HRMS (ESI-TOF) (m/z): [M]+ calcd for C7H8INO2S+, 296.9553; found, 296.9551.
- 2-((1-Chloroethyl)thio)-3-iodopyridine (5b)
- Yellow liquid (136 mg, 91% yield); Rf = 0.5 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.50 (dt, J = 4.7, 1.2 Hz, 1H), 7.98 (dt, J = 7.7, 1.2 Hz, 1H), 6.82 (ddd, J = 7.8, 4.7, 0.8 Hz, 1H), 6.19 (q, J = 6.9 Hz, 1H), 2.01 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 158.8, 148.4, 146.2, 121.1, 93.2, 61.9, 25.7. HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C7H7ClINNaS+, 321.8925; found, 321.8917.
- Ethyl(imino)(3-iodopyridin-2-yl)-λ6-sulfanone (5c)
- Yellow liquid (132 mg, 89% yield); Rf = 0.5 (Hexane/EtOAc = 3:1); 1H NMR (500 MHz, CDCl3) δ 8.51 (dd, J = 4.6, 1.5 Hz, 1H), 8.32 (dd, J = 7.9, 1.5 Hz, 1H), 7.11 (dd, J = 7.9, 4.6 Hz, 1H), 3.71 (ddt, J = 70.8, 14.2, 7.2 Hz, 2H), 1.43 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 159.3, 150.4, 147.1, 126.4, 85.1, 46.5, 7.7. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C7H10IN2OS+, 296.9553; found, 296.9551.
- 2-(Ethylthio)-5-(4-methylthiophen-2-yl)pyridine (5e)
- Yellow liquid (92 mg, 78% yield); Rf = 0.4 (Hexane/EtOAc = 5:1); 1H NMR (500 MHz, CDCl3) δ 8.88 (s, 1H), 8.58 (d, J = 4.5 Hz, 1H), 8.06 (dt, J = 7.9, 2.0 Hz, 1H), 7.39 (dd, J = 7.9, 4.9 Hz, 1H), 7.08 (d, J = 1.2 Hz, 1H), 2.52 (q, J = 7.4 Hz, 3H), 2.36 (d, J = 1.0 Hz, 2H), 1.01 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 149.3, 147.9, 142.2, 141.9, 137.4, 131.1, 128.9, 123.3, 120.7, 29.9, 16.0, 14.5. HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C12H14NS2+, 236.0557; found, 236.0562.
- 1,2-dibenzyldisulfane (5f) [61]
- White solid (107 mg, 87% yield), MP: 71–72 °C; Rf = 0.5 (Hexane/EtOAc = 20:1); 1H NMR (400 MHz, CDCl3) δ 7.36–7.29 (m, 5H), 7.29–7.23 (m, 5H), 3.62 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 137.4 (2C), 129.4 (4C), 128.5 (4C), 127.4 (2C), 43.4 (2C).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dvoral, C.A.; Schmitz, W.D.; Poon, D.J.; Pryde, D.C.; Lawson, J.P.; Amos, R.A.; Meyers, A.I. The Synthesis of Streptogramin Antibiotics: (−)-Griseoviridin and Its C-8 Epimer. Angew. Chem. Int. Ed. 2000, 39, 1664–1666. [Google Scholar] [CrossRef]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F.; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Rogers, J.D.; Holland, S.D.; Larson, P.; Amin, R.D.; Haesen, R.; Freeman, A.; Seiberling, M.; Merz, M.; Cheng, H. Pharmacokinetics and Bioavailability of Montelukast Sodium (MK-0476) in Healthy Young and Elderly Volunteers. Pharm. Res. 1996, 13, 445–448. [Google Scholar] [CrossRef]
- Kahan, F.M.; Kropp, H.; Sumdelof, J.G.; Bormbaum, J. Thienamycin: Development of Imipenem-Cilastatin. J. Antimicrob. Chemother. 1983, 12, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Mitsudo, T. Metal-Catalyzed Carbon−Sulfur Bond Formation. Chem. Rev. 2000, 100, 3205–3220. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ding, Y. Recent Advances in the Synthesis of Thioether. Mini-Rev. Org. Chem. 2017, 14, 407–431. [Google Scholar] [CrossRef]
- Xiao, F.; Chen, S.; Li, C.; Huang, H.; Deng, G. Copper-Catalyzed Three-Component One-Pot Synthesis of Aryl Sulfides with Sulfur Powder under Aqueous Conditions. Adv. Syn. Catal. 2016, 358, 3881–3883. [Google Scholar] [CrossRef]
- Huang, H.; Qu, Z.; Ji, X.; Deng, G.J. Three-component Bs-heterocycliation for Synthesis of 2-Aminobenzo[4,5]thieno[3,2-d]thiazoles. Org. Chem. Front. 2019, 6, 1146–1150. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Yang, S.; An, Y.; Wu, W.; Jiang, H. Assembly of 3-Sulfenylbenzofurans and 3-Sulfenylindoles by Palladium-Catalyzed Cascade Annulation/Arylthiolation Reaction. J. Org. Chem. 2016, 81, 2875–2887. [Google Scholar] [CrossRef]
- Nguyen, T.B. Recent Advances in Organic Reactions Involving Elemental Sulfur. Adv. Syn. Catal. 2017, 359, 1066–1130. [Google Scholar] [CrossRef]
- Qiao, Z.; Wei, J.; Jiang, X. Direct Cross-Coupling Access to Diverse Aromatic Sulfide: Palladium-Catalyzed Double C–S Bond Construction Using Na2S2O3 as a Sulfurating Reagent. Org. Lett. 2014, 16, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.T.; Camara, K.; Han, Z.S.; Xu, Y.; Lee, H.; Busacca, C.A.; Senanayake, C.H. The Reaction of Grignard Reagents with Bunte Salts: A Thiol-Free Synthesis of Sulfides. Org. Lett. 2014, 16, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Jiang, X. Controllable Sulfoxidation and Sulfenylation with Organic Thiosulfate Salts via Dual Electron- and Energy-Transfer Photocatalysis. ACS Catal. 2017, 7, 7587–7592. [Google Scholar] [CrossRef]
- Luo, F.; Pan, C.; Li, L.; Chen, F.; Cheng, J. Copper-Mediated Methylthiolation of Aryl Halides with DMSO. Chem. Commun. 2011, 47, 5304–5306. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Luo, S.H.; Wu, H.Q.; Chen, L.Q.; Jiang, K.; Hao, Z.F.; Wang, Z.Y. Copper(I)-Catalyzed Alkyl- and Arylsulfenylation of 3,4-Dihalo-2(5H)-Furanones (X=Br, Cl) with Sulfoxides under Mild Conditions. Adv. Synth. Catal. 2017, 359, 2961–2971. [Google Scholar] [CrossRef]
- Wang, F.; Rao, W.; Wang, S. Nickel-Catalyzed Reductive Thiolation of Unactivated Alkyl Bromides and Arenesulfonyl Cyanides. J. Org. Chem. 2021, 86, 8970–8979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhou, Y.; Huang, J.; Tu, C.; Zhou, X.; Yin, G. Cesium Carbonate-Promoted Synthesis of Aryl Methyl Sulfides Using S-Methylisothiourea Sulfate under Transition-Metal-Free Conditions. Org. Biomol. Chem. 2018, 16, 6316–6321. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, P.; Anbarasan, P. Palladium Catalyzed Aryl (Alkyl)Thiolation of Unactivated Arenes. Org. Lett. 2014, 16, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D.G.; Janine Cossy, J. Synthesis of Aryl Sulfides: Metal-Free C–H Sulfenylation of Electron-Rich Arenes. Org. Lett. 2015, 17, 3898–3901. [Google Scholar] [CrossRef]
- Arisawa, M.; Yamagushi, M. Rhodium-Catalyzed Synthesis of Organosulfur Compounds Involving S-S Bond Cleavage of Disulfides and Sulfur. Molecules 2020, 25, 3595. [Google Scholar] [CrossRef]
- Bao, Y.; Yang, X.; Dai, Z.; Ji, S.; Zhou, Q.; Yang, F. Iodine-Promoted Tunable Synthesis of 2-Naphthyl Thioethers and 1-Naphthyl Thioethers. Adv. Syn. Catal. 2019, 361, 2154–2158. [Google Scholar] [CrossRef]
- Duan, Y.; Guo, Z.; Zheng, T.; Lu, Y.; XU, J.; Liu, J.; Yang, F. Iodine-Promoted Reductive Sulfenylation Using Ketones as Hydride Donors. J. Org. Chem. 2024, 89, 5851–5856. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Tian, L.; Ji, X.; Deng, G.J.; Huang, H. Copper-Catalyzed 1,2-Sulfonyletherification of 1,3-Dienes. Org. Lett. 2024, 26, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Liu, H.; Chen, H.; Huang, H. Nickel-catalyzed Direct Formation of the C–S Bonds of aryl Sulfides from Arylsulfonyl Chlorides and Aryl Iodides using Mn as a Reducing Agent. Org. Chem. Front. 2017, 4, 31–36. [Google Scholar] [CrossRef]
- Zhong, S.; Zhou, Z.; Zhao, F.; Mao, G.; Deng, G.J.; Huang, H. Deoxygenative C–S Bond Coupling with Sulfinates via Nickel/Photoredox Dual Catalysis. Org. Lett. 2022, 24, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Ge, N.; Jiang, X. CO2-promoted Oxidative Cross-coupling Reaction for C–S bond Formation via Masked Strategy in an Odourless Way. Chem. Commun. 2015, 51, 10295–10298. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Cui, W.; Song, X.; Xu, G.; Jiang, M.; Sun, K.; Lv, J.; Yang, D. Sulfonylation of Aryl Halides by Visible Light/Copper Catalysis. Org. Lett. 2021, 23, 3663–3668. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Gui, Y.; Yu, B.; Jin, Y.; Tian, S.K. By-Product-Catalyzed Redox-Neutral Sulfenylation/Deiodination/Aromatization of Cyclic Alkenyl Iodides with Sulfonyl Hydrazides. Adv. Syn.Catal. 2016, 358, 3368–3372. [Google Scholar] [CrossRef]
- Saini, Y.; Khungar, B. Recyclable Imidazolium Ion-tagged Nickel Catalyst for Microwave-assisted C–S Cross-coupling in Water using Sulfonyl Hydrazide as the Sulfur Source. New J. Chem. 2018, 42, 12796–12801. [Google Scholar] [CrossRef]
- Bulmer, G.; Mann, F.G. The Chemistry of Xanthic Acid Derivatives. Part I. The Preparation and Comparative Properties of Isomeric Xanthates and Dithiocarbonates. J. Chem. Soc. 1945, 666–674. [Google Scholar] [CrossRef]
- Zard, S.Z. The Xanthate Route to Ketones: When the Radical Is Better than the Enolate. Acc. Chem. Res. 2018, 51, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Debien, L.; Quiclet-Sire, B.; Zard, S.Z. Allylic Alcohols: Ideal Radical Allylating Agents? Acc. Chem. Res. 2015, 48, 1237–1253. [Google Scholar] [CrossRef] [PubMed]
- Soleiman-Beigi, M.; Arzehgar, Z. A Novel Method for the Direct Synthesis of Symmetrical and Unsymmetrical Sulfides and Disulfides from Aryl Halides and Ethyl Potassium Xanthogenate. Synlett 2018, 29, 986–992. [Google Scholar] [CrossRef]
- SoKolov, A.I.; Mikhaylov, A.A.; Baleeva, N.S.; Baranov, M.S. Xanthates as Thiol Surrogates for Nucleophilic Substitution with Aryl Halides. Eur. J. Org. Chem. 2021, 30, 4350–4357. [Google Scholar] [CrossRef]
- Akkilagunta, V.K.; Kakulapati, R.R. Synthesis of Unsymmetrical Sulfides Using Ethyl Potassium Xanthogenate and Recyclable Copper Catalyst under Ligand-Free Conditions. J. Org. Chem. 2011, 76, 6819–6824. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.A.; Bugaenko, D.I.; Bogdanov, A.V.; Karchava, A.V. Visible-Light-Driven Thioesterification of Aryl Halides with Potassium Thiocarboxylates: Transition-Metal Catalyst-Free Incorporation of Sulfur Functionalities into an Aromatic Ring. J. Org. Chem. 2022, 87, 8170–8182. [Google Scholar] [CrossRef] [PubMed]
- Bugaenko, D.T.; Volkov, A.A.; Karchava, A.V. A Thiol-Free Route to Alkyl Aryl Thioethers. J. Org. Chem. 2023, 88, 9968–9972. [Google Scholar] [CrossRef]
- Degani, I.; Fochi, R.; Regondi, V. Phase-Transfer Synthesis of Symmetrical S,S-dialkyl Dithiocarbonates via o-Methyl S-alkyl Dithiocarbonates. Synthesis 1980, 5, 375–378. [Google Scholar] [CrossRef]
- He, R.; Liu, Y.; Feng, Y.; Chen, L.; Huang, Y.; Xie, F.; Li, Y. Access to Thienopyridine and Thienoquinoline Derivatives via Site-selective C–H Bond Functionalization and Annulationm. Org. Lett. 2022, 24, 3167–3172. [Google Scholar] [CrossRef]
- Luo, X.; He, R.; Liu, Q.; Gao, Y.; Li, J.; Chen, X.; Zhu, Z.; Huang, Y.; Li, Y. Metal-free Oxidative Esterification of Ketones and Potassium Xanthates: Selective Synthesis of α-Ketoesters and Esters. J. Org. Chem. 2020, 85, 5220–5230. [Google Scholar] [CrossRef]
- Feng, Y.; Nie, J.; Xie, S.; He, Z.; Hong, H.; Li, J.; Huang, Y.; Chen, L.; Li, Y. Potassium Xanthate–promoted Reductive Sulfuration Reaction: From Aldehydes to Thiol, Disulfide, and Thioester Derivatives. Chem. Commun. 2024, 59, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Karasawa, A.; Oku, A. A new Synthetic Route to Vinyl Sulfides Utilizing the Reaction of (Phenylthio)carbenes with Nitrile Anions. J. Org. Chem. 1986, 51, 842–846. [Google Scholar] [CrossRef]
- Hua, Y.; Zhang, W.; Wang, X.; Ge, Z.; Li, R. Synthesis of novel five-membered endocyclic sulfoximines-1-aryl-3-(arylimino)-3,5-dihydro-1,4,2-dithiazole 1-oxides. Tetrahedron 2017, 73, 4387–4391. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Chen, Z.; Li, J.; Ji, X.; Chen, L.; Huang, Y.; Liu, Q.; Li, Y. Synthesis of Substituted Thiophenes through Dehydration and Heterocyclization of Alkynols. J. Org. Chem. 2022, 87, 3555–3566. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, L.; Liu, X.; Li, B.; Sun, N. Copper-promoted Hydration and Annulation of 2-Fluorophenylacetylene Derivatives: From Alkynes to Benzo[b]furans and benzo[b]thiophenes. Beilstein J. Org. Chem. 2014, 10, 2886–2891. [Google Scholar] [CrossRef]
- Khan, M.Y.; Cho, M.S.; Kwark, Y.J. Dual Roles of a Xanthate as a Radical Source and Chain Transfer Agent in the Photoinitiated RAFT Polymerization of Vinyl Acetate. Macromolecules 2014, 47, 1929–1934. [Google Scholar] [CrossRef]
- Erian, A.W.; Reid, D.L.; Warkentin, J. Reactions of Dimethoxycarbene with Xanthates. J. Sulfur Chem. 2005, 26, 203–209. [Google Scholar] [CrossRef]
- Liu, Q.; Ci, C.; Zhao, H.; Xie, R.; Jiang, H.; Zhang, M. Direct Access to Functional Phenazines via Oxidative Annulation of Anilines and o-Phenylenediamines with a Reusable cobalt Catalyst. Green Chem. 2023, 25, 678–683. [Google Scholar] [CrossRef]
- Tan, Z.; Liang, Y.; Yang, L.; Cao, L.; Jiang, H.; Zhang, M. Site-Specific Oxidative C−H Chalcogenation of (Hetero)Aryl-Fused Cyclic Amines Enabled by Nanocobalt Oxides. Org. Lett. 2018, 20, 6554–6558. [Google Scholar] [CrossRef]
- Shen, W.; Wu, Q.; Gong, X.; Ao, G.; Liu, F. A Facile Method for Hydroxytrifluoromethylation of Alkenes with Langlois Reagent and DMSO. Green Chem. 2019, 21, 2983–2987. [Google Scholar] [CrossRef]
- Cao, X.H.; Pan, X.; Zhou, P.J.; Asekun, O.T. Manganese(iii)-mediated direct Csp2–H Radical Trifluoromethylation of Coumarins with Sodium Trifluoromethanesulfinate. Chem. Commun. 2014, 50, 3359–3362. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Mase, T. A General Palladium-Catalyzed Coupling of Aryl Bromides/Triflates and Thiols. Org. Lett. 2004, 6, 4587–4590. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, J.; Sun, W.; Pan, Y.; Ding, A.; Liu, W.; Guo, H. Thioxanthone-TfOH Complex (9-HTXTF) Photoredox Enabled Reduction of Sulfoxides. Tetrahedron Lett. 2024, 139, 155007. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, J.; Seo, H.; Park, S.; Chae, J. Copper(II)-Catalyzed Single-Step Synthesis of Aryl Thiols from Aryl Halides and 1,2-Ethanedithiol. Adv. Synth. Catal. 2015, 357, 2205–2212. [Google Scholar] [CrossRef]
- Ryota, I.; Miki, B.; Kei, M.; Junichiro, Y. Ni-Catalyzed Aryl Sulfide Synthesis through an Aryl Exchange Reaction. J. Am. Chem. Soc. 2021, 143, 10333–10340. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, W.; Ma, D. L-Proline-Promoted CuI-Catalyzed C-S Bond Formation between Aryl Iodides and Thiols. Synth. Commun. 2007, 37, 25–35. [Google Scholar] [CrossRef]
- Anuradha, N.; Imran, K.; Somraj, G.; Govindasamy, S. Visible-Light-Driven Halogen-Bond-Assisted Direct Synthesis of Heteroaryl Thioethers Using Transition-Metal-Free One-Pot C–I Bond Formation/C–S Cross-Coupling Reaction. J. Org. Chem. 2021, 86, 2570–2581. [Google Scholar] [CrossRef]
- Benjamin, H.; Moritz, B.; Paul, K. Thiolation of Pyridine-2-sulfonamides using Magnesium Thiolates. Synthesis 2019, 51, 4452–4462. [Google Scholar] [CrossRef]
- Forrest, H.S.; Walker, J. Chemotherapeutic agents of the Sulphone type. Part V. 2: 5-Disubstituted Derivatives of Pyridine. J. Chem. Soc. 1948, 1939–1945. [Google Scholar] [CrossRef]
- Beugelmansm, R.; Bois-Choussy, M.; Boudet, B. Etude des Reactions de srn1-partie 10: Action de Sulfanions sur les Halogenures d’aryle Fonctionnalises. Synthese Directe de Benzothiophenes et Thienopyridines. Tetrahedron 1983, 39, 4153–4161. [Google Scholar] [CrossRef]
- Obe, M.; Tanaka, K.; Nishiyama, K.; Ando, W. Aerobic Oxidation of Thiols to Disulfides Catalyzed by Diaryl Tellurides under Photosensitized Conditions. J. Org. Chem. 2011, 76, 4173–4177. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | EtOCS2K (mmol) | Solvent (mL) | T (°C) | T (h) | Yield (%) |
| 1 | EtOCS2K (1) | DMF (2) | 150 | 6 | 77 |
| 2 | EtOCS2K (1) | o-xylene (2) | 150 | 6 | <5 |
| 3 | EtOCS2K (1) | DMSO (2) | 150 | 6 | 95 |
| 4 | EtOCS2K (1) | DMAc (2) | 150 | 6 | 57 |
| 5 | EtOCS2K (1) | NMP (2) | 150 | 6 | 45 |
| 6 | EtOCS2K (1) | DMSO (1) | 150 | 6 | 94 |
| 7 | EtOCS2K (0.8) | DMSO (1) | 150 | 6 | 78 |
| 8 | EtOCS2K (1) | DMSO (1) | 130 | 6 | 95 |
| 9 | EtOCS2K (1) | DMSO (1) | 100 | 6 | 94 |
| 10 | EtOCS2K (1) | DMSO (1) | 80 | 6 | 89 |
| 11 | EtOCS2K (1) | DMSO (1) | 100 | 3 | 94 |
| 12 | EtOCS2K (1) | DMSO (1) | 100 | 1 | 93 |
| 13 | EtOCS2K (1) | DMSO (1) | 100 | 0.5 | 87 |
 | |||||
|---|---|---|---|---|---|
| Entry | EtOCS2K (mmol) | Additive (mmol) | Solvent | Time (h) | Yield (%) |
| 1 | EtOCS2K (1.2) | I2 (1.5) | DMF | 24 | 37 |
| 2 | EtOCS2K (1.2) | I2 (1.5) | DMF | 30 | 45 |
| 3 | EtOCS2K (1.2) | I2 (1.5) | DMF | 36 | 92 |
| 4 | EtOCS2K (0.8) | I2 (1.5) | DMF | 48 | 69 |
| 5 | EtOCS2K (1.2) | I2 (0.7) | DMF | 36 | 10 |
| 6 | EtOCS2K (1.2) | NH4I (1.5) | DMF | 36 | <5 |
| 7 | EtOCS2K (1.2) | HI (1.5) | DMF | 36 | 15 |
| 8 | EtOCS2K (1.2) | I2 (1.5) | DMSO | 36 | <5 |
| 9 | EtOCS2K (1.2) | I2 (1.5) | NMP | 36 | <5 |
| 10 | EtOCS2K (1.2) | I2 (1.5) | DMAc | 36 | 43 |
| 11 c | EtOCS2K (1.2) | I2 (1.5) | DMF | 36 | 39 |
| 12 | EtOCS2K (1.2) | - | DMF | 36 | <5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, J.; He, Z.; Xie, S.; Li, Y.; He, R.; Chen, L.; Luo, X. Expedient Synthesis of Alkyl and Aryl Thioethers Using Xanthates as Thiol-Free Reagents. Molecules 2024, 29, 2485. https://doi.org/10.3390/molecules29112485
Nie J, He Z, Xie S, Li Y, He R, Chen L, Luo X. Expedient Synthesis of Alkyl and Aryl Thioethers Using Xanthates as Thiol-Free Reagents. Molecules. 2024; 29(11):2485. https://doi.org/10.3390/molecules29112485
Chicago/Turabian StyleNie, Jinli, Ziqing He, Sijie Xie, Yibiao Li, Runfa He, Lu Chen, and Xiai Luo. 2024. "Expedient Synthesis of Alkyl and Aryl Thioethers Using Xanthates as Thiol-Free Reagents" Molecules 29, no. 11: 2485. https://doi.org/10.3390/molecules29112485
APA StyleNie, J., He, Z., Xie, S., Li, Y., He, R., Chen, L., & Luo, X. (2024). Expedient Synthesis of Alkyl and Aryl Thioethers Using Xanthates as Thiol-Free Reagents. Molecules, 29(11), 2485. https://doi.org/10.3390/molecules29112485