
Drug-Loaded Mesoporous Silica Nanoparticles Enhance Antitumor Immunotherapy by Regulating MDSCs


Abstract

1. Introduction
2. Results
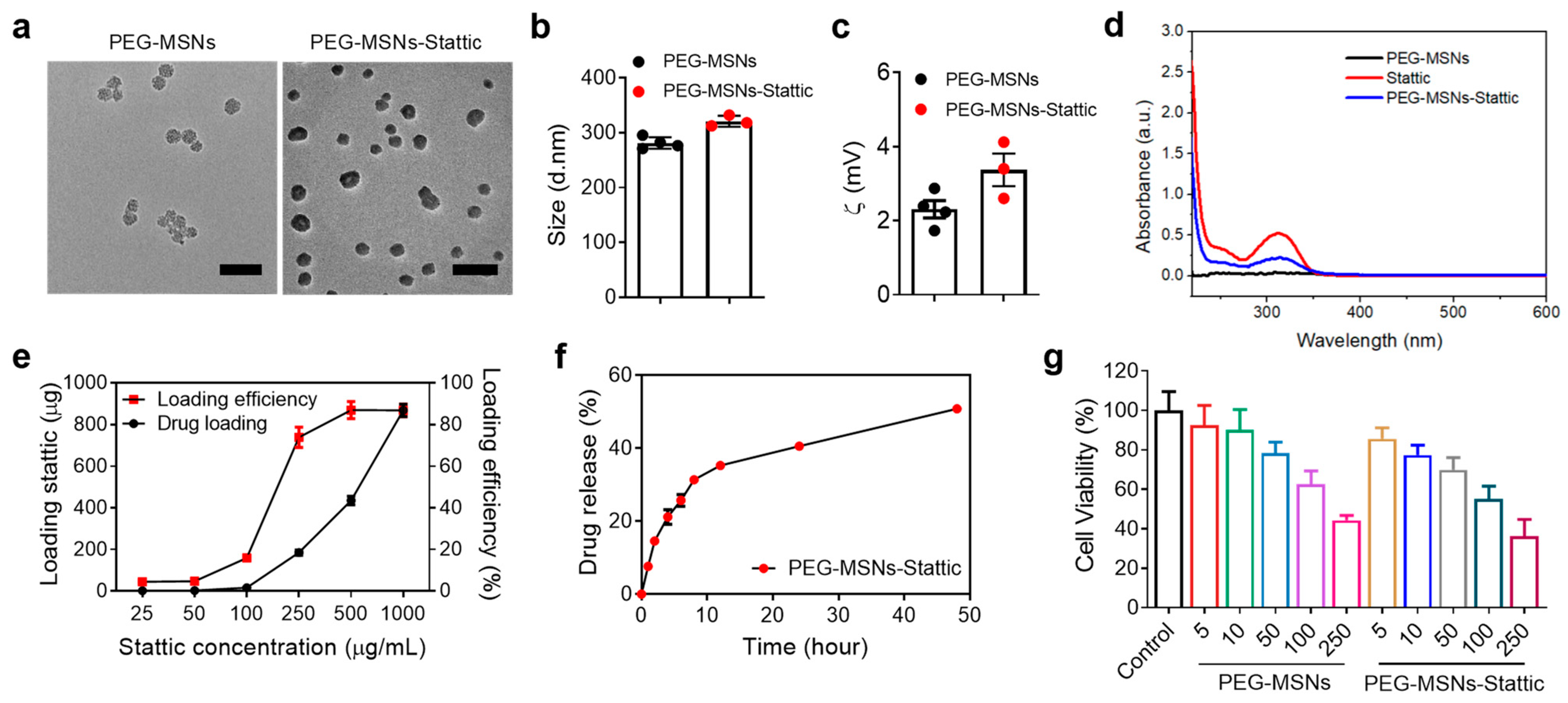
2.1. Characterization of PEG-MSNs and Their Toxicity, Drug Loading, and Release In Vitro
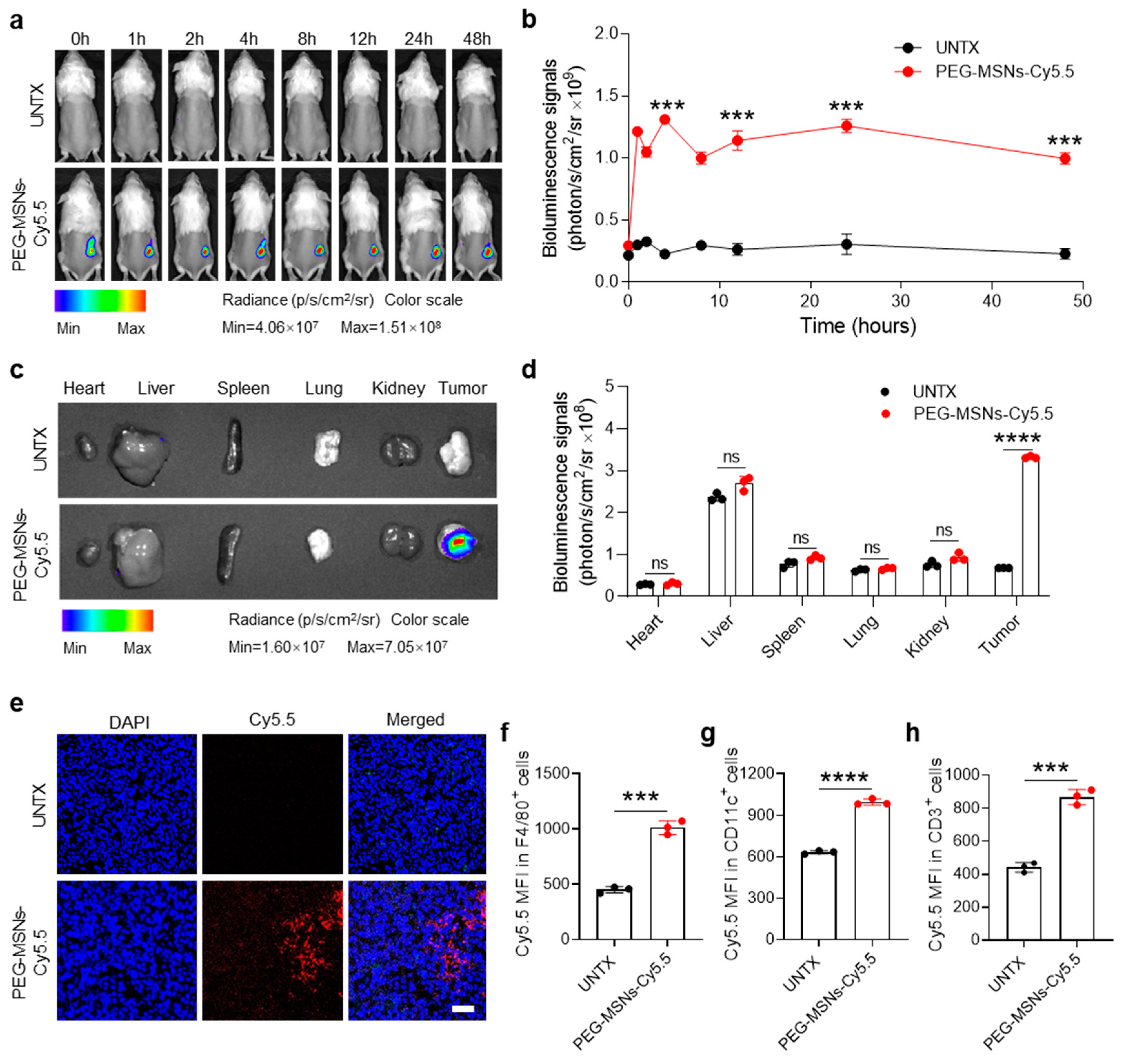
2.2. Biodistribution of PEG-MSNs in Tumors
2.3. PEG-MSNs-Stattic in Combination with aPD-L1 Blockade Inhibited Tumor Growth
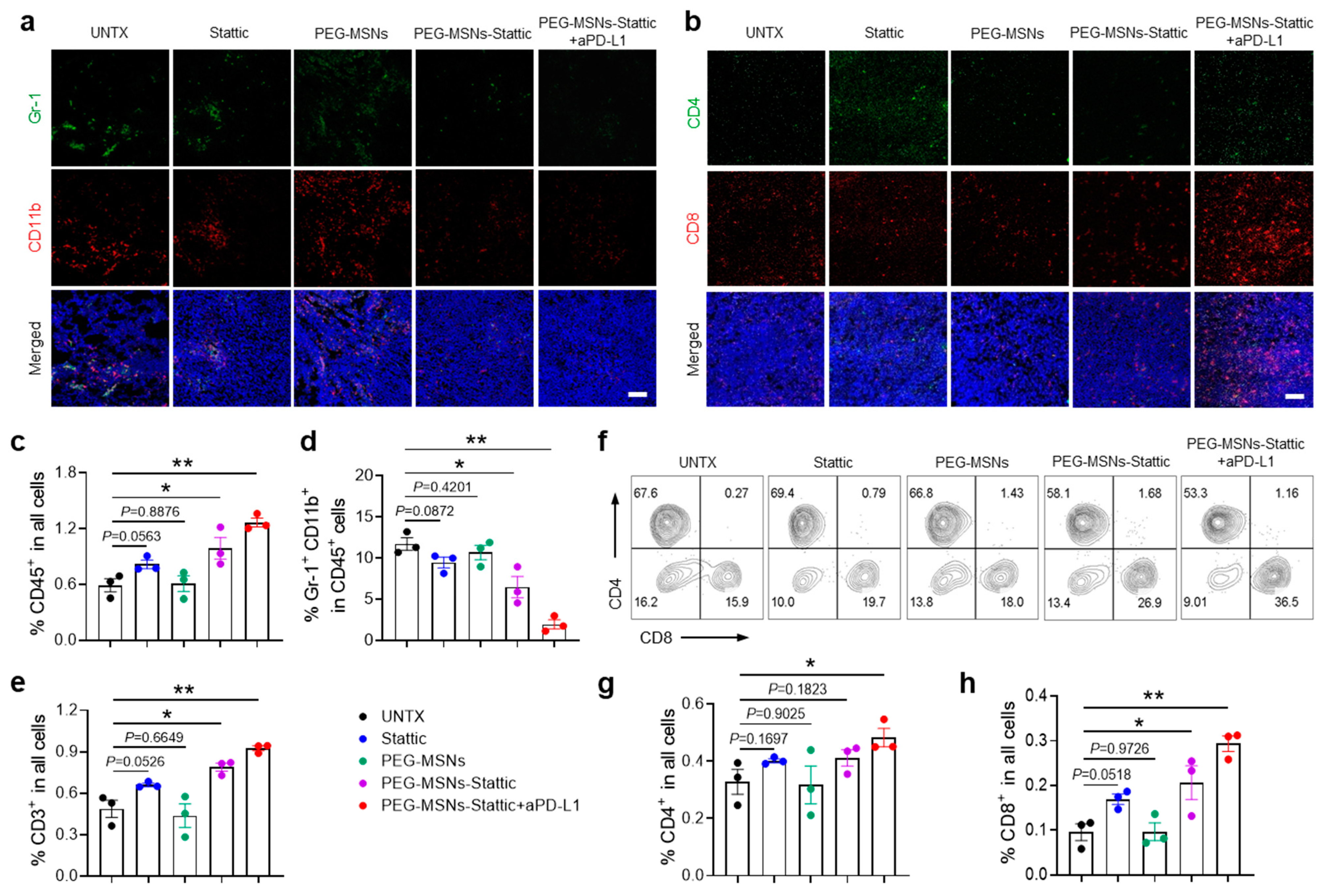
2.4. PEG-MSNs-Stattic Enhanced ICB Therapy by Reducing MDSC Numbers in Tumors
3. Materials and Methods
3.1. Materials
3.2. Mice and Cell Lines
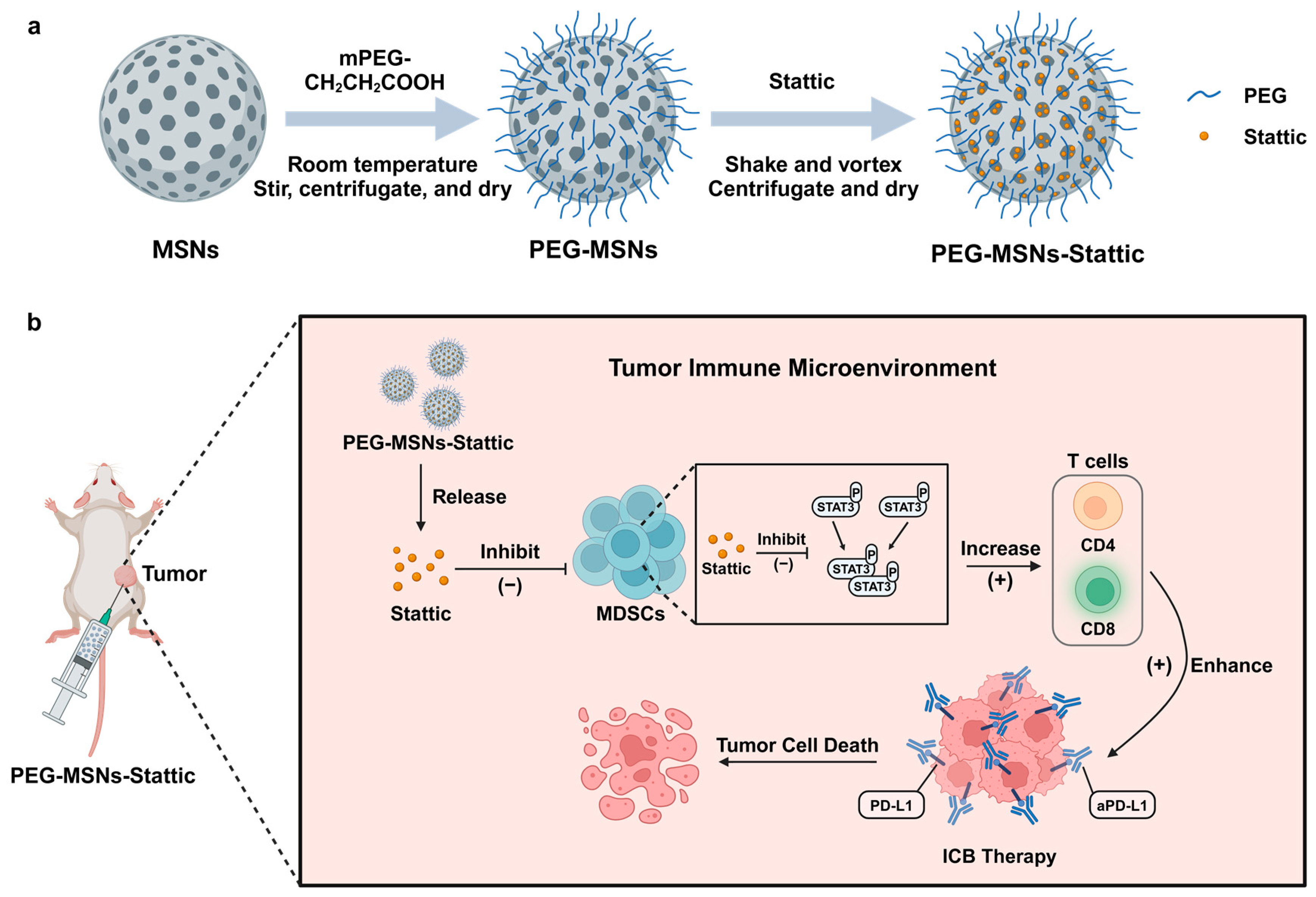
3.3. Preparation and Characterization of PEG-MSNs and PEG-MSNs-Stattic
3.4. Drug Loading and Drug Release Evaluation
3.5. In Vitro Cytotoxicity Assessment
3.6. Establishment and Treatment of Tumor Models
3.7. Biodistribution of PEG-MSNs in Tumors
3.8. Immunofluorescence Analysis of Tumors
3.9. Flow Cytometry Analysis of Tumors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A.J.C.C.J.C. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Yang, Q.; Sun, R.; Zhang, Y.; Ma, Q.; Shen, Y.; Wang, B.; Chen, Y.; Xu, J.; Tian, B.J.N.R. Nanoparticle accumulation in liver may induce resistance to immune checkpoint blockade therapy. Nano Res. 2023, 16, 5237–5246. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, Y.; Ma, D.; Cheng, W.; Liu, J.; Yong, T.; Chen, H.; Wang, C. Immunotherapy: Reshape the Tumor Immune Microenvironment. Front. Immunol. 2022, 13, 844142. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int. Immunopharmacol. 2020, 88, 106939. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Dai, H.; Bai, J.; Xu, J.; Ma, Q.; Fei, Z.; Zhou, X.; Leong, K.W.; Wang, C.J.F.R. Degradation-resistant implanted biomaterials establish an immunosuppressive microenvironment that induces T cell exhaustion by recruiting myeloid cells. Fundam. Res. 2022, 2, 648–658. [Google Scholar] [CrossRef]
- Bullock, K.; Richmond, A. Suppressing MDSC Recruitment to the Tumor Microenvironment by Antagonizing CXCR2 to Enhance the Efficacy of Immunotherapy. Cancers 2021, 13, 6293. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, T.; Zhu, W.; Xie, S.; Zhao, Z.; Feng, B.; Guo, H.; Yang, R. Targeting MDSC for Immune-Checkpoint Blockade in Cancer Immunotherapy: Current Progress and New Prospects. Clin. Med. Insights Oncol. 2021, 15, 11795549211035540. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Bitsch, R.; Kurzay, A.; Ozbay Kurt, F.; De La Torre, C.; Lasser, S.; Lepper, A.; Siebenmorgen, A.; Muller, V.; Altevogt, P.; Utikal, J.; et al. STAT3 inhibitor Napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J. Immunother. Cancer 2022, 10, e004384. [Google Scholar] [CrossRef]
- Hofer, F.; Di Sario, G.; Musiu, C.; Sartoris, S.; De Sanctis, F.; Ugel, S. A Complex Metabolic Network Confers Immunosuppressive Functions to Myeloid-Derived Suppressor Cells (MDSCs) within the Tumour Microenvironment. Cells 2021, 10, 2700. [Google Scholar] [CrossRef]
- Hellsten, R.; Lilljebjörn, L.; Johansson, M.; Leandersson, K.; Bjartell, A. The STAT3 inhibitor galiellalactone inhibits the generation of MDSC-like monocytes by prostate cancer cells and decreases immunosuppressive and tumorigenic factors. Prostate 2019, 79, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Mohrherr, J.; Uras, I.Z.; Moll, H.P.; Casanova, E. STAT3: Versatile Functions in Non-Small Cell Lung Cancer. Cancers 2020, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Furqan, M.; Akinleye, A.; Mukhi, N.; Mittal, V.; Chen, Y.; Liu, D. STAT inhibitors for cancer therapy. J. Hematol. Oncol. 2013, 6, 90. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Jeelani, P.G.; Mulay, P.; Venkat, R.; Ramalingam, C. Multifaceted Application of Silica Nanoparticles. A Review. Silicon-Neth. 2020, 12, 1337–1354. [Google Scholar] [CrossRef]
- Dai, H.; Fan, Q.; Wang, C. Recent applications of immunomodulatory biomaterials for disease immunotherapy. Exploration 2022, 2, 20210157. [Google Scholar] [CrossRef]
- Dai, H.X.; Fan, Q.; Fei, Z.Y.; Ma, Q.L.; Yang, Q.Y.; Chu, J.C.; Zhou, X.F.; Zhang, Y.; Dong, Z.L.; Xu, F.; et al. Systemically administered silica nanoparticles result in diminished T cell response in lung. Nano Today 2022, 42, 101332. [Google Scholar] [CrossRef]
- Hao, N.; Liu, H.; Li, L.; Chen, D.; Li, L.; Tang, F. In vitro degradation behavior of silica nanoparticles under physiological conditions. J. Nanosci. Nanotechnol. 2012, 12, 6346–6354. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.G.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, J.; Shi, J.; Zhu, Z.; Zhang, L.; Bu, W.; Guo, L.; Chen, Y. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials 2010, 31, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, H.J.; Feng, H.Y.; Yang, S.C.; Liu, X.L.; Lai, X.; Lu, Q.; Lovell, J.F.; Chen, H.Z.; Fang, C. Enhanced Drug Delivery by Nanoscale Integration of a Nitric Oxide Donor To Induce Tumor Collagen Depletion. Nano Lett. 2019, 19, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Shi, H.; Li, Z.; Shen, H.; Ma, K.; Li, B.; Shen, S.; Jin, Y. A multifunctional mesoporous silica nanocomposite for targeted delivery, controlled release of doxorubicin and bioimaging. Colloids Surf. B Biointerfaces 2013, 110, 138–147. [Google Scholar] [CrossRef]
- Pan, Y.; Zhou, F.; Zhang, R.; Claret, F.X. Stat3 inhibitor Stattic exhibits potent antitumor activity and induces chemo- and radio-sensitivity in nasopharyngeal carcinoma. PLoS ONE 2013, 8, e54565. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Kong, M.; Tang, J.; Qiao, Q.; Wu, T.; Qi, Y.; Tan, S.; Gao, X.; Zhang, Z. Biodegradable Hollow Mesoporous Silica Nanoparticles for Regulating Tumor Microenvironment and Enhancing Antitumor Efficiency. Theranostics 2017, 7, 3276–3292. [Google Scholar] [CrossRef]
- Zolnik, B.S.; González-Fernández, A.; Sadrieh, N.; Dobrovolskaia, M.A. Minireview: Nanoparticles and the Immune System. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef]
- Mahdavi Gorabi, A.; Sadat Ravari, M.; Sanaei, M.J.; Davaran, S.; Kesharwani, P.; Sahebkar, A. Immune checkpoint blockade in melanoma: Advantages, shortcomings and emerging roles of the nanoparticles. Int. Immunopharmacol. 2022, 113, 109300. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents & Resources | Source | Identifier |
|---|---|---|
| Antibodies | ||
| FITC anti-mouse F4/80 | Biolegend | Cat # 123108; RRID: AB_893502 |
| PE anti-mouse CD45 | Biolegend | Cat # 103106; RRID: AB_312971 |
| FITC anti-mouse CD11c | Biolegend | Cat # 117306; RRID: AB_313775 |
| PE anti-mouse CD3 | Biolegend | Cat # 201412; RRID: AB_10804049 |
| PE anti-mouse IFN-γ | Biolegend | Cat # 507806; RRID: AB_2830472 |
| PE anti-mouse/human CD11b | Biolegend | Cat # 260042; RRID: AB_2536482 |
| FITC anti-mouse CD45 | Biolegend | Cat # 103108; RRID: AB_312973 |
| APC anti-mouse Ly-6G/Ly-6C (Gr-1) | Biolegend | Cat # 108412; RRID: AB_313377 |
| PE anti-mouse/human CD11b | Biolegend | Cat # 101208; RRID: AB_312791 |
| Percp/Cyanine5.5 anti-mouse Ki-67 | Biolegend | Cat # 652424; RRID: AB_2629531 |
| APC anti-human CD8 | Biolegend | Cat # 344722; RRID: AB_2829639 |
| Experimental Models: Cell Lines | ||
| RAW 264.7 | Cell Bank of Shanghai Institutes for Biological Sciences | N/A |
| CT26-Luc | Cell Bank of Shanghai Institutes for Biological Sciences | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Amna, N.; Shi, Y.; Sun, R.; Weng, C.; Chen, J.; Dai, H.; Wang, C. Drug-Loaded Mesoporous Silica Nanoparticles Enhance Antitumor Immunotherapy by Regulating MDSCs. Molecules 2024, 29, 2436. https://doi.org/10.3390/molecules29112436
Xu C, Amna N, Shi Y, Sun R, Weng C, Chen J, Dai H, Wang C. Drug-Loaded Mesoporous Silica Nanoparticles Enhance Antitumor Immunotherapy by Regulating MDSCs. Molecules. 2024; 29(11):2436. https://doi.org/10.3390/molecules29112436
Chicago/Turabian StyleXu, Changlin, Nida Amna, Yuchen Shi, Rong Sun, Chenhui Weng, Jiaoyu Chen, Huaxing Dai, and Chao Wang. 2024. "Drug-Loaded Mesoporous Silica Nanoparticles Enhance Antitumor Immunotherapy by Regulating MDSCs" Molecules 29, no. 11: 2436. https://doi.org/10.3390/molecules29112436
APA StyleXu, C., Amna, N., Shi, Y., Sun, R., Weng, C., Chen, J., Dai, H., & Wang, C. (2024). Drug-Loaded Mesoporous Silica Nanoparticles Enhance Antitumor Immunotherapy by Regulating MDSCs. Molecules, 29(11), 2436. https://doi.org/10.3390/molecules29112436