
Novel 9-Methylanthracene Derivatives as p53 Activators for the Treatment of Glioblastoma Multiforme

 and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Antiproliferative Activity Analysis
2.3. Mechanistic Analyses of 13e against U87 Cells
2.4. Molecular Docking of 13e
3. Materials and Methods
3.1. Cytotoxicity Evaluation
3.2. MTT Assay
3.3. Cell Cycle Analysis
3.4. Western Blotting
3.5. Data and Statistical Analysis
3.6. Chemistry Section
3.6.1. General Methods
3.6.2. Synthesis of Compound 4
3.6.3. Synthesis of Compound 7
3.6.4. Synthesis of Compound 9
3.6.5. Synthesis of Compound 11
3.6.6. General Synthesis Procedure for XI-011, 12a–12l, 13a–13l, and 14a–14s
3.7. Computational Modeling Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Osborn, A.; Louis, D.; Poussaint, T.; Linscott, L.; Salzman, K. The 2021 World Health Organization classification of tumors of the central nervous system: What neuroradiologists need to know. Am. J. Neuroradiol. 2022, 43, 928–937. [Google Scholar] [CrossRef] [PubMed]
- King, J.L.; Benhabbour, S.R. Glioblastoma Multiforme—A Look at the Past and a Glance at the Future. Pharmaceutics 2021, 13, 1053. [Google Scholar] [CrossRef]
- Mair, M.J.; Geurts, M.; van den Bent, M.J.; Berghoff, A.S. A basic review on systemic treatment options in WHO grade II–III gliomas. Cancer Treat. Rev. 2021, 92, 102124. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef] [PubMed]
- Lübtow, M.M.; Oerter, S.; Quader, S.; Jeanclos, E.; Cubukova, A.; Krafft, M.; Haider, M.S.; Schulte, C.; Meier, L.; Rist, M. In Vitro Blood–Brain Barrier Permeability and Cytotoxicity of an Atorvastatin-Loaded Nanoformulation Against Glioblastoma in 2D and 3D Models. Mol. Pharm. 2020, 17, 1835–1847. [Google Scholar] [CrossRef]
- Di Filippo, L.D.; Azambuja, J.H.; Dutra, J.A.P.; Luiz, M.T.; Duarte, J.L.; Nicoleti, L.R.; Saad, S.T.O.; Chorilli, M. Improving temozolomide biopharmaceutical properties in glioblastoma multiforme (GBM) treatment using GBM-targeting nanocarriers. Eur. J. Pharm. Biopharm. 2021, 168, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wei, D.; Dai, X.; Stevens, M.F.; Bradshaw, T.D.; Luo, Y.; Zhang, J. C8-substituted imidazotetrazine analogs overcome temozolomide resistance by inducing DNA adducts and DNA damage. Front. Oncol. 2019, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Rumiato, E.; Bertorelle, R.; Saggioro, D.; Farina, P.; Della Puppa, A.; Zustovich, F.; Berti, F.; Sacchetto, V.; Marcato, R. Clinical and genetic factors associated with severe hematological toxicity in glioblastoma patients during radiation plus temozolomide treatment: A prospective study. Am. J. Clin. Oncol. 2015, 38, 514–519. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Xiao, Z.-Z.; Wang, Z.-F.; Lan, T.; Huang, W.-H.; Zhao, Y.-H.; Li, Z.-Q. Carmustine as a supplementary therapeutic option for glioblastoma: A systematic review and meta-analysis. Front. Neurol. 2020, 11, 552557. [Google Scholar] [CrossRef]
- Weller, M.; Le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 2020, 87, 102029. [Google Scholar] [CrossRef] [PubMed]
- Seystahl, K.; Hentschel, B.; Loew, S.; Gramatzki, D.; Felsberg, J.; Herrlinger, U.; Westphal, M.; Schackert, G.; Thon, N.; Tatagiba, M. Bevacizumab versus alkylating chemotherapy in recurrent glioblastoma. J. Cancer Res. Clin. Oncol. 2020, 146, 659–670. [Google Scholar] [CrossRef]
- Jakobsen, J.; Urup, T.; Grunnet, K.; Toft, A.; Johansen, M.; Poulsen, S.; Christensen, I.; Muhic, A.; Poulsen, H. Toxicity and efficacy of lomustine and bevacizumab in recurrent glioblastoma patients. J. Neuro-Oncol. 2018, 137, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Momta, H.; Tonooka, A.; Noguchi, H.; Yamamoto, K.; Wanibuchi, M.; Minamida, Y.; Hasegawa, T.; Houkin, K. Glioblastoma simultaneously present with adjacent meningioma: Case report and review of the lit-erature. J. Neuro-Oncol. 2010, 99, 147. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef]
- Manea, A.J.; Ray, S.K. Advanced bioinformatics analysis and genetic technologies for targeting autophagy in glioblastoma multiforme. Cells 2023, 12, 897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, Z.; Li, Y.; Gong, Y.; Lyu, X.; Lou, J.; Zhang, D.; Meng, X.; Zhao, Y. Structure-based discovery of MDM2/4 dual inhibitors that exert antitumor activities against MDM4-overexpressing cancer cells. J. Med. Chem. 2022, 65, 6207. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.J.; Si, D.J.; Sun, X.J.; Wang, M.Y.; Yang, Y.B.; Wang, B.; Wen, H.M.; Li, W.; Liu, J. Structure-based dis-covery of novel α-aminoketone derivatives as dual p53-MDM2/MDMX inhibitors for the treatment of cancer. Eur. J. Med. Chem. 2023, 252, 115282. [Google Scholar] [CrossRef]
- Wang, S.; Chen, F.-E. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur. J. Med. Chem. 2022, 236, 114334. [Google Scholar] [CrossRef]
- Zhang, S.; Lou, J.; Li, Y.; Zhou, F.; Yan, Z.; Lyu, X.; Zhao, Y. Recent progress and clinical development of inhibitors that block MDM4/p53 protein–protein interactions. J. Med. Chem. 2021, 64, 10621–10640. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, C. A small-molecule p53 activator induces apoptosis through inhibiting MDMX expression in breast cancer cells. Neoplasia 2011, 13, 611–619, IN6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, G.; Yang, Y.; Wang, Y.; Guo, M.; Yin, Q.; Yan, C.; Tian, J.; Fu, F.; Wang, H. A small-molecule inhibitor of MDMX suppresses cervical cancer cells via the inhibition of E6-E6AP-p53 axis. Pharmacol. Res. 2022, 177, 106128. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, S.; Yao, H.; Hu, Y.; Wang, Y.; Jiang, J.; Li, X.; Fu, F.; Yin, Q.; Wang, H. Identification of a novel heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) ligand that disrupts HnRNPA2B1/nucleic acid interactions to inhibit the MDMX-p53 axis in gastric cancer. Pharmacol. Res. 2023, 189, 106696. [Google Scholar] [CrossRef]
- Mansor, S.F. Manipulation of p53 protein in bladder cancer treatment. IIUM Med. J. Malays. 2021, 20, 137. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Structure | U87 (% Inhib. @ 10 µM) | AlogP | Cmpd | Structure | U87 (% Inhib. @ 10 µM) | AlogP |
|---|---|---|---|---|---|---|---|
| XI-011 |  | 91.8 | 4.65 | 12h |  | 9.9 | 4.17 |
| 12a |  | 15.2 | 3.65 | 12i |  | 2.7 | 3.58 |
| 12b |  | 9.0 | 3.16 | 12j |  | 6.8 | 2.58 |
| 12c | 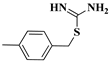 | 11.8 | 2.65 | 12k |  | 10.7 | 2.65 |
| 12d |  | 14.8 | 1.44 | 12l |  | 11.3 | 3.19 |
| 12e |  | 1.5 | 3.16 | 13a |  | 57.9 | 4.62 |
| 12f |  | 13.1 | 3.62 | DOX | 81.1 | ||
| 12g |  | 14.5 | 3.14 | DMSO | 4.7 | ||
| Cmpd | Structure | U87 (% Inhib. @ 10 µM) | AlogP | Cmpd | Structure | U87 (% Inhib. @ 10 µM) | AlogP |
|---|---|---|---|---|---|---|---|
| 13a |  | 57.9 | 4.62 | 13h |  | 0.7 | 4.56 |
| 13b |  | 19.3 | 4.58 | 13i |  | 31.2 | 5.18 |
| 13c |  | 35.4 | 4.93 | 13j |  | 13.3 | 6.83 |
| 13d |  | 93.3 | 5.17 | 13k |  | 28.1 | 8.73 |
| 13e |  | 88.6 | 4.92 | 13l |  | 86.7 | 7.24 |
| 13f |  | 65.1 | 5.47 | DOX | 81.1 | ||
| 13g | 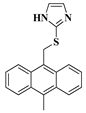 | 41.2 | 4.97 | DMSO | 4.7 | ||
| Cmpd | Structure | U87 (% Inhib. @ 10 μM) | AlogP | Cmpd | Structure | U87 (% Inhib. @ 10 μM) | AlogP |
|---|---|---|---|---|---|---|---|
| XI-011 |  | 91.8 | 4.65 | 14k |  | 14.2 | 4.99 |
| 14a |  | 79.0 | 5.06 | 14l |  | 0.7 | 4.09 |
| 14b |  | 94.7 | 5.58 | 14m |  | 36.8 | 4.99 |
| 14c |  | 83.6 | 4.72 | 14n |  | 86.5 | 5.51 |
| 14d |  | 17.3 | 5.35 | 14o |  | 50.7 | 4.96 |
| 14e |  | 19.3 | 5.00 | 14p |  | 19.0 | 3.64 |
| 14f |  | 43.6 | 7.66 | 14q |  | 12.5 | 3.67 |
| 14g |  | 28.0 | 8.17 | 14r |  | 2.1 | 6.27 |
| 14h |  | 64.6 | 5.48 | 14s |  | 6.9 | 4.35 |
| 14i |  | 76.4 | 4.68 | DOX | 81.1 | ||
| 14j |  | 11.8 | 4.13 | DMSO | 4.7 | ||
| Cmpd | IC50 (μM) | |||
|---|---|---|---|---|
| U87 | Hela | MCF-7 | HHL-5 | |
 (XI-011) (XI-011) | 0.69 ± 0.02 | 1.51 ± 0.06 | 4.87 ± 0.30 | 0.55 ± 0.03 |
 (13d) (13d) | 1.62 ± 0.02 | 5.66 ± 0.29 | 4.74 ± 0.20 | 0.85 ± 0.10 |
 (13e) (13e) | 0.53 ± 0.03 | 1.63 ± 0.25 | 0.97 ± 0.11 | 1.11 ± 0.08 |
 (14a) (14a) | 1.24 ± 0.24 | 3.64 ± 0.27 | 4.56 ± 0.03 | 1.51 ± 0.16 |
 (14b) (14b) | 1.35 ± 0.10 | 2.66 ± 0.19 | 4.55 ± 0.27 | 1.62 ± 0.13 |
 (14n) (14n) | 1.84 ± 0.12 | 7.74 ± 0.81 | 7.60 ± 1.37 | 1.67 ± 0.16 |
| Dox | 0.44 ± 0.06 | 0.54± 0.05 | 1.32 ± 0.16 | 0.32 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Wang, Y.; Li, X.; Sun, Z.; Qiang, S.; Wang, H.; Liu, Y. Novel 9-Methylanthracene Derivatives as p53 Activators for the Treatment of Glioblastoma Multiforme. Molecules 2024, 29, 2396. https://doi.org/10.3390/molecules29102396
Feng Y, Wang Y, Li X, Sun Z, Qiang S, Wang H, Liu Y. Novel 9-Methylanthracene Derivatives as p53 Activators for the Treatment of Glioblastoma Multiforme. Molecules. 2024; 29(10):2396. https://doi.org/10.3390/molecules29102396
Chicago/Turabian StyleFeng, Yuxin, Yingjie Wang, Xiaoxue Li, Ziqiang Sun, Sihan Qiang, Hongbo Wang, and Yi Liu. 2024. "Novel 9-Methylanthracene Derivatives as p53 Activators for the Treatment of Glioblastoma Multiforme" Molecules 29, no. 10: 2396. https://doi.org/10.3390/molecules29102396
APA StyleFeng, Y., Wang, Y., Li, X., Sun, Z., Qiang, S., Wang, H., & Liu, Y. (2024). Novel 9-Methylanthracene Derivatives as p53 Activators for the Treatment of Glioblastoma Multiforme. Molecules, 29(10), 2396. https://doi.org/10.3390/molecules29102396