
Discovery and Synthesis of Hydroxy-l-Proline Blockers of the Neutral Amino Acid Transporters SLC1A4 (ASCT1) and SLC1A5 (ASCT2)


Abstract
1. Introduction
2. Results
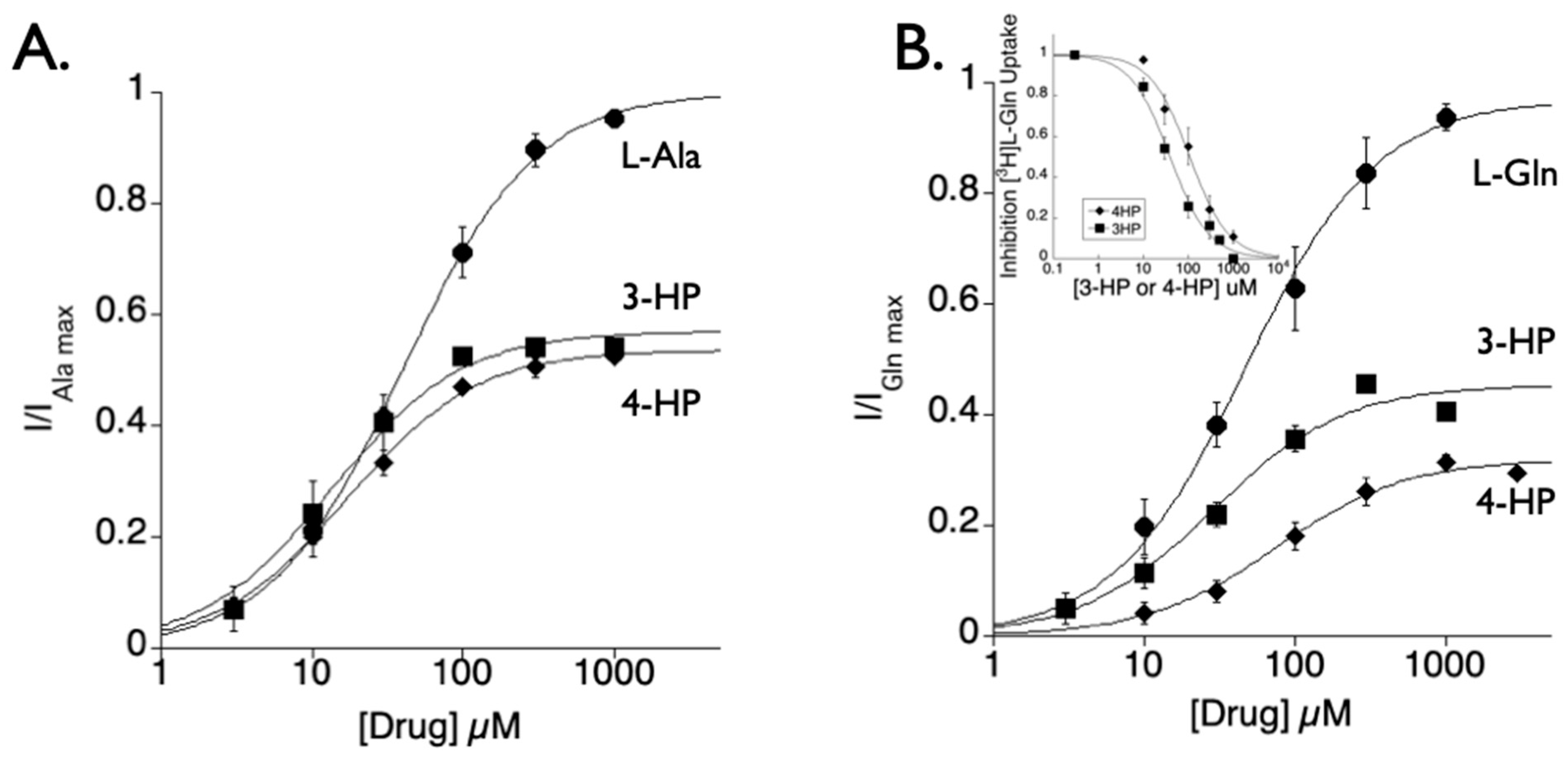
2.1. Hydroxy Proline Scaffold
2.2. Design of SLC1A4 and SLC1A5 Inhibitors
Analogs of Hydroxy-l-Proline: Design
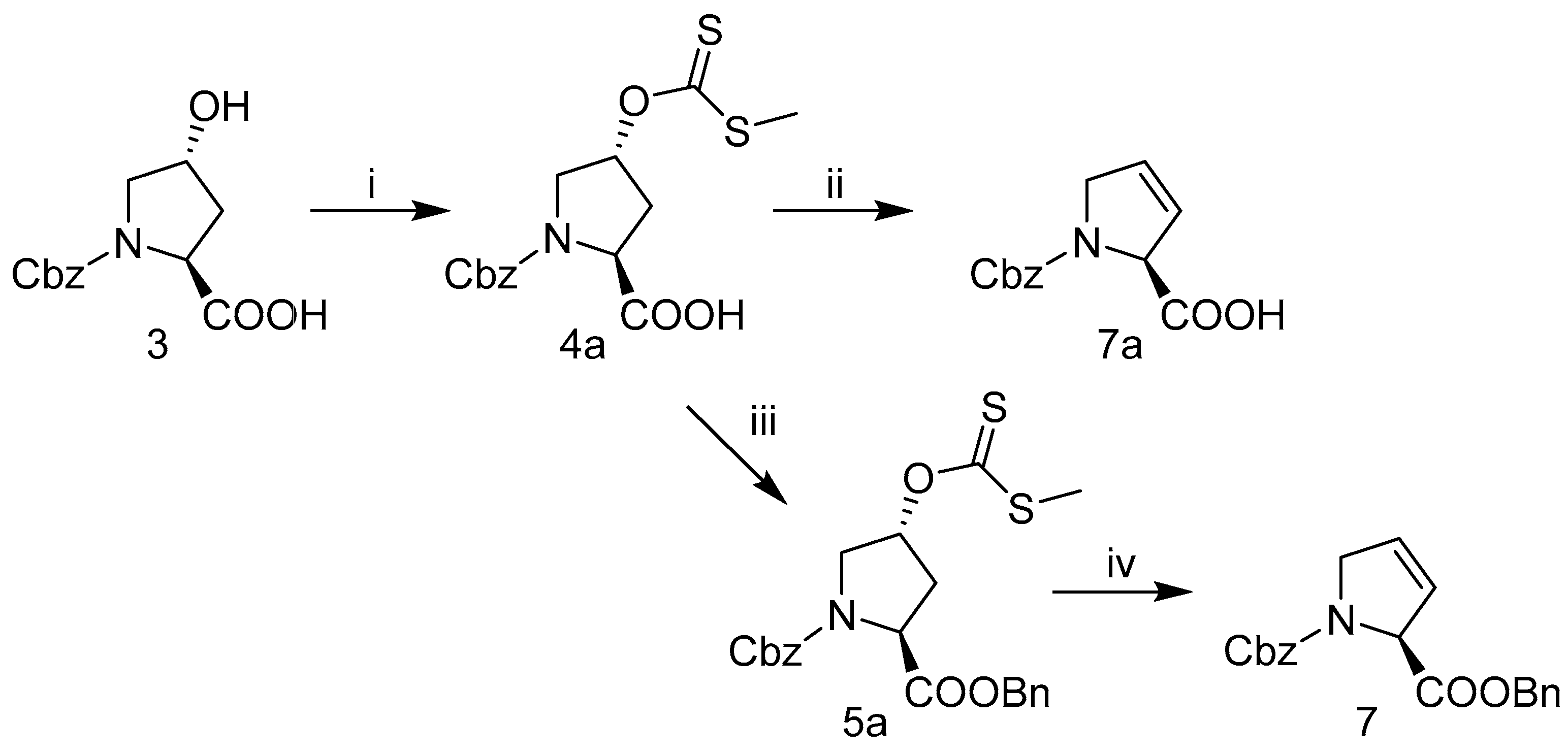
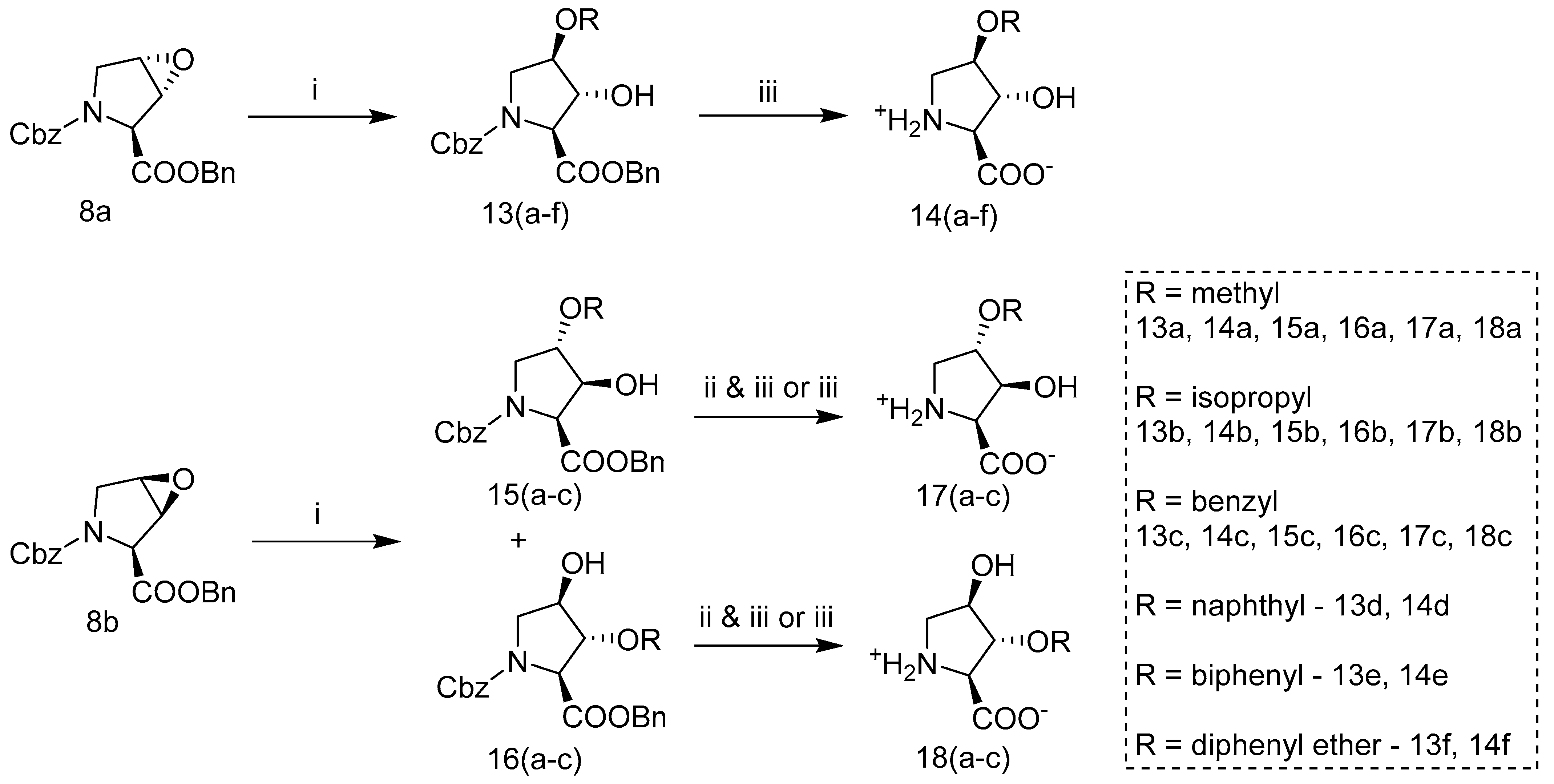
2.3. Synthesis of Hydroxyproline Analogs
2.3.1. N-Cbz-(Cis- or Trans)-3,4-Epoxy-l-Proline Benzyl Ester
2.3.2. Alternative Synthesis of N-Cbz-3,4-Dehydro-l-Proline-Benzyl Ester 7 or N-Cbz-3,4-Dehydro-l-Proline 7a
2.3.3. Methyl-, Phenyl-, or Phenol Ether Hydroxyprolines
2.3.4. Alkoxy Hydroxy-Pyrrolidine Carboxylic Acids (AHPCs)
2.4. Pharmacology, Functional Screening of Substituted Hydroxyprolines
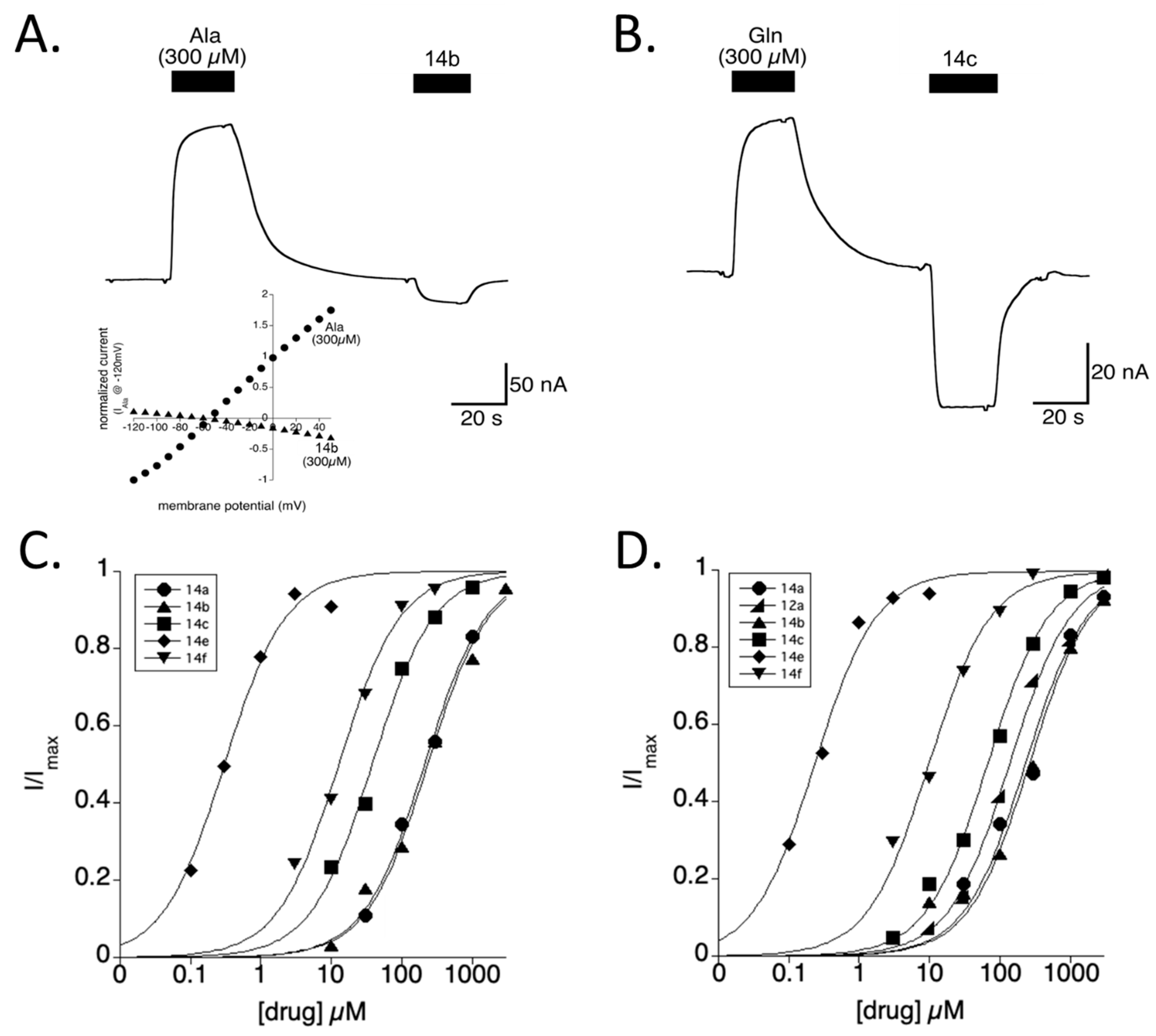
2.4.1. Hydroxyproline Analogs Inhibit SLC1A4 and SLC1A5
2.4.2. BPOHP Pharmacology
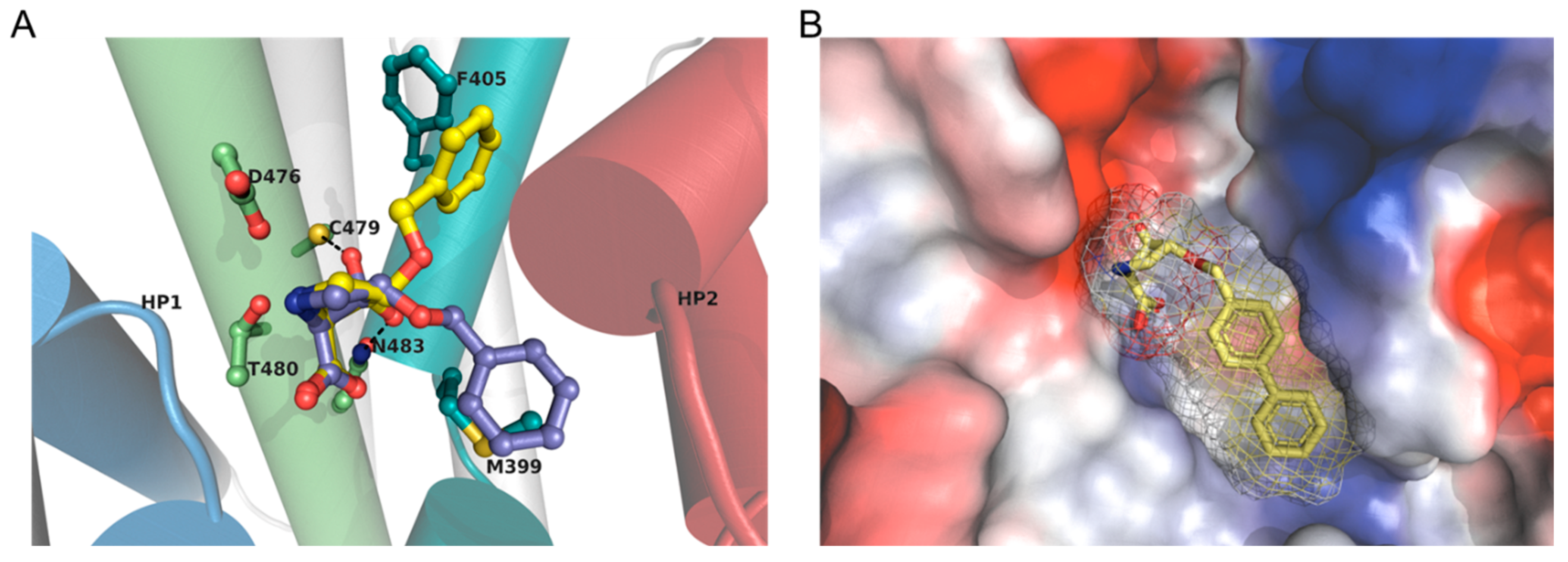
2.5. Computational Docking
3. Methods
3.1. Chemicals and Reagents
3.2. Expression and Functional Testing of Amino Acid Transporters Expressed in Xenopus Laevis Oocytes
3.3. Computational Models and Docking
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AHPC | Alkoxy hydroxy-pyrrolidine carboxylic acids |
| 3-HP | Trans-3-hydroxy-l-proline |
| 4-HP | Trans-4-hydroxy-l-proline |
| ASC | Ala, Ser, Cys-selective transporter |
| BPOHP | Biphenyl-O-hydroxy-proline, structure 14 |
| gCOSY | Gradient correlation spectroscopy |
| mCPBA | meta-chloroperoxybenzoic acid |
| NMDA | N-methyl d-aspartate |
| PDB | Protein Data Bank |
| SAR | Structure–activity relationship |
| SLC | Solute carrier superfamily |
| TBOA | l-threo-β-benzyloxyaspartate |
| TEVC | Two-electrode voltage clamp |
| TM | Transmembrane helix |
References
- Arriza, J.L.; Kavanaugh, M.P.; Fairman, W.A.; Wu, Y.N.; Murdoch, G.H.; North, R.A.; Amara, S.G. Cloning and Expression of a Human Neutral Amino Acid Transporter with Structural Similarity to the Glutamate Transporter Gene Family. J. Biol. Chem. 1993, 268, 15329–15332. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, S.; Tamarappoo, B.K.; Kilberg, M.S.; Puranam, R.S.; McNamara, J.O.; Guadaño-Ferraz, A.; Fremeau, R.T. Cloning and Expression of a Novel Na(+)-Dependent Neutral Amino Acid Transporter Structurally Related to Mammalian Na+/Glutamate Cotransporters. J. Biol. Chem. 1993, 268, 15351–15355. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya-Tate, N.; Endou, H.; Kanai, Y. Cloning and Functional Characterization of a System ASC-like Na+-Dependent Neutral Amino Acid Transporter. J. Biol. Chem. 1996, 271, 14883–14890. [Google Scholar] [CrossRef] [PubMed]
- Kekuda, R.; Prasad, P.D.; Fei, Y.-J.; Torres-Zamorano, V.; Sinha, S.; Yang-Feng, T.L.; Leibach, F.H.; Ganapathy, V. Cloning of the Sodium-Dependent, Broad-Scope, Neutral Amino Acid Transporter Bo from a Human Placental Choriocarcinoma Cell Line. J. Biol. Chem. 1996, 271, 18657–18661. [Google Scholar] [CrossRef] [PubMed]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A Guide to Plasma Membrane Solute Carrier Proteins. FEBS J. 2021, 288, 2784–2835. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The Concise Guide to Pharmacology 2019/20: Transporters. Br. J. Pharmacol 2019, 176, S397–S493. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Gallo, L.; Jadhav, A.; Hawkins, R.; Parker, C.G. The Druggability of Solute Carriers. J. Med. Chem. 2020, 63, 3834–3867. [Google Scholar] [CrossRef]
- Christensen, H.N. Role of Amino Acid Transport and Countertransport in Nutrition and Metabolism. Physiol. Rev. 1990, 70, 43–77. [Google Scholar] [CrossRef] [PubMed]
- Scopelliti, A.J.; Font, J.; Vandenberg, R.J.; Boudker, O.; Ryan, R.M. Structural Characterisation Reveals Insights into Substrate Recognition by the Glutamine Transporter ASCT2/SLC1A5. Nat. Commun. 2018, 9, 38. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. ASCT-1 Is a Neutral Amino Acid Exchanger with Chloride Channel Activity. J. Biol. Chem. 1996, 271, 27991–27994. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Flux Coupling in a Neuronal Glutamate Transporter. Nature 1996, 383, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Clémençon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 High-Affinity Glutamate and Neutral Amino Acid Transporter Family. Mol. Asp. Med. 2013, 34, 108–120. [Google Scholar] [CrossRef]
- Grewer, C.; Grabsch, E. New Inhibitors for the Neutral Amino Acid Transporter ASCT2 Reveal Its Na+-Dependent Anion Leak. J. Physiol. 2004, 557, 747–759. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino Acid Transporters ASCT2 and LAT1 in Cancer: Partners in Crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Finger, R.E.; Onan, M.C.; Bode, B.P. ASCT2 Silencing Regulates Mammalian Target-of-Rapamycin Growth and Survival Signaling in Human Hepatoma Cells. Am. J. Physiol. Cell Physiol. 2007, 293, C55–C63. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional Transport of Amino Acids Regulates MTOR and Autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, C.; Rajapakshe, K.; Dong, J.; Shi, Y.; Tsouko, E.; Mukhopadhyay, R.; Jasso, D.; Dawood, W.; Coarfa, C.; et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol. Cancer Res. 2017, 15, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc Suppression of MiR-23a/b Enhances Mitochondrial Glutaminase Expression and Glutamine Metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Bröer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 Controls Glutamine Uptake and Tumour Growth in Triple-Negative Basal-like Breast Cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The Glutamine Transporter ASCT2 (SLC1A5) Promotes Tumor Growth Independently of the Amino Acid Transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological Blockade of ASCT2-Dependent Glutamine Transport Leads to Antitumor Efficacy in Preclinical Models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 Mediates Glutamine Transport Required for Lung Cancer Cell Growth and Survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef] [PubMed]
- Heimer, G.; Marek-Yagel, D.; Eyal, E.; Barel, O.; Oz Levi, D.; Hoffmann, C.; Ruzzo, E.K.; Ganelin-Cohen, E.; Lancet, D.; Pras, E.; et al. SLC1A4 Mutations Cause a Novel Disorder of Intellectual Disability, Progressive Microcephaly, Spasticity and Thin Corpus Callosum. Clin. Genet. 2015, 88, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Damseh, N.; Simonin, A.; Jalas, C.; Picoraro, J.A.; Shaag, A.; Cho, M.T.; Yaacov, B.; Neidich, J.; Al-Ashhab, M.; Juusola, J.; et al. Mutations in SLC1A4, Encoding the Brain Serine Transporter, Are Associated with Developmental Delay, Microcephaly and Hypomyelination. J. Med. Genet. 2015, 52, 541–547. [Google Scholar] [CrossRef]
- Srour, M.; Hamdan, F.F.; Gan-Or, Z.; Labuda, D.; Nassif, C.; Oskoui, M.; Gana-Weisz, M.; Orr-Urtreger, A.; Rouleau, G.A.; Michaud, J.L. A Homozygous Mutation in SLC1A4 in Siblings with Severe Intellectual Disability and Microcephaly. Clin. Genet. 2015, 88, E1–E4. [Google Scholar] [CrossRef]
- Ratz-Mitchem, M.L.; Leary, G.; Grindeland, A.; Silvius, D.; Guter, J.; Kavanaugh, M.P.; Gunn, T.M. Generation and Characterization of a Knock-in Mouse Model for Spastic Tetraplegia, Thin Corpus Callosum, and Progressive Microcephaly (SPATCCM). Mamm. Genome 2023, 34, 572–585. [Google Scholar] [CrossRef]
- Kaplan, E.; Zubedat, S.; Radzishevsky, I.; Valenta, A.C.; Rechnitz, O.; Sason, H.; Sajrawi, C.; Bodner, O.; Konno, K.; Esaki, K.; et al. ASCT1 (Slc1a4) Transporter Is a Physiologic Regulator of Brain d-Serine and Neurodevelopment. Proc. Natl. Acad. Sci. USA 2018, 115, 9628–9633. [Google Scholar] [CrossRef]
- Foster, A.C.; Farnsworth, J.; Lind, G.E.; Li, Y.X.; Yang, J.Y.; Dang, V.; Penjwini, M.; Viswanath, V.; Staubli, U.; Kavanaugh, M.P. d-Serine Is a Substrate for Neutral Amino Acid Transporters ASCT1/SLC1A4 and ASCT2/SLC1A5, and Is Transported by Both Subtypes in Rat Hippocampal Astrocyte Cultures. PLoS ONE 2016, 11, e0156551. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.; Artoul, S.; Segal, A.C.; Kolodney, G.; Radzishevsky, I.; Dikopoltsev, E.; Foltyn, V.N.; Inoue, R.; Mori, H.; Billard, J.M.; et al. Neuronal d-Serine and Glycine Release via the Asc-1 Transporter Regulates NMDA Receptor-Dependent Synaptic Activity. J. Neurosci. 2013, 33, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Balu, D.; Wolosker, H. d-Serine, the Shape-Shifting NMDA Receptor Co-Agonist. Neurochem. Res. 2020, 45, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Balu, D.T. d-Serine as the Gatekeeper of NMDA Receptor Activity: Implications for the Pharmacologic Management of Anxiety Disorders. Transl. Psychiatry 2020, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Orzylowski, M.; Fujiwara, E.; Mousseau, D.D.; Baker, G.B. An Overview of the Involvement of d-Serine in Cognitive Impairment in Normal Aging and Dementia. Front. Psychiatry 2021, 12, 754032. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, M.W.; Oliet, S.H.R.; Panatier, A. NMDARs, coincidence detectors of astrocytic and neuronal activities. Int. J. Mol. Sci. 2021, 22, 7258. [Google Scholar] [CrossRef]
- Perez, E.J.; Tapanes, S.A.; Loris, Z.B.; Balu, D.T.; Sick, T.J.; Coyle, J.T.; Liebl, D.J. Enhanced Astrocytic d-Serine Underlies Synaptic Damage after Traumatic Brain Injury. J. Clin. Investig. 2017, 127, 3114–3125. [Google Scholar] [CrossRef]
- Li, S.; Uno, Y.; Rudolph, U.; Cobb, J.; Liu, J.; Anderson, T.; Levy, D.; Balu, D.T.; Coyle, J.T. Astrocytes in Primary Cultures Express Serine Racemase, Synthesize d-Serine and Acquire A1 Reactive Astrocyte Features. Biochem. Pharmacol. 2018, 151, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Tapanes, S.A.; Arizanovska, D.; Díaz, M.M.; Folorunso, O.O.; Harvey, T.; Brown, S.E.; Radzishevsky, I.; Close, L.N.; Jagid, J.R.; Graciolli Cordeiro, J.; et al. Inhibition of Glial d-Serine Release Rescues Synaptic Damage after Brain Injury. Glia 2022, 70, 1133–1152. [Google Scholar] [CrossRef]
- Albers, T.; Marsiglia, W.; Thomas, T.; Gameiro, A.; Grewer, C. Defining Substrate and Blocker Activity of Alanine-Serine-Cysteine Transporter 2 (ASCT2) Ligands with Novel Serine Analogs. Mol. Pharmacol. 2012, 81, 356–365. [Google Scholar] [CrossRef]
- Schulte, M.L.; Khodadadi, A.B.; Cuthbertson, M.L.; Smith, J.A.; Manning, H.C. 2-Amino-4-Bis(Aryloxybenzyl)Aminobutanoic Acids: A Novel Scaffold for Inhibition of ASCT2-Mediated Glutamine Transport Dedicated to the Memory of Eric S. Dawson, Ph.D. Bioorg. Med. Chem. Lett. 2016, 26, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Nγ-Aryl Glutamine Analogues as Probes of the ASCT2 Neutral Amino Acid Transporter Binding Site. Bioorg Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Tanui, R.; Gameiro, A.; Eisenberg, G.; Colas, C.; Schlessinger, A.; Grewer, C. Structure Activity Relationships of Benzylproline-Derived Inhibitors of the Glutamine Transporter ASCT2. Bioorg Med. Chem. Lett. 2017, 27, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Ndaru, E.; Garibsingh, R.A.A.; Shi, Y.Y.; Wallace, E.; Zakrepine, P.; Wang, J.; Schlessinger, A.; Grewer, C. Novel Alanine Serine Cysteine Transporter 2 (ASCT2) Inhibitors Based on Sulfonamide and Sulfonic Acid Ester Scaffolds. J. Gen. Physiol. 2019, 151, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Pinilla-Tenas, J.; Barber, A.; Lostao, M.P. Transport of Proline and Hydroxyproline by the Neutral Amino-Acid Exchanger ASCT1. J. Membr. Biol. 2003, 195, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J.; Stanley, M.S.; Anderson, M.W.; Cotman, C.W.; Chamberlin, A.R. Conformationally Defined Neurotransmitter Analogs. Selective Inhibition of Glutamate Uptake by One Pyrrolidine-2,4-Dicarboxylate Diastereomer. J. Med. Chem. 1991, 34, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Kenneth Robinson, J.; Lee, V.; Claridge, T.D.W.; Baldwin, J.E.; Schofield, C.J. Synthesis of (2S, 3R, 4S), (2S, 3S, 4R)-Epoxyprolines and Aminohydroxyprolines. Tetrahedron 1998, 54, 981–996. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Kozlowski, J.; Wilhelm, R.S. Chemistry of Higher Order Mixed Organocuprates. 2. Reactions of Epoxides. J. Am. Chem. Soc. 1982, 104, 2305–2307. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Wilhelm, R.S.; Kozlowski, J.A.; Parker, D. Substitution Reactions of Secondary Halides and Epoxides with Higher Order, Mixed Organocuprates, R2Cu(CN)Li2: Synthetic, Stereochemical, and Mechanistic Aspects. J. Org. Chem. 1984, 49, 3928–3938. [Google Scholar] [CrossRef]
- Herdeis, C.; Aschenbrenner, A.; Kirfel, A.; Schwabenländer, F. Synthesis of and from S-Pyroglutamic Acid. Regio- and Diastereoselective Ring Opening of Its Derivatives. Tetrahedron Asymmetry 1997, 8, 2421–2432. [Google Scholar] [CrossRef]
- Sajiki, H. Selective Inhibition of Benzyl Ether Hydrogenolysis with Pd/C Due to the Presence of Ammonia, Pyridine or Ammonium Acetate. Tetrahedron Lett. 1995, 36, 3465–3468. [Google Scholar] [CrossRef]
- Sajiki, H.; Hattori, K.; Hirota, K. The Formation of a Novel Pd/C−Ethylenediamine Complex Catalyst: Chemoselective Hydrogenation without Deprotection of the O. -Benzyl and N. -Cbz Groups. J. Org. Chem. 1998, 63, 7990–7992. [Google Scholar] [CrossRef]
- Boudker, O.; Ryan, R.M.; Yernool, D.; Shimamoto, K.; Gouaux, E. Coupling Substrate and Ion Binding to Extracellular Gate of a Sodium-Dependent Aspartate Transporter. Nature 2007, 445, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Canul-Tec, J.C.; Assal, R.; Cirri, E.; Legrand, P.; Brier, S.; Chamot-Rooke, J.; Reyes, N. Structure and Allosteric Inhibition of Excitatory Amino Acid Transporter 1. Nature 2017, 544, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Leary, G.P.; Holley, D.C.; Stone, E.F.; Lyda, B.R.; Kalachev, L.V.; Kavanaugh, M.P. The Central Cavity in Trimeric Glutamate Transporters Restricts Ligand Diffusion. Proc. Natl. Acad. Sci. USA 2011, 108, 14980–14985. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.-M.; Taly, J.-F.; Notredame, C. T-Coffee: A Web Server for the Multiple Sequence Alignment of Protein and RNA Sequences Using Structural Information and Homology Extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The Role of ASCT2 in Cancer: A Review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, K.; Sakai, R.; Takaoka, K.; Yumoto, N.; Nakajima, T.; Amara, S.G.; Shigeri, Y. Characterization of Novel l-Threo -β-Benzyloxyaspartate Derivatives, Potent Blockers of the Glutamate Transporters. Mol. Pharmacol. 2004, 65, 1008–1015. [Google Scholar] [CrossRef]
- Greenfield, A.; Grosanu, C.; Dunlop, J.; McIlvain, B.; Carrick, T.; Jow, B.; Lu, Q.; Kowal, D.; Williams, J.; Butera, J. Synthesis and Biological Activities of Aryl-Ether-, Biaryl-, and Fluorene-Aspartic Acid and Diaminopropionic Acid Analogs as Potent Inhibitors of the High-Affinity Glutamate Transporter EAAT-2. Bioorg Med. Chem. Lett. 2005, 15, 4985–4988. [Google Scholar] [CrossRef]
- Garaeva, A.A.; Oostergetel, G.T.; Gati, C.; Guskov, A.; Paulino, C.; Slotboom, D.J. Cryo-EM Structure of the Human Neutral Amino Acid Transporter ASCT2. Nat. Struct. Mol. Biol. 2018, 25, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Garaeva, A.A.; Guskov, A.; Slotboom, D.J.; Paulino, C. A One-Gate Elevator Mechanism for the Human Neutral Amino Acid Transporter ASCT2. Nat. Commun. 2019, 10, 3427. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Plotnikova, O.; Bonin, P.D.; Subashi, T.A.; McLellan, T.J.; Dumlao, D.; Che, Y.; Dong, Y.Y.; Carpenter, E.P.; West, G.M.; et al. Cryo-EM Structures of the Human Glutamine Transporter SLC1A5 (ASCT2) in the Outward-Facing Conformation. eLife 2019, 8, e48120. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Grewer, C.; Otte, N.J.; Gameiro, A.; Albers, T.; Singh, K.; Shere, H.; Bonomi, M.; Holst, J.; Schlessinger, A. Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput. Biol. 2015, 11, e1004477. [Google Scholar] [CrossRef] [PubMed]
- Garibsingh, R.-A.A.; Ndaru, E.; Garaeva, A.A.; Shi, Y.; Zielewicz, L.; Zakrepine, P.; Bonomi, M.; Slotboom, D.J.; Paulino, C.; Grewer, C.; et al. Rational Design of ASCT2 Inhibitors Using an Integrated Experimental-Computational Approach. Proc. Natl. Acad. Sci. USA 2021, 118, e2104093118. [Google Scholar] [CrossRef] [PubMed]
- Lyda, B.R.; Natale, N.R.; Esslinger, C.S.; Kavanaugh, M.P. Novel Inhibitors of the Amino Acid Transporters ASCT1 and ASCT2. US20130065935A1, 14 March 2013. [Google Scholar]
- Lyda, B.R. Synthesis of N-β-Aryl-Aspartamides, N-α-Arylamide-Aspartates, and Hydroxy-l-Proline Derivatives as Inhibitors of Amino Acid for Evaluating the Glutamine / Glutamate Cycle. Ph.D. Dissertation, The University of Montana, Missoula, MT, USA, 11 February 2011. [Google Scholar]
- Bendahan, A.; Armon, A.; Madani, N.; Kavanaugh, M.P.; Kanner, B.I. Arginine 447 Plays a Pivotal Role in Substrate Interactions in a Neuronal Glutamate Transporter. J. Biol. Chem. 2000, 275, 37436–37442. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Fairweather, S.; Bröer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- van Geldermalsen, M.; Quek, L.-E.; Turner, N.; Freidman, N.; Pang, A.; Guan, Y.F.; Krycer, J.R.; Ryan, R.; Wang, Q.; Holst, J. Benzylserine Inhibits Breast Cancer Cell Growth by Disrupting Intracellular Amino Acid Homeostasis and Triggering Amino Acid Response Pathways. BMC Cancer 2018, 18, 689. [Google Scholar] [CrossRef]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef]
- Chiu, M.; Sabino, C.; Taurino, G.; Bianchi, M.G.; Andreoli, R.; Giuliani, N.; Bussolati, O. GPNA Inhibits the Sodium-Independent Transport System l for Neutral Amino Acids. Amino Acids 2017, 49, 1365–1372. [Google Scholar] [CrossRef]
- Freidman, N.J.; Briot, C.; Ryan, R.M. Characterizing Unexpected Interactions of a Glutamine Transporter Inhibitor with Members of the SLC1A Transporter Family. J. Biol. Chem. 2022, 298, 102178. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Dominici, S.; Piaggi, S.; Belcastro, E.; Chiu, M.; Taurino, G.; Pacini, S.; Bussolati, O.; Pompella, A. γ-Glutamyltransferase Enzyme Activity of Cancer Cells Modulates l-γ-Glutamyl-p-Nitroanilide (GPNA) Cytotoxicity. Sci. Rep. 2019, 9, 891. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Coles, G.; Vennitti, J.; Zhang, Z.; Comb, W.C.; Xing, S.; Javed, K.; Bröer, A.; Bröer, S. Quantitative Modelling of Amino Acid Transport and Homeostasis in Mammalian Cells. Nat. Commun. 2021, 12, 5282. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | Structure | SLC1A4 Ki (µM) | SLC1A5 Ki (µM) | Compound No. | Structure | SLC1A4 Ki (µM) | SLC1A5 Ki (µM) |
|---|---|---|---|---|---|---|---|
| 1 | 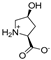 | 805 ± 148 | 872 ± 201 | 14a | 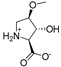 | 214 ± 32 | 235 ± 69 |
| 26 |  | No Block | 926 ± 252 | 14b |  | 237 ± 40 | 268 ± 70 |
| 27 |  | No Block | 1077 ± 322 | 14c | 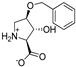 | 39 ± 5 | 69 ± 7 |
| 10d |  | 332 ± 74 | 1128 ± 318 | 17a | 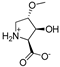 | 481 ± 77 | 600 ± 176 |
| 10c | 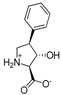 | ~600 | ~600 | 17b | 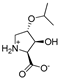 | 505 ± 105 | ~400 |
| 10b | 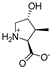 | 564 ± 149 | 922 ± 316 | 17c |  | 493 ± 61 | 828 ± 248 |
| 10a |  | 1050 ± 110 | 1149 ± 283 | 14d | 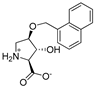 | ~10 | ~10 |
| 12a | 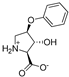 | 150 ± 43 | 141 ± 20 | 14e | 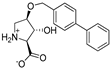 | 0.31 ± 0.04 | 0.24 ± 0.04 |
| 12b | 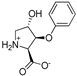 | 594 ± 191 | 502 ± 110 | 14f | 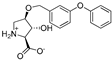 | 13 ± 2 | 10 ± 2 |
| 12c |  | ~150 | ~150 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyda, B.R.; Leary, G.P.; Farnsworth, J.; Seaver, B.; Silvius, D.; Kavanaugh, M.P.; Esslinger, C.S.; Natale, N.R. Discovery and Synthesis of Hydroxy-l-Proline Blockers of the Neutral Amino Acid Transporters SLC1A4 (ASCT1) and SLC1A5 (ASCT2). Molecules 2024, 29, 2330. https://doi.org/10.3390/molecules29102330
Lyda BR, Leary GP, Farnsworth J, Seaver B, Silvius D, Kavanaugh MP, Esslinger CS, Natale NR. Discovery and Synthesis of Hydroxy-l-Proline Blockers of the Neutral Amino Acid Transporters SLC1A4 (ASCT1) and SLC1A5 (ASCT2). Molecules. 2024; 29(10):2330. https://doi.org/10.3390/molecules29102330
Chicago/Turabian StyleLyda, Brent R., Gregory P. Leary, Jill Farnsworth, Benjamin Seaver, Derek Silvius, Michael P. Kavanaugh, C. Sean Esslinger, and Nicholas R. Natale. 2024. "Discovery and Synthesis of Hydroxy-l-Proline Blockers of the Neutral Amino Acid Transporters SLC1A4 (ASCT1) and SLC1A5 (ASCT2)" Molecules 29, no. 10: 2330. https://doi.org/10.3390/molecules29102330
APA StyleLyda, B. R., Leary, G. P., Farnsworth, J., Seaver, B., Silvius, D., Kavanaugh, M. P., Esslinger, C. S., & Natale, N. R. (2024). Discovery and Synthesis of Hydroxy-l-Proline Blockers of the Neutral Amino Acid Transporters SLC1A4 (ASCT1) and SLC1A5 (ASCT2). Molecules, 29(10), 2330. https://doi.org/10.3390/molecules29102330