
Molecular Mechanism of Interaction between DNA Aptamer and Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus 2 Variants Revealed by Steered Molecular Dynamics Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
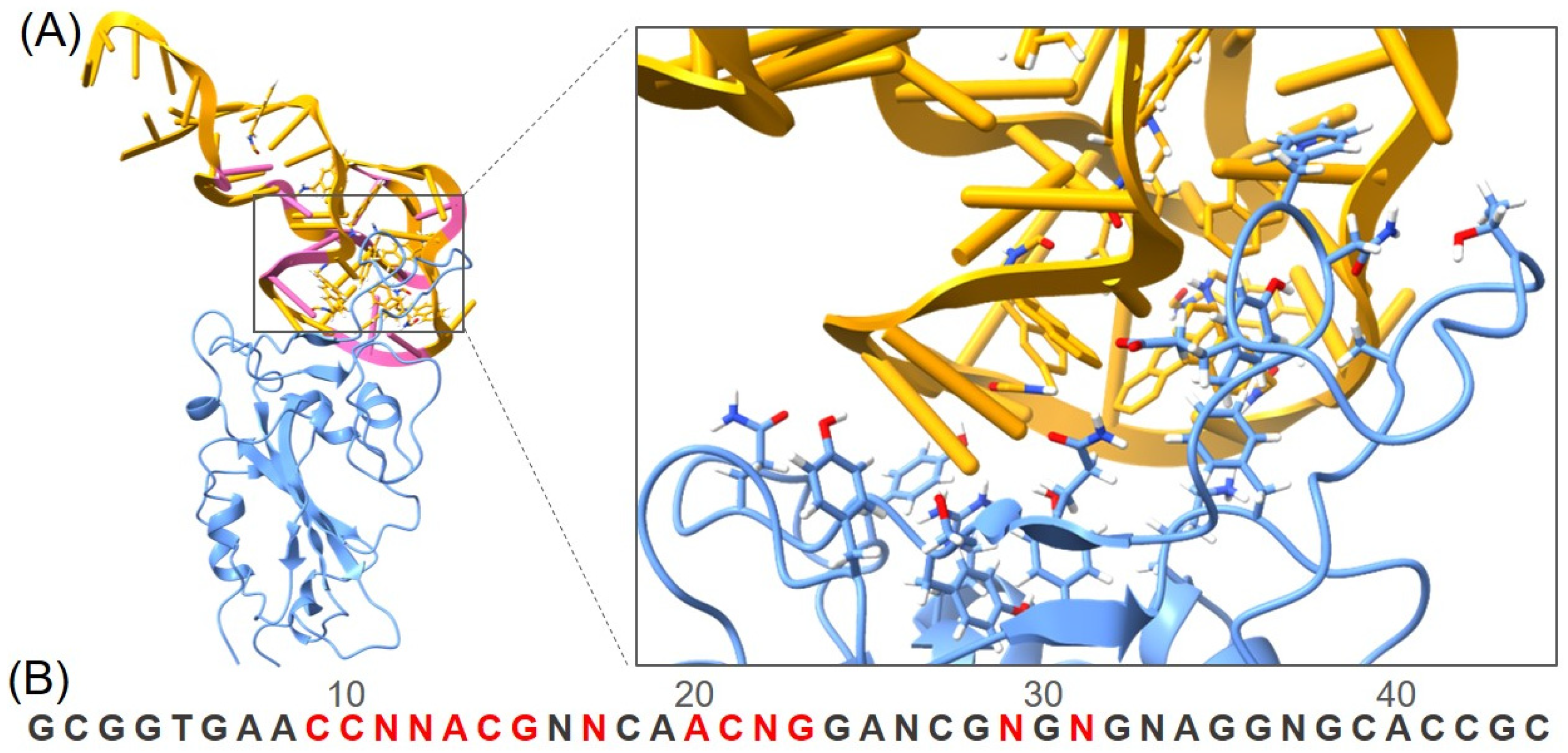
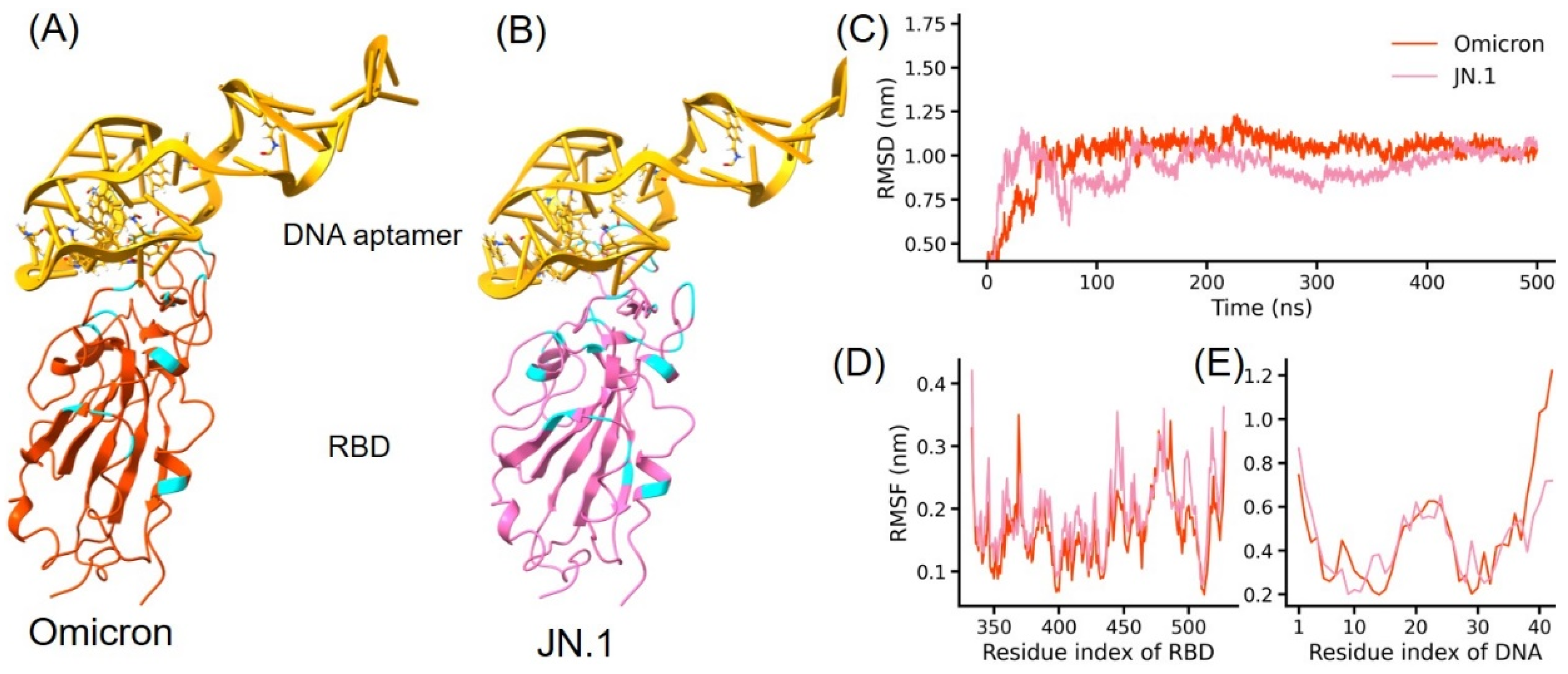
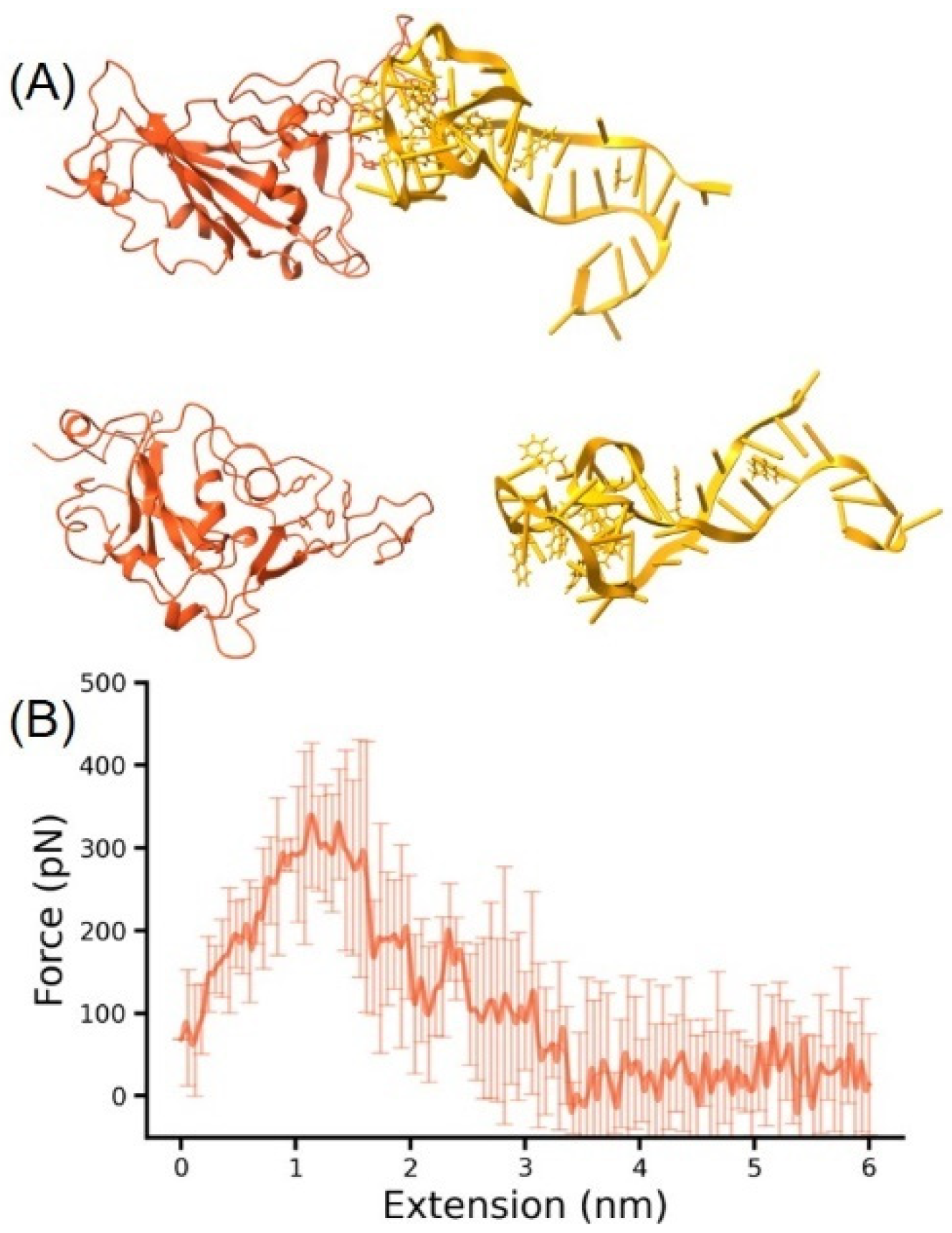
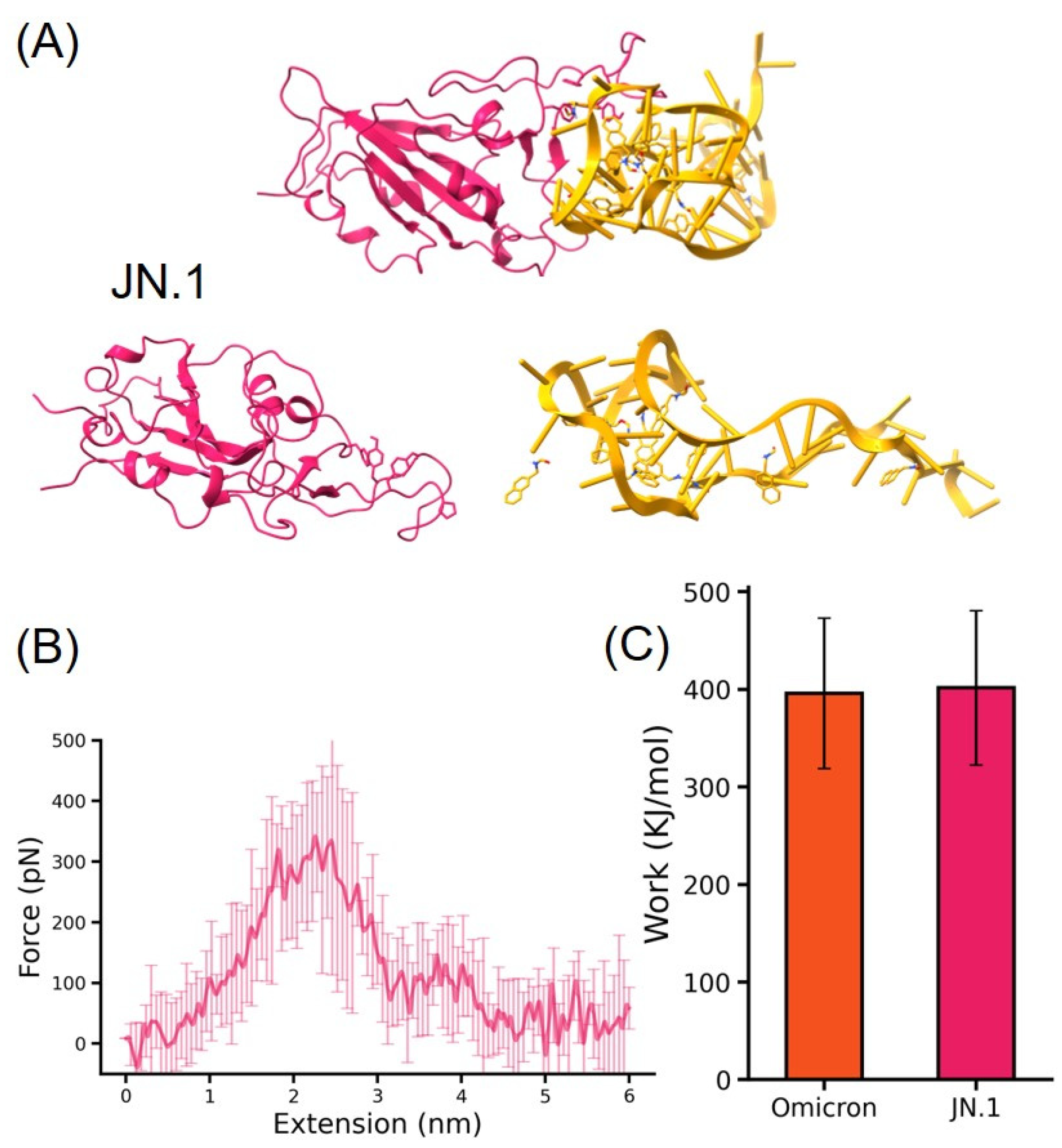
2. Result and Discussion
3. Materials and Methods
3.1. Structure Modeling
3.2. MD Simulation Force Field Preparation
3.3. MD Simulation for RBD-DNA Aptamer Unbinding
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.-M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Elliott, P.; Bodinier, B.; Eales, O.; Wang, H.; Haw, D.; Elliott, J.; Whitaker, M.; Jonnerby, J.; Tang, D.; Walters, C.E.; et al. Rapid increase in Omicron infections in England during December 2021: REACT-1 study. Science 2022, 375, eabn8347. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and functional impact by SARS-CoV-2 Omicron spike mutations. Cell Rep. 2022, 39, 110729. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; Hinay, A.A., Jr.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J.; et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect. Dis. 2024, 24, e82. [Google Scholar] [CrossRef]
- Wang, B.; Guo, C.; Chen, G.; Park, B.; Xu, B. Following aptamer–ricin specific binding by single molecule recognition and force spectroscopy measurements. Chem. Commun. 2012, 48, 1644–1646. [Google Scholar] [CrossRef]
- Sun, M.; Wu, Z.; Zhang, J.; Chen, M.; Lu, Y.; Yang, C.; Song, Y. Spherical neutralizing aptamer suppresses SARS-CoV-2 Omicron escape. Nano Today 2022, 44, 101499. [Google Scholar] [CrossRef]
- Ji, C.; Wei, J.; Zhang, L.; Hou, X.; Tan, J.; Yuan, Q.; Tan, W. Aptamer–Protein Interactions: From Regulation to Biomolecular Detection. Chem. Rev. 2023, 123, 12471–12506. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, Z.; Liu, D.; Jiang, H.; Zhang, Z.-K.; Lu, A.; Zhang, B.-T.; Yu, Y.; Zhang, G. Structural Biology for the Molecular Insight between Aptamers and Target Proteins. Int. J. Mol. Sci. 2021, 22, 4093. [Google Scholar] [CrossRef]
- Adachi, T.; Nakamura, Y. Aptamers: A Review of Their Chemical Properties and Modifications for Therapeutic Application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.-L.; Wu, J.; Qi, J.; Zeng, Z.; Wan, Q.; Chen, Z.; Manandhar, P.; Cavener, V.S.; Boyle, N.R.; et al. Neutralizing Aptamers Block S/RBD-ACE2 Interactions and Prevent Host Cell Infection. Angew. Chem. Int. Ed. 2021, 60, 10273–10278. [Google Scholar] [CrossRef]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the Chemistry of DNA for in Vitro Selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar] [CrossRef]
- Kacherovsky, N.; Yang, L.F.; Dang, H.V.; Cheng, E.L.; Cardle, I.I.; Walls, A.C.; McCallum, M.; Sellers, D.L.; DiMaio, F.; Salipante, S.J.; et al. Discovery and Characterization of Spike N-Terminal Domain-Binding Aptamers for Rapid SARS-CoV-2 Detection. Angew. Chem. Int. Ed. 2021, 60, 21211–21215. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, B.; Ding, X.; Zheng, P. Probing nanomechanical interactions of SARS-CoV-2 variants Omicron and XBB with common surfaces. Chem. Commun. 2023, 59, 11268–11271. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Brooks, C.L.; MacKerell, A.D.; Post, C.B.; Nilsson, L. Biomolecular dynamics in the 21st century. Biochim. Biophys. Acta. Gen. Subj. 2024, 1868, 130534. [Google Scholar] [CrossRef] [PubMed]
- Sang, P.; Chen, Y.-Q.; Liu, M.-T.; Wang, Y.-T.; Yue, T.; Li, Y.; Yin, Y.-R.; Yang, L.-Q. Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants. Int. J. Mol. Sci. 2022, 23, 14796. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.-P.; Yang, Y.-C.; Chen, H.-Y. Unraveling the binding mechanisms of SARS-CoV-2 variants through molecular simulations. Heliyon 2024, 10, e27193. [Google Scholar] [CrossRef]
- Pederzoli, M.; Wasif Baig, M.; Kývala, M.; Pittner, J.; Cwiklik, L. Photophysics of BODIPY-Based Photosensitizer for Photodynamic Therapy: Surface Hopping and Classical Molecular Dynamics. J. Chem. Theory Comput. 2019, 15, 5046–5057. [Google Scholar] [CrossRef]
- Morriss-Andrews, A.; Shea, J.-E. Simulations of Protein Aggregation: Insights from Atomistic and Coarse-Grained Models. J. Phys. Chem. Lett. 2014, 5, 1899–1908. [Google Scholar] [CrossRef]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Durner, E.; Schoeler, C.; Malinowska, K.H.; Carvalho, B.G.; Bayer, E.A.; Luthey-Schulten, Z.; Gaub, H.E.; Nash, M.A. Mechanisms of Nanonewton Mechanostability in a Protein Complex Revealed by Molecular Dynamics Simulations and Single-Molecule Force Spectroscopy. J. Am. Chem. Soc. 2019, 141, 14752–14763. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, D.; Yang, W.T.; Marszalek, P.E. Piecewise All-Atom SMD Simulations Reveal Key Secondary Structures in Luciferase Unfolding Pathway. Biophy. J. 2020, 119, 2251–2261. [Google Scholar] [CrossRef]
- Tian, F.; Li, G.; Zheng, B.; Liu, Y.; Shi, S.; Deng, Y.; Zheng, P. Verification of sortase for protein conjugation by single-molecule force spectroscopy and molecular dynamics simulations. Chem. Commun. 2020, 56, 3943–3946. [Google Scholar] [CrossRef]
- Zheng, B.; Xiao, Y.; Tong, B.; Mao, Y.; Ge, R.; Tian, F.; Dong, X.; Zheng, P. S373P Mutation Stabilizes the Receptor-Binding Domain of the Spike Protein in Omicron and Promotes Binding. JACS Au 2023, 3, 1902–1910. [Google Scholar] [CrossRef]
- Gunnoo, M.; Cazade, P.-A.; Orlowski, A.; Chwastyk, M.; Liu, H.; Ta, D.T.; Cieplak, M.; Nash, M.; Thompson, D. Steered molecular dynamics simulations reveal the role of Ca2+ in regulating mechanostability of cellulose-binding proteins. Phys. Chem. Chem. Phys. 2018, 20, 22674–22680. [Google Scholar] [CrossRef] [PubMed]
- Rico, F.; Gonzalez, L.; Casuso, I.; Puig-Vidal, M.; Scheuring, S. High-Speed Force Spectroscopy Unfolds Titin at the Velocity of Molecular Dynamics Simulations. Science 2013, 342, 741–743. [Google Scholar] [CrossRef]
- Zhao, P.; Xu, C.-Q.; Sun, C.; Xia, J.; Sun, L.; Li, J.; Xu, H. Exploring the difference of bonding strength between silver(i) and chalcogenides in block copolymer systems. Polym. Chem. 2020, 11, 7087–7093. [Google Scholar] [CrossRef]
- Chen, G.; Wang, H.; Bumba, L.; Masin, J.; Sebo, P.; Li, H. The adenylate cyclase toxin RTX domain follows a series templated folding mechanism with implications for toxin activity. J. Biol. Chem. 2023, 299, 105150. [Google Scholar] [CrossRef] [PubMed]
- Baier, D.; Müller, T.; Mohr, T.; Windberger, U. Red Blood Cell Stiffness and Adhesion Are Species-Specific Properties Strongly Affected by Temperature and Medium Changes in Single Cell Force Spectroscopy. Molecules 2021, 26, 2771. [Google Scholar] [CrossRef] [PubMed]
- Sen Mojumdar, S.; Scholl, Z.N.; Dee, D.R.; Rouleau, L.; Anand, U.; Garen, C.; Woodside, M.T. Partially native intermediates mediate misfolding of SOD1 in single-molecule folding trajectories. Nat. Commun. 2017, 8, 1881. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Caballero, A.; Echelman, D.J.; Tapia-Rojo, R.; Haldar, S.; Eckels, E.C.; Fernandez, J.M. Protein folding modulates the chemical reactivity of a Gram-positive adhesin. Nat. Chem. 2021, 13, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.S.; Gruber, S.; Hausch, A.; Gomes, P.; Milles, L.F.; Nicolaus, T.; Schendel, L.C.; Navajas, P.L.; Procko, E.; Lietha, D.; et al. A tethered ligand assay to probe SARS-CoV-2:ACE2 interactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2114397119. [Google Scholar] [CrossRef]
- Bauer, M.S.; Gruber, S.; Hausch, A.; Melo, M.C.R.; Gomes, P.S.F.C.; Nicolaus, T.; Milles, L.F.; Gaub, H.E.; Bernardi, R.C.; Lipfert, J. Single-molecule force stability of the SARS-CoV-2–ACE2 interface in variants-of-concern. Nat. Nanotechnol. 2024, 19, 399–405. [Google Scholar] [CrossRef]
- Zhu, R.; Canena, D.; Sikora, M.; Klausberger, M.; Seferovic, H.; Mehdipour, A.R.; Hain, L.; Laurent, E.; Monteil, V.; Wirnsberger, G.; et al. Force-tuned avidity of spike variant-ACE2 interactions viewed on the single-molecule level. Nat. Commun. 2022, 13, 7926. [Google Scholar] [CrossRef]
- Köhler, M.; Karner, A.; Leitner, M.; Hytönen, V.P.; Kulomaa, M.; Hinterdorfer, P.; Ebner, A. pH-Dependent Deformations of the Energy Landscape of Avidin-like Proteins Investigated by Single Molecule Force Spectroscopy. Molecules 2014, 19, 12531–12546. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Tian, F.; Zheng, P. Interdomain Linker Effect on the Mechanical Stability of Ig Domains in Titin. Int. J. Mol. Sci. 2022, 23, 9836. [Google Scholar] [CrossRef]
- Suay-Corredera, C.; Pricolo, M.R.; Velazquez-Carreras, D.; Pathak, D.; Nandwani, N.; Pimenta-Lopes, C.; Sanchez-Ortiz, D.; Urrutia-Irazabal, I.; Vilches, S.; Dominguez, F.; et al. Nanomechanical Phenotypes in Cardiac Myosin-Binding Protein C Mutants That Cause Hypertrophic Cardiomyopathy. Acs Nano 2021, 15, 10203–10216. [Google Scholar] [CrossRef]
- Lei, H.; Ma, Q.; Li, W.; Wen, J.; Ma, H.; Qin, M.; Wang, W.; Cao, Y. An ester bond underlies the mechanical strength of a pathogen surface protein. Nat. Commun. 2021, 12, 5082. [Google Scholar] [CrossRef]
- Zhang, S.; Qian, H.-J.; Liu, Z.; Ju, H.; Lu, Z.-Y.; Zhang, H.; Chi, L.; Cui, S. Towards Unveiling the Exact Molecular Structure of Amorphous Red Phosphorus by Single-Molecule Studies. Angew. Chem. Int. Ed. 2019, 58, 1659–1663. [Google Scholar] [CrossRef]
- Shi, S.; Wang, Z.; Deng, Y.; Tian, F.; Wu, Q.; Zheng, P. Combination of Click Chemistry and Enzymatic Ligation for Stable and Efficient Protein Immobilization for Single-Molecule Force Spectroscopy. CCS Chem. 2022, 4, 598–604. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Z.; Yang, Z.; Li, G.; Zheng, P. pH-Dependent PdS3 Site in α3DIV Revealed by Single-Molecule Force Spectroscopy. J. Phys. Chem. B 2023, 127, 2934–2940. [Google Scholar] [CrossRef]
- Yuan, G.; Ma, Q.; Wu, T.; Wang, M.; Li, X.; Zuo, J.; Zheng, P. Multistep Protein Unfolding Scenarios from the Rupture of a Complex Metal Cluster Cd3S9. Sci. Rep. 2019, 9, 10518–10525. [Google Scholar] [CrossRef]
- Song, G.; Tian, F.; Liu, H.; Li, G.; Zheng, P. Pioglitazone Inhibits Metal Cluster Transfer of mitoNEET by Stabilizing the Labile Fe–N Bond Revealed at Single-Bond Level. J. Phys. Chem. Lett. 2021, 12, 3860–3867. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2015, 12, 405–413. [Google Scholar] [CrossRef]
- Ravikumar, Y.; Koonyosying, P.; Srichairatanakool, S.; Ponpandian, L.N.; Kumaravelu, J.; Srichairatanakool, S. In Silico Molecular Docking and Dynamics Simulation Analysis of Potential Histone Lysine Methyl Transferase Inhibitors for Managing β-Thalassemia. Molecules 2023, 28, 7266. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, X.; Xu, C.; Zheng, B.; Yu, H.; Zheng, P. Molecular Mechanism of Interaction between DNA Aptamer and Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus 2 Variants Revealed by Steered Molecular Dynamics Simulations. Molecules 2024, 29, 2215. https://doi.org/10.3390/molecules29102215
Ding X, Xu C, Zheng B, Yu H, Zheng P. Molecular Mechanism of Interaction between DNA Aptamer and Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus 2 Variants Revealed by Steered Molecular Dynamics Simulations. Molecules. 2024; 29(10):2215. https://doi.org/10.3390/molecules29102215
Chicago/Turabian StyleDing, Xuan, Chao Xu, Bin Zheng, Hanyang Yu, and Peng Zheng. 2024. "Molecular Mechanism of Interaction between DNA Aptamer and Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus 2 Variants Revealed by Steered Molecular Dynamics Simulations" Molecules 29, no. 10: 2215. https://doi.org/10.3390/molecules29102215
APA StyleDing, X., Xu, C., Zheng, B., Yu, H., & Zheng, P. (2024). Molecular Mechanism of Interaction between DNA Aptamer and Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus 2 Variants Revealed by Steered Molecular Dynamics Simulations. Molecules, 29(10), 2215. https://doi.org/10.3390/molecules29102215