
Influence of Equatorial Co-Ligands on the Reactivity of LFeIIIOIPh

Abstract
:
1. Introduction
2. Results and Discussion
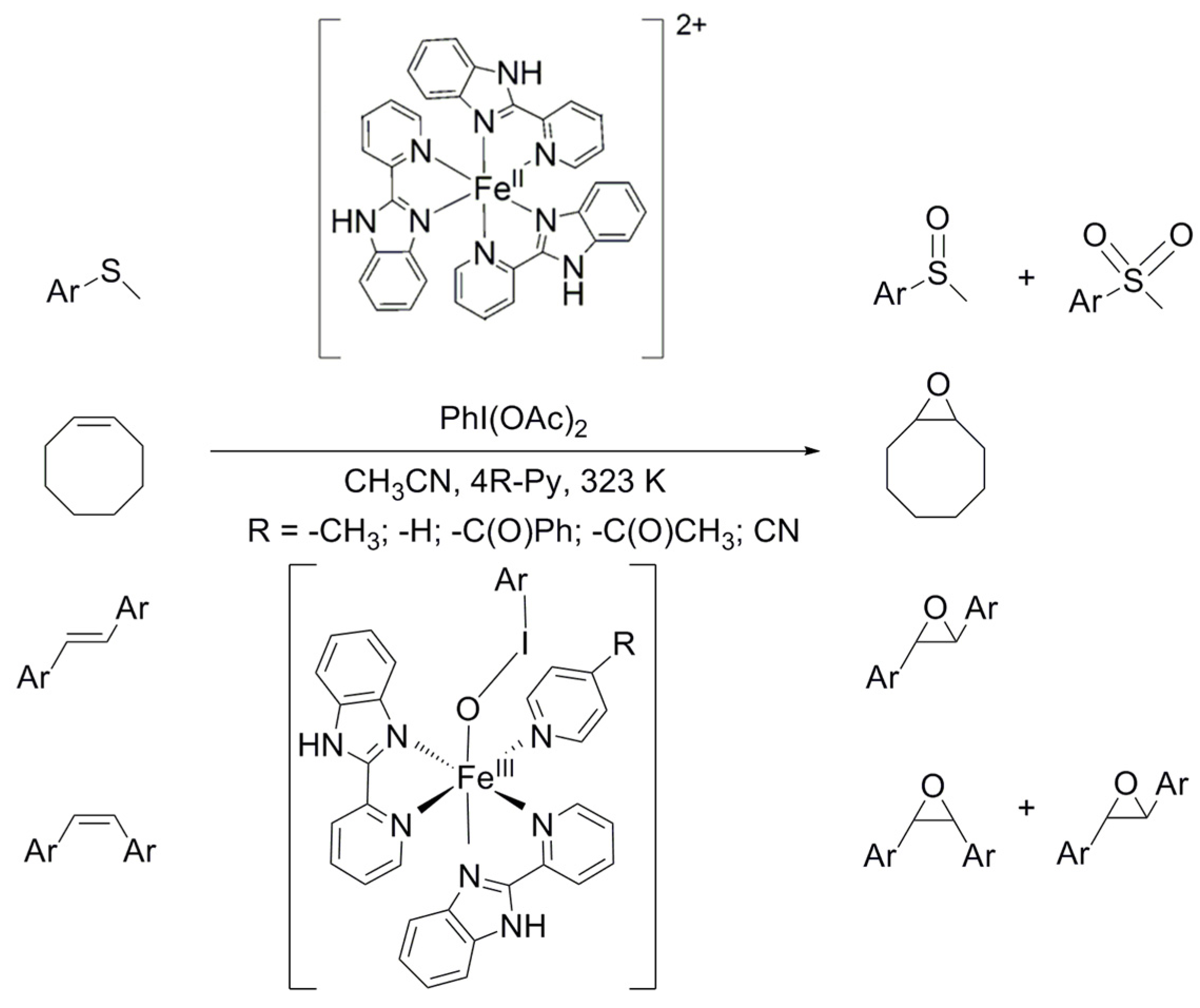
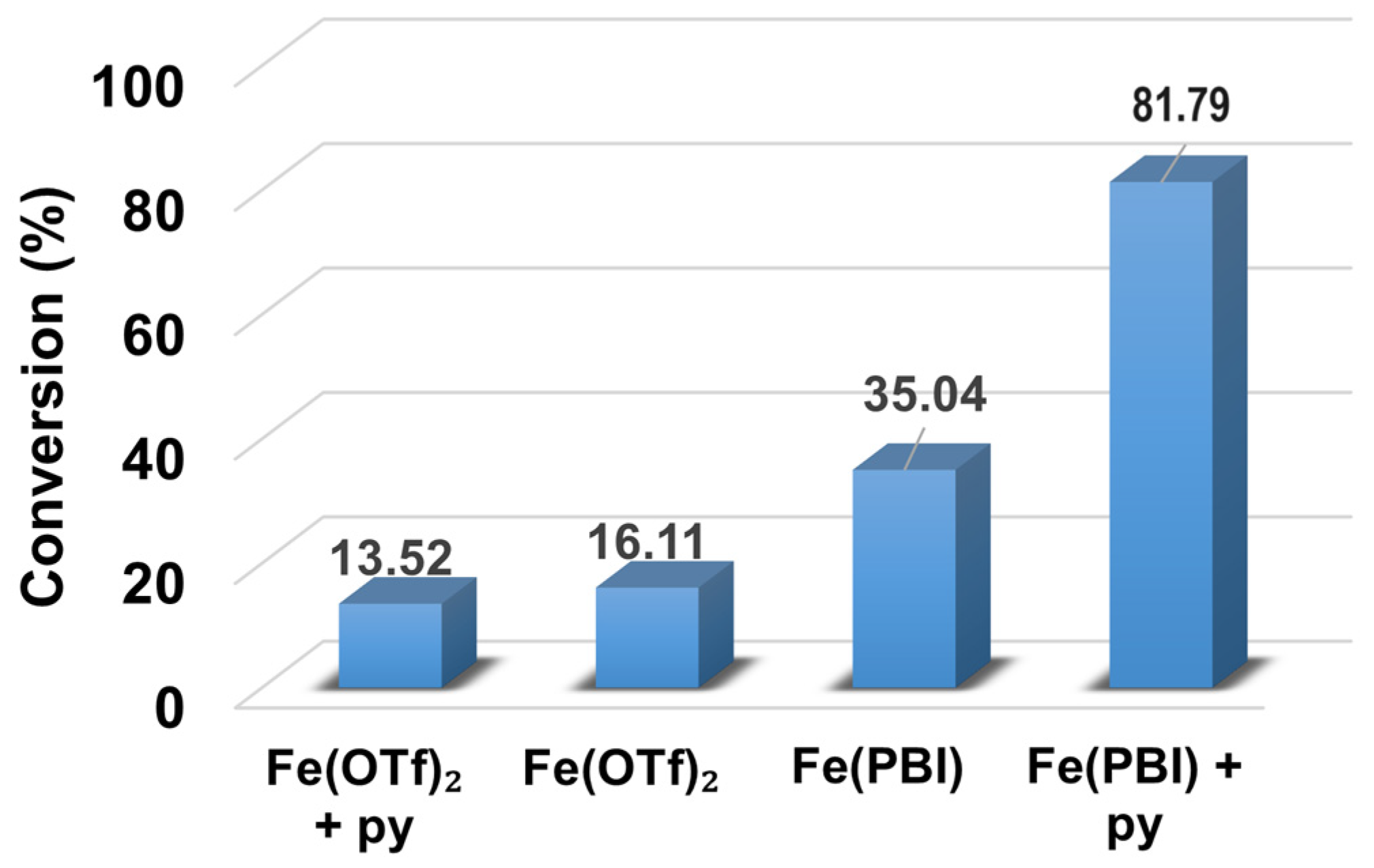
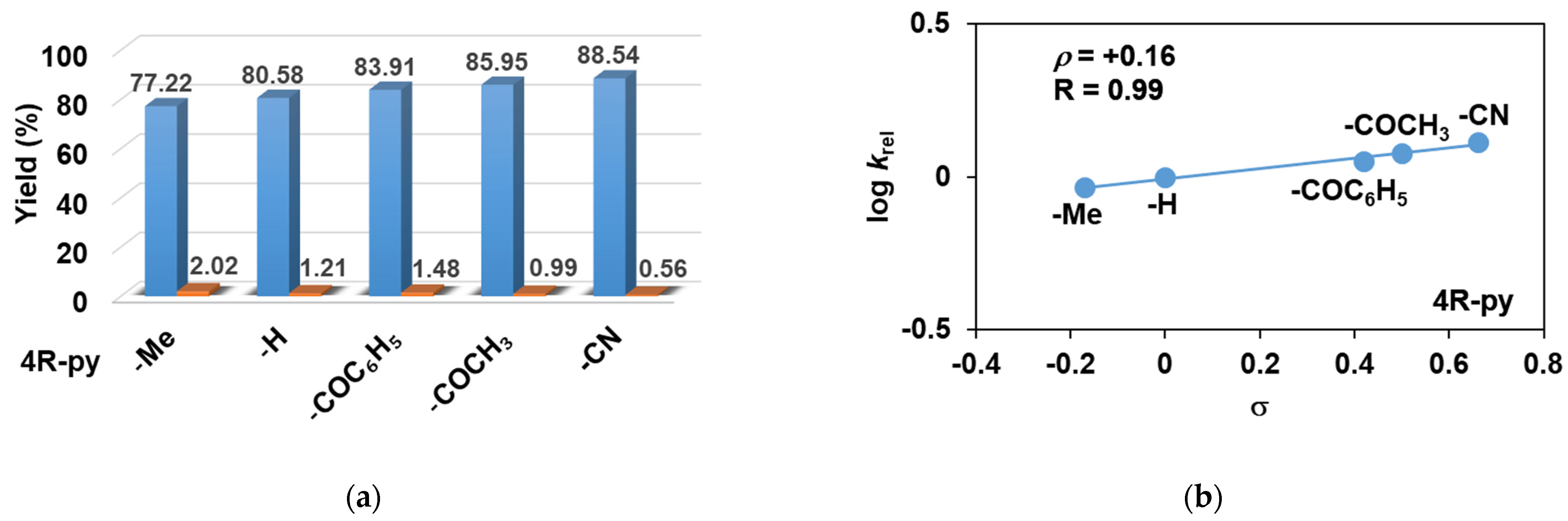
2.1. Influence of Equatorial Ligands on the FeIIIOIPh-Mediated Catalytic Sulfoxidation Reactions
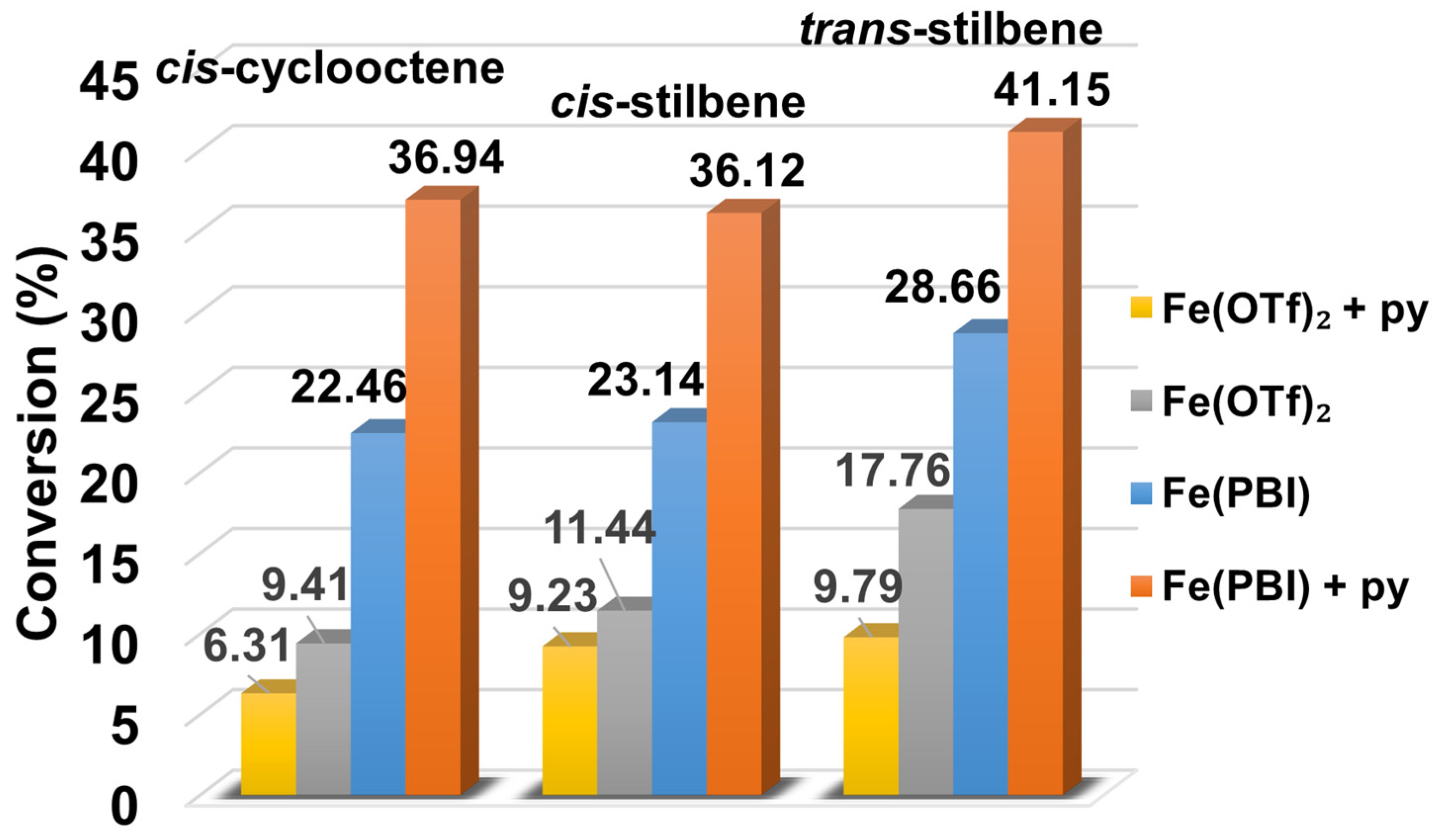
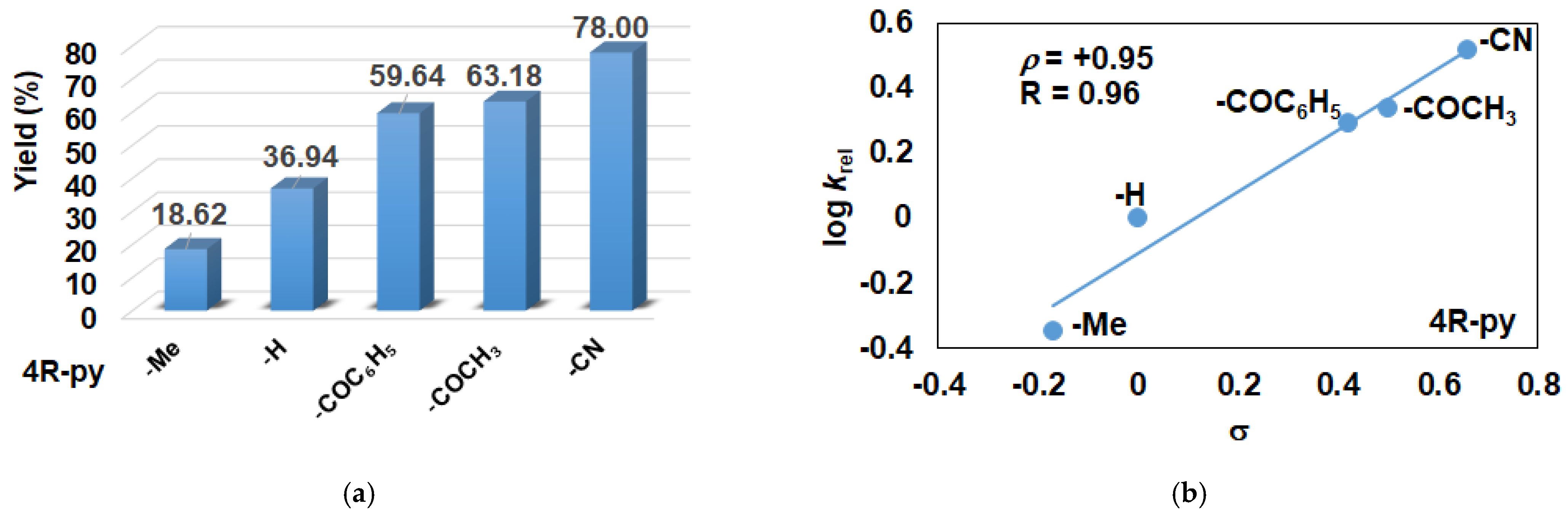
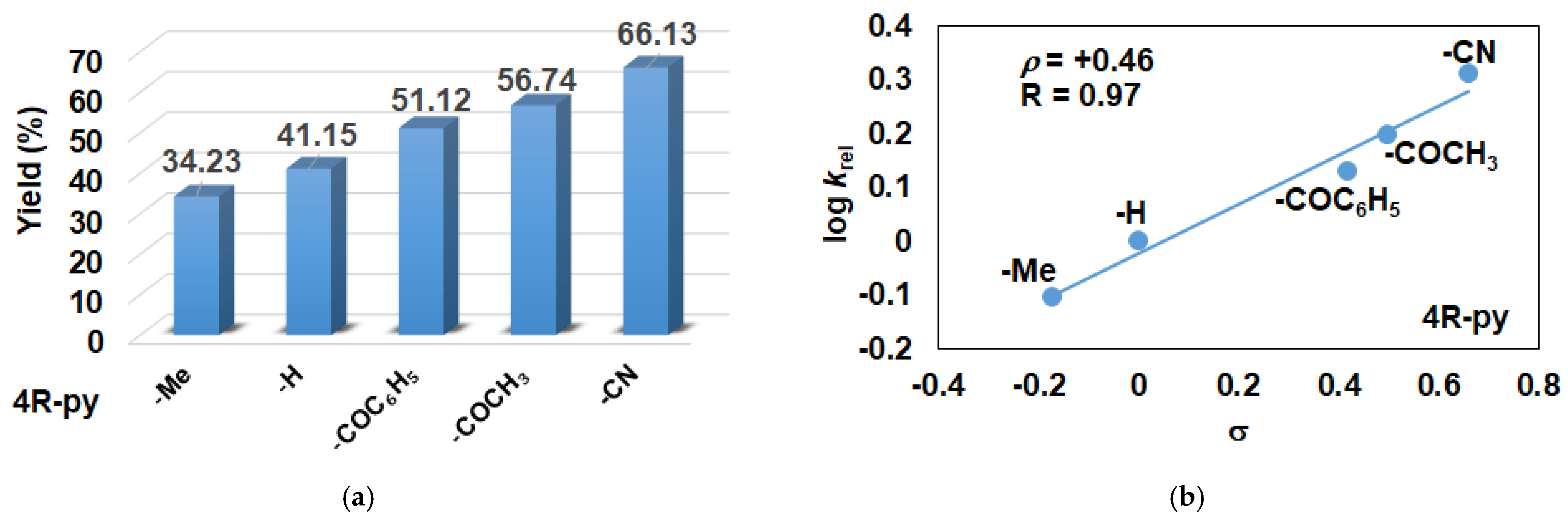
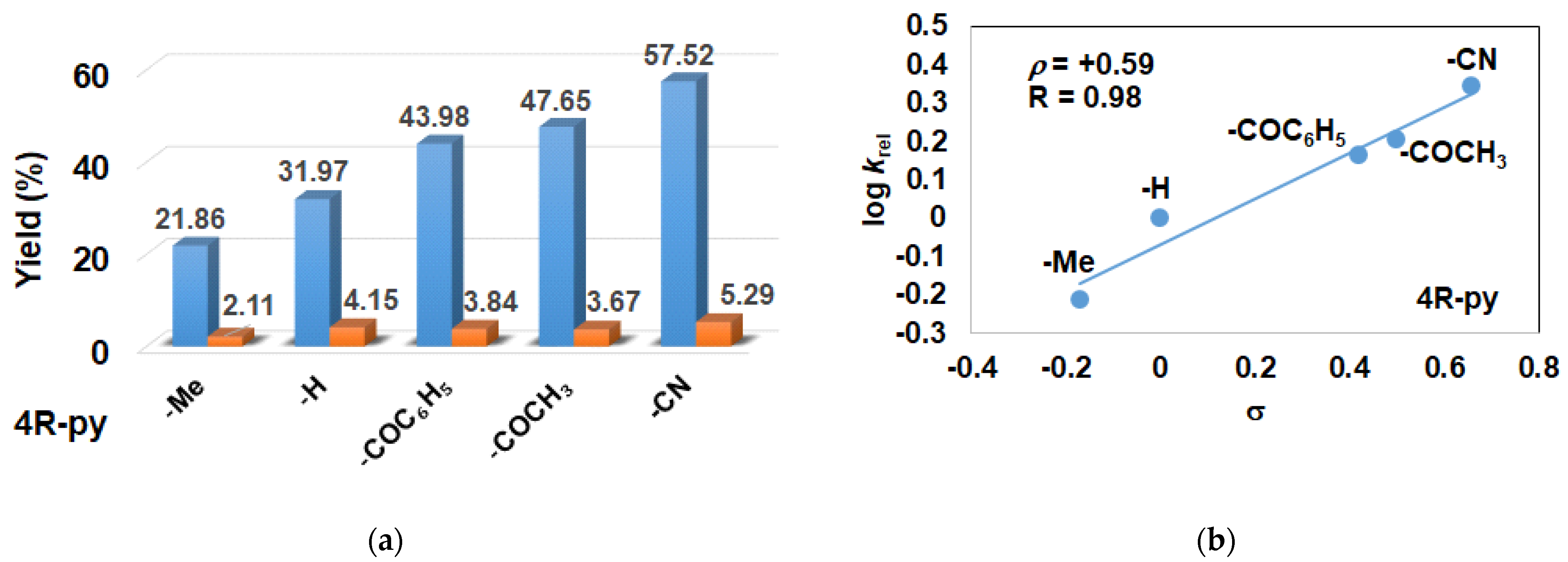
2.2. Influence of Equatorial Ligands on the FeIIIOIPh-Mediated Catalytic Epoxidation Reactions
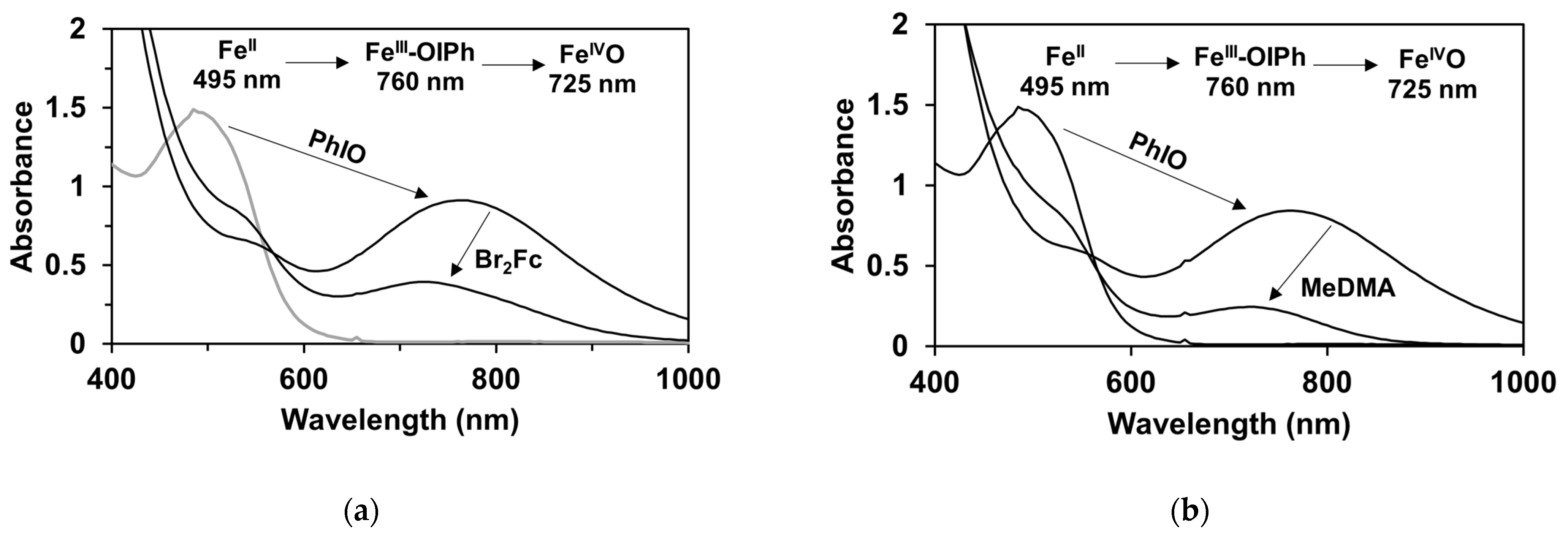
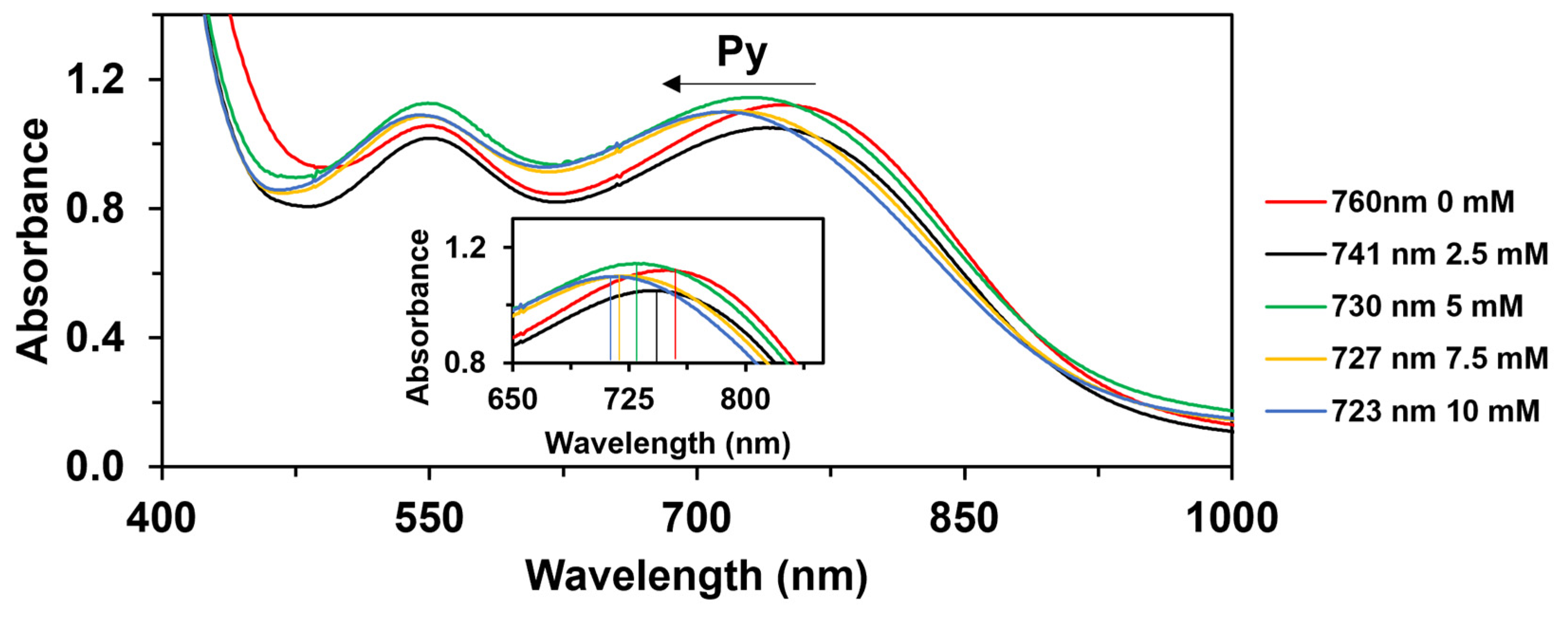
2.3. Catalytic Oxidation of Cis-Cyclooctene Followed by UV-Vis Measurements (Mechanistic Studies)
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Que, L., Jr.; Ho, R.Y.N. Dioxygen Activation by Enzymes with Mononuclear Non-Heme Iron Active Sites. Chem. Rev. 1996, 96, 2607–2624. [Google Scholar] [CrossRef]
- Green, M.T. C-H bond activation in heme proteins: The role of thiolate ligation in cytochrome P450. Curr. Opin. Chem. Biol. 2009, 13, 84–88. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef]
- Krebs, C.; Fujimori, D.G.; Walsh, C.T.; Bollinger, J.M., Jr. Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef]
- Costas, M.; Chen, K.; Que, L., Jr. Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 2000, 200, 517–544. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef]
- Shaik, S.; Lai, W.; Chen, H.; Wang, Y. The Valence Bond Way: Reactivity Patterns of Cytochrome P450 Enzymes and Synthetic Analogs. Acc. Chem. Res. 2010, 43, 1154–1165. [Google Scholar] [CrossRef]
- Fontecave, M.; Ménage, S.; Duboc-Toia, C. Functional Models of Non-Heme Diiron Enzymes. Coord. Chem. Rev. 1998, 178–180, 1555–1572. [Google Scholar] [CrossRef]
- Nehru, K.; Seo, M.S.; Kim, J.; Nam, W. Oxidative N-Dealkylation Reactions by Oxoiron(IV) Complexes of Nonheme and Heme Ligands. Inorg. Chem. 2007, 46, 293–298. [Google Scholar] [CrossRef]
- Nam, W. Dioxygen Activation by Metalloenzymes and Models. Acc. Chem. Res. 2007, 40, 465. [Google Scholar] [CrossRef]
- Nam, W. High-valent Iron(IV)-Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res. 2007, 40, 522–531. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.R.; Que, L., Jr. High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar] [CrossRef]
- Kaizer, J.; Klinker, E.J.; Oh, N.Y.; Rohde, J.U.; Song, W.J.; Stubna, A.; Kim, J.; Munck, E.; Nam, W.; Que, L., Jr. Nonheme FeIVO Complexes That Can Oxidize the C-H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc. 2004, 126, 472–473. [Google Scholar] [CrossRef] [PubMed]
- Klinker, E.J.; Kaizer, J.; Brennessel, W.W.; Woodrum, N.L.; Cramer, C.J.; Que, L., Jr. Structures of Nonheme Oxoiron(IV) Complexes from X-ray Crystallography, NMR Spectroscopy, and DFT Calculations. Angew. Chem. Int. Ed. 2005, 44, 3690–3694. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Lee, Y.-M.; Fukuzumi, S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Oloo, W.N.; Que, L., Jr. Bioinspired Nonheme Iron Catalysts for C–H and C═C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Lakk-Bogáth, D.; Csonka, R.; Speier, G.; Reglier, M.; Simaan, A.J.; Naubron, J.V.; Giorgi, M.; Lazar, K.; Kaizer, J. Oxoiron(IV) Complex Derived from Chiral Pentadentate Ligand asN4Py. Inorg. Chem. 2016, 55, 10090–10093. [Google Scholar] [CrossRef]
- Kumar, R.; Pandey, B.; Sen, A.; Ansari, M.; Sharma, S.; Rajaraman, G. Role of oxidation state, ferryl-oxygen, and ligand architecture on the reactivity of popular high-valent FeIV=O species: A theoretical perspective. Coord. Chem. Rev. 2020, 419, 213397. [Google Scholar] [CrossRef]
- Guo, M.; Corona, T.; Ray, K.; Nam, W. Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent Sci. 2018, 5, 13–28. [Google Scholar] [CrossRef]
- Roy, L. Theoretical Identification of the Factors Governing the Reactivity of C−H Bond Activation by Non-Heme Iron(IV)-Oxo Complexes. ChemPlusChem 2019, 84, 893–906. [Google Scholar] [CrossRef]
- Lee, J.L.; Ross, D.L.; Barman, S.K.; Ziller, J.W.; Borovik, A.S. C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg. Chem. 2021, 60, 13759–13783. [Google Scholar] [CrossRef] [PubMed]
- Warm, K.; Paskin, A.; Kuhlmann, U.; Bill, E.; Swart, M.; Haumann, M.; Dau, H.; Hildebrandt, P.; Ray, K. A Pseudotetrahedral Terminal Oxoiron(IV) Complex: Mechanistic Promiscuity in C-H Bond Oxidation Reactions. Angew. Chem. Int. Ed. 2021, 60, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Kupper, C.; Mondal, B.; Serrano-Plana, J.; Klawitter, I.; Neese, F.; Costas, M.; Ye, S.; Meyer, F. Nonclassical Single-State Reactivity of an Oxo-Iron(IV) Complex Confined to Triplet Pathways. J. Am. Chem. Soc. 2017, 139, 8939–8949. [Google Scholar] [CrossRef]
- Deutscher, J.; Gerschel, P.; Warm, K.; Kuhlmann, U.; Mebs, S.; Haumann, M.; Dau, H.; Hildebrandt, P.; Apfel, U.P.; Ray, K. A Bioinspired Oxoiron(IV) Motif Supported on a N2S2 macrocyclic Ligand. Chem. Commun. 2021, 57, 2947–2950. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Mallick, D.; Shaik, S. Kinetic Isotope Effect Determination Probes the Spin of the Transition State, Its Stereochemistry, and Its Ligand Sphere in Hydrogen Abstraction Reactions of Oxoiron(IV) Complexes. Acc. Chem. Res. 2018, 51, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Eguía, B.N.; Serrano-Plana, J.; Company, A.; Costas, M. Catalytic O2 activation with Synthetic Models of α-Ketoglutarate Dependent Oxygenases. Chem. Commun. 2020, 56, 14369–14372. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Alili, A.; Barman, P.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Interplay Between Steric and Electronic Effects: A Joint Spectroscopy and Computational Study of Nonheme Iron(IV)-Oxo Complexes. Chem. Eur. J. 2019, 25, 5086–5098. [Google Scholar] [CrossRef]
- Rasheed, W.; Draksharapu, A.; Banerjee, S.; Young, V.G., Jr.; Fan, R.; Guo, Y.; Ozerov, M.; Nehrkorn, J.; Krzystek, J.; Telser, J.; et al. Crystallographic Evidence for a Sterically Induced Ferryl Tilt in a Non-Heme Oxoiron(IV) Complex that Makes it a Better Oxidant. Angew. Chem. Int. Ed. 2018, 57, 9387–9391. [Google Scholar] [CrossRef]
- Mukherjee, G.; Lee, C.W.Z.; Nag, S.S.; Alili, A.; Cantú Reinhard, F.G.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Dramatic rate-Enhancement of oxygen atom transfer by an Iron(IV)-oxo species by equatorial ligand field perturbations. Dalton Trans. 2018, 47, 14945–14957. [Google Scholar] [CrossRef]
- Singh, R.; Ganguly, G.; Malinkin, S.O.; Demeshko, S.; Meyer, F.; Nordlander, E.; Paine, T.K. A Mononuclear Nonheme Iron(IV)-Oxo Complex of a Substituted N4Py Ligand: Effect of Ligand Field on Oxygen Atom Transfer and C-H Bond Cleavage Reactivity. Inorg. Chem. 2019, 58, 1862–1876. [Google Scholar] [CrossRef]
- Geng, C.; Ye, S.; Neese, F. Does a higher metal oxidation state necessarily imply higher reactivity toward H-atom transfer? A computational study of C–H bond oxidation by high-valent iron-oxo and -nitrido complexes. Dalton Trans. 2014, 43, 6079–6086. [Google Scholar] [CrossRef] [PubMed]
- Lennartson, A.; McKenzie, C.J. An Iron(III) Iodosylbenzene Complex: A Masked Non-Heme FeVO. Angew. Chem. Int. Ed. 2012, 51, 6767–6770. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Wang, B.; Seo, M.S.; Lee, Y.M.; Kim, M.J.; Kim, H.R.; Ogura, T.; Garcia-Serres, R.; Clémancey, M.; Latour, J.M.; et al. Highly Reactive Nonheme Iron(III) Iodosylarene Complexes in Alkane Hydroxylation and Sulfoxidation Reactions. Angew. Chem. Int. Ed. 2014, 53, 6388–6392. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lee, Y.M.; Seo, M.S.; Nam, W. Mononuclear Nonheme Iron(III)-Iodosylarene and High-Valent Iron-Oxo Complexes in Olefin Epoxidation Reactions. Angew. Chem. Int. Ed. 2015, 54, 11740–11744. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Li, X.-X.; Cho, K.-B.; Sun, W.; Xia, C.; Nam, W.; Wang, Y. Mutable Properties of Nonheme Iron(III)–Iodosylarene Complexes Result in the Elusive Multiple-Oxidant Mechanism. J. Am. Chem. Soc. 2017, 139, 7444–7447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Pandey, B.; Rajaraman, G.J. Comparative oxidative ability of iron(III)-iodosylarene vs. high-valent iron(IV/V)-oxo species: Is lower oxidation state a key to enhance selectivity in organic transformations? Indian Chem. Soc. 2019, 96, 825–836. [Google Scholar]
- Wang, C.; Kurahashi, T.; Inomata, K.; Hada, M.; Fujii, H. Oxygen-Atom Transfer from Iodosylarene Adducts of a Manganese(IV) Salen Complex: Effect of Arenes and Anions on I(III) of the Coordinated Iodosylarene. Inorg. Chem. 2013, 52, 9557–9566. [Google Scholar] [CrossRef]
- Wang, C.; Kurahashi, T.; Fujii, H. Structure and Reactivity of an Iodosylarene Adduct of a Manganese(IV)-Salen Complex. Angew. Chem. 2012, 124, 7929–7931. [Google Scholar] [CrossRef]
- Guo, M.; Dong, H.; Li, J.; Cheng, B.; Huang, Y.-Q.; Feng, Y.-Q.; Lei, A. Spectroscopic Observation of Iodosylarene Metalloporphyrin Adducts and Manganese(V)-Oxo Porphyrin Species in a Cytochrome P450 Analogue. Nat. Commun. 2012, 3, 1190. [Google Scholar] [CrossRef]
- Guo, M.; Lee, Y.-M.; Fukuzumi, S.; Nam, W. Biomimetic metal-oxidant adducts as active oxidants in oxidation reactions. Coord. Chem. Rev. 2021, 435, 213807. [Google Scholar] [CrossRef]
- Guo, M.; Lee, Y.-M.; Seo, M.S.; Kwon, Y.-J.; Li, X.-X.; Ohta, T.; Kim, W.-S.; Sarangi, R.; Fukuzumi, S.; Nam, W. Mn(III)-Iodosylarene Porphyrins as an Active Oxidant in Oxidation Reactions: Synthesis, Characterization, and Reactivity Studies. Inorg. Chem. 2018, 57, 10232–10240. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Choi, S.K.; Lim, M.H.; Rohde, J.-U.; Kim, I.; Kim, J.; Kim, C.; Que, L., Jr. Reversible formation of iodosylbenzene-iron porphyrin intermediates in the reaction of oxoiron(IV) porphyrin pi-cation radicals and iodobenzene. Angew. Chem. Int. Ed. 2003, 42, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Hill, E.A.; Kelty, M.L.; Filatov, A.S.; Anderson, J.S. Isolable iodosylarene and iodoxyarene adducts of Co and their O-atom transfer and C–H activation reactivity. Chem. Sci. 2018, 9, 4493–4499. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Ohta, T.; Cho, J.J. Structure and Reactivity of a Mononuclear Nonheme Manganese(III)–Iodosylarene Complex. Am. Chem. Soc. 2018, 140, 16037–16041. [Google Scholar]
- Török, P.; Lakk-Bogáth, D.; Kaizer, J. Stoichiometric Alkane and Aldehyde Hydrtoxylation Reactions Mediated by In Situ Generated Iron(III)-Iodosylbenzene Adduct. Molecules 2023, 28, 1855. [Google Scholar] [CrossRef] [PubMed]
- Lakk-Bogáth, D.; Szávuly, M.; Török, P.; Kaizer, J. Catalytic and Stoichiometric Baeyer-Villiger Oxidation Mediated by Nonheme Peroxi-Diiron(III), Acylperoxo, and Iodosylbenzene Iron(III) Intermediates. Molecules 2022, 27, 2814. [Google Scholar] [CrossRef] [PubMed]
- Török, P.; Lakk-Bogáth, D.; Kaizer, J. Mechanisms of Sulfoxidation and Epoxidation Mediated by Iron(III)-Iodosylbenzene Adduct: Electron-Transfer vs Oxygen-Transfer Mechanism. Molecules 2023, 28, 4745. [Google Scholar] [CrossRef] [PubMed]
- Pap, J.S.; Draksharapu, A.; Giorgi, M.; Browne, W.R.; Kaizer, J.; Speier, G. Stabilisation of μ-peroxido-bridged Fe(III) intermediates with non-symmetric bidentate N-donor ligands. Chem. Commun. 2014, 50, 1326–1329. [Google Scholar] [CrossRef]
- Adam, W.; Hajra, S.; Herderich, M.; Saha-Moller, C.R. A highly chemoselective oxidation of alcohols to carbonyl products with iodosobenzene diacetate mediated by chromium(III)(salen) complexes: Synthetic and mechanistic aspects. Org. Lett. 2000, 2, 2773–2776. [Google Scholar] [CrossRef]
- In, J.H.; Park, S.E.; Song, R.; Nam, W. Iodobenzene diacetate as an efficient terminal oxidant in iron(III) porphyrin complex-catalyzed oxygenation reactions. Inorg. Chim. Acta 2003, 343, 373–376. [Google Scholar] [CrossRef]
- Carreno, C.M. Applications of sulfoxides to asymmetric synthesis of biologically active compounds. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar] [CrossRef]
- Otocka, S.; Kwiatkowska, M.; Madalinska, L.; Kielbasinski, P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef] [PubMed]
- Joergensen, K.A. Transition-metal-catalyzed epoxidations. Coord. Chem. Rev. 1989, 89, 431–458. [Google Scholar] [CrossRef]
- Adam, W.; Roschmann, K.J.; Chantu, R.; Saha-Möller, C.R.; Seebach, D. cis-Stilbene and (1α,2β,3α)-(2-Ethenyl-3-methoxycyclopropyl)benzene as Mechanistic Probes in the MnIII(salen)-Catalyzed Epoxidation: Influence of the Oxygen Source and the Counterion on the Diastereoselectivity of the Competitive Concerted and Radical-Type Oxygen Transfer. J. Am. Chem. Soc. 2002, 124, 5068–5073. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Coligand | Conversion (%) 2,3 | Products Yields (%) 2 | Selectivity (%) PhS(O)Me | TOF (1/h) 4 | krel 5 | |
|---|---|---|---|---|---|---|---|
| PhS(O)Me | PhS(O)2Me | ||||||
| 1 | - | 35.04 | 34.96 | 0.08 | 99.7 | 8.75 | - |
| 2 | 4-Me-Py | 79.24 | 77.22 | 2.02 | 97.45 | 19.81 | 0.92 |
| 3 | Py | 81.79 | 80.58 | 1.21 | 98.52 | 20.45 | 1 |
| 4 | 4-COC6H5-Py | 85.39 | 83.91 | 1.48 | 98.26 | 21.35 | 1.13 |
| 5 | 4-COCH3-Py | 86.94 | 85.95 | 0.99 | 98.86 | 21.74 | 1.20 |
| 6 | 4-CN-Py | 89.10 | 88.54 | 0.56 | 99.37 | 22.28 | 1.30 |
| Entry | Co-Ligand | Yield (%) 2 Epoxide | TON 3 | TOF (1/h) 4 | krel 5 |
|---|---|---|---|---|---|
| 1 | - | 22.46 | 22.46 | 5.62 | - |
| 2 | 4-Me-Py | 18.62 | 18.62 | 4.65 | 0.44 |
| 3 | Py | 36.94 | 36.94 | 9.24 | 1 |
| 4 | 4-COC6H5-Py | 59.64 | 59.64 | 14.91 | 1.97 |
| 5 | 4-COCH3-Py | 63.18 | 63.18 | 15.79 | 2.17 |
| 6 | 4-CN-Py | 78.00 | 78.00 | 19.50 | 3.28 |
| Entry | Co-Ligand | Yield (%) 2 trans-Epoxide | TON 3 | TOF (1/h) 4 | krel 5 |
|---|---|---|---|---|---|
| 1 | - | 36.66 | 36.66 | 9.17 | - |
| 2 | 4-Me-Py | 34.23 | 34.23 | 8.55 | 0.79 |
| 3 | Py | 41.15 | 41.15 | 10.29 | 1 |
| 4 | 4-COC6H5-Py | 51.12 | 51.12 | 12.78 | 1.35 |
| 5 | 4-COCH3-Py | 56.74 | 56.74 | 14.19 | 1.58 |
| 6 | 4-CN-Py | 66.13 | 66.13 | 16.53 | 2.04 |
| Entry | Co-Ligand | Conv. (%) 2,3 | Products Yields (%) 2 | Selectivity (%) trans-Stilbene Epoxide | TOF (1/h) 4 | krel 5 | |
|---|---|---|---|---|---|---|---|
| cis-Stilbene Epoxide | trans-Stilbene Epoxide | ||||||
| 1 | - | 24.14 | 3.18 | 19.96 | 81.76 | 5.78 | - |
| 2 | 4-Me-Py | 23.97 | 2.11 | 21.86 | 91.19 | 5.99 | 0.61 |
| 3 | Py | 36.12 | 4.15 | 31.97 | 88.51 | 9.3 | 1 |
| 4 | 4-COC6H5-Py | 47.82 | 3.84 | 43.98 | 91.96 | 11.95 | 1.45 |
| 5 | 4-COCH3-Py | 51.32 | 3.67 | 47.65 | 92.85 | 12.83 | 1.60 |
| 6 | 4-CN-Py | 62.81 | 5.29 | 57.52 | 91.58 | 15.70 | 2.20 |
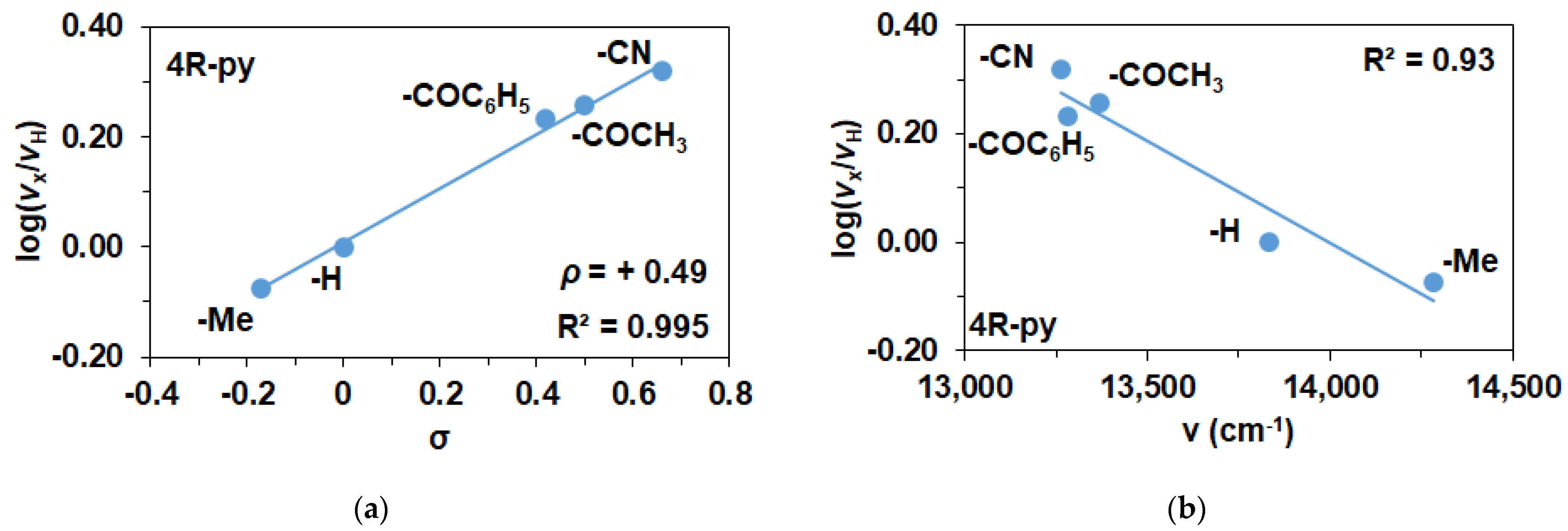
| Entry | Coligand | λ (nm) | ν (cm−1) | σ | v0 (10−4 Ms−1) 1 | log(vX/vH) |
|---|---|---|---|---|---|---|
| 1 | - | 760 | 13,158 | - | - | - |
| 2 | 4-Me-Py | 700 | 14,286 | −0.17 | 1.26 | −0.076 |
| 3 | Py | 723 | 13,831 | 0 | 1.5 | 0.000 |
| 4 | 4-COC6H5-Py | 748 | 13,369 | 0.42 | 2.56 | 0.232 |
| 5 | 4-COCH3-Py | 753 | 13,280 | 0.5 | 2.72 | 0.258 |
| 6 | 4-CN-Py | 754 | 13,263 | 0.66 | 3.12 | 0.318 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lakk-Bogáth, D.; Pintarics, D.; Török, P.; Kaizer, J. Influence of Equatorial Co-Ligands on the Reactivity of LFeIIIOIPh. Molecules 2024, 29, 58. https://doi.org/10.3390/molecules29010058
Lakk-Bogáth D, Pintarics D, Török P, Kaizer J. Influence of Equatorial Co-Ligands on the Reactivity of LFeIIIOIPh. Molecules. 2024; 29(1):58. https://doi.org/10.3390/molecules29010058
Chicago/Turabian StyleLakk-Bogáth, Dóra, Dénes Pintarics, Patrik Török, and József Kaizer. 2024. "Influence of Equatorial Co-Ligands on the Reactivity of LFeIIIOIPh" Molecules 29, no. 1: 58. https://doi.org/10.3390/molecules29010058
APA StyleLakk-Bogáth, D., Pintarics, D., Török, P., & Kaizer, J. (2024). Influence of Equatorial Co-Ligands on the Reactivity of LFeIIIOIPh. Molecules, 29(1), 58. https://doi.org/10.3390/molecules29010058