
1-Hydroxyalkylphosphonium Salts—Synthesis and Properties

 , , ,
, , ,
Abstract
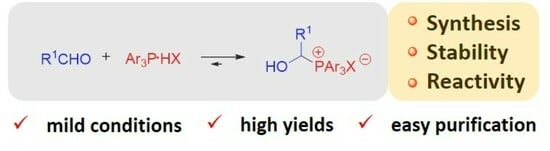
:
1. Introduction
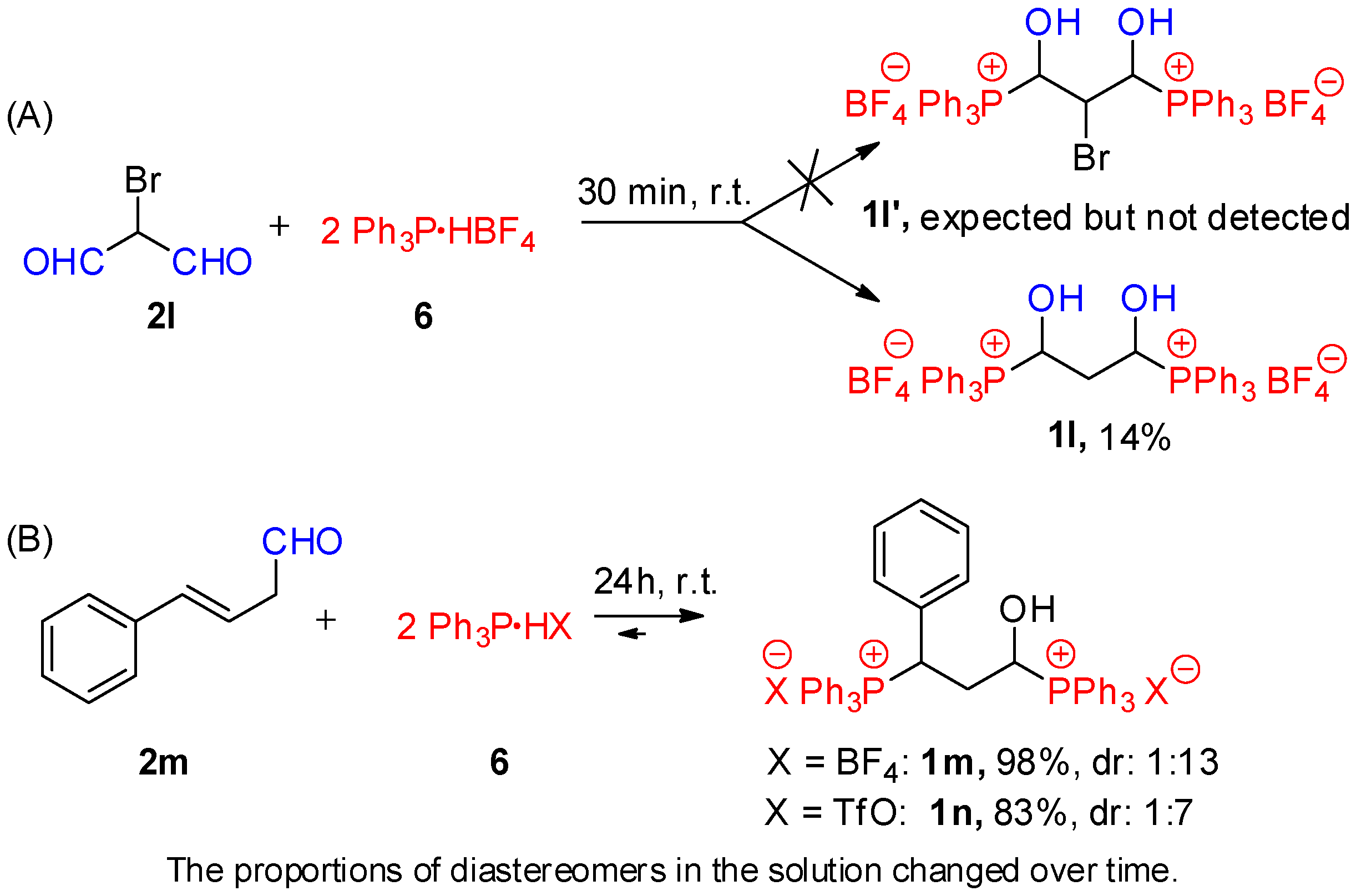
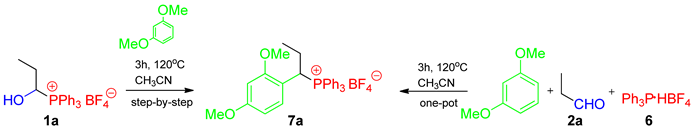
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of 1-Hydroxyalkylphosphonium Salts
3.3. Synthesis of 1-Hydroxyalkylphosphonium Salts on a Gram-Scale
3.4. Synthesis of 1-Hydroxyalkylphosphonium Salts in the Presence of PPh3 and HBF4·Et2O
- 1-hydroxypropyltriphenylphosphonium tetrafluoroborate (1a). White crystals (379.6 mg, 93% yield), mp 125–127 °C. 1H NMR (400 MHz, CDCl3) δ 7.83–7.62 (m, 15H, 3×Ph), 5.64 (dd, J = 10.2, 3.0 Hz, 1H, CαH), 5.31 (br s, 1H, OH), 1.91–1.80 (m, 2H, CH2), and 1.20 (td, J = 7.2, 0.9 Hz, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.2, 134.3 (d, J = 8.5 Hz), 130.5 (d, J = 12.0 Hz), 117.2 (d, J = 81.1 Hz), 69.9 (d, J = 60.9 Hz, CαH), 25.7 (d, J = 4.9 Hz, CH2), and 10.3 (d, J = 14.4 Hz, CH3); 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.5 ppm; IR (ATR) 3407, 1438, 1110, 1088, 1066, 996, and 976 cm−1. HRMS (TOF-ESI) calcd. for C21H22OP+ [M+] 321.1408, found 321.1422.
- 1-hydroxypropyltriphenylphosphonium bromide (1b) [24]. White crystals (353.1 mg, 88% yield), mp 150–152 °C (lit.: mp 157–159 °C [24]). 1H NMR (400 MHz, CDCl3) δ 7.95–7.71 (m, 9H, Ph), 7.70–7.58 (m, 6H, Ph), 5.96–5.88 (m, 1H, CαH), 1.91–1.77 (m, 2H, CH2), and 1.24 (td, J = 7.2, 1.1 Hz, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 134.9 (d, J = 3.0 Hz), 134.4 (d, J = 9.0 Hz), 130.3 (d, J = 12.1 Hz), 117.7 (d, J = 80.6 Hz), 68.4 (d, J = 60.4 Hz, CαH), 25.7 (d, J = 6.2 Hz, CH2), and 10.7 (d, J = 14.5 Hz, CH3); 31P{1H} NMR (161.9 MHz, CDCl3) δ 20.9 ppm; IR (ATR) 3072, 1438, 1111, 754 cm−1.
- 1-hydroxypropyltriphenylphosphonium triflate (1c). White crystals (371.6 mg, 79% yield), mp 132–134 °C. 1H NMR (400 MHz, CDCl3) δ 7.84–7.70 (m, 9H, Ph), 7.70–7.62 (m, 6H, Ph), 5.76–5.67 (m, 1H, CαH), 1.91–1.76 (m, 2H, CH2), and 1.21 (td, J = 7.4, 1.2 Hz, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.1 (d, J = 3.0 Hz), 134.4 (d, J = 8.9 Hz), 130.5 (d, J = 12.0 Hz), 120.7 (q, J = 319.6 Hz, CF3), 117.5 (d, J = 80.7 Hz), 69.7 (d, J = 60.4 Hz, CαH), 25.8 (d, J = 5.4 Hz, CH2), and 10.4 (d, J = 14.7 Hz, CH3); 31P{1H} NMR (161,9 MHz, CDCl3) δ 21.0 ppm; IR (ATR) 3266, 1438, 1292, 1244, 1224, 1155, 1109, and 1028 cm−1. HRMS (TOF-ESI) calcd. For C21H22OP+ [M+] 321.1408, found 321.1407.
- 1-hydroxypropyltris(4-methoxyphenyl)phosphonium tetrafluoroborate (1d). Resin (328.8 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ 7.70–7.53 (m, 6H, aromatic), 7.20–7.07 (m, 6H, aromatic), 5.42 (dd, J = 10.5, 3.0 Hz, 1H, CαH), 3.90 (s, 9H, 3xOCH3), 1.87–1.68 (m, 2H, CH2), and 1.17 (td, J = 7.2, 0.4 Hz, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 164.7 (d, J = 3.0 Hz), 136.1 (d, J = 10.5 Hz), 116.1 (d, J = 13.2 Hz), 108.0 (d, J = 89.2 Hz), 70.1 (d, J = 65.2 Hz, CαH), 55.9 (OCH3), 25.4 (CH2), and 10.3 (d, J = 14.3 Hz, CH3); 31P{1H} NMR (161.9 MHz, CDCl3) δ 19.9 ppm; IR (ATR) 3442, 1593, 1503, 1263, 1110, 1055, and 1016 cm−1. HRMS (TOF-ESI) calcd. for C24H28O4P+ [M+] 411.1725, found 411.1721.
- hydroxymethyltriphenylphosphonium tetrafluoroborate (1f) [16,17]. White crystals (361.1 mg, 95% yield), mp 132–134 °C C (lit.: mp 128–130 °C [16], 130–131 °C [17]). 1H NMR (400 MHz, CDCl3) δ 7.85–7.64 (m, 15H, 3xPh), 5.47 (s, 2H, CH2); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.4 (d, J = 3.0 Hz), 134.1 (d, J = 9.5 Hz), 130.5 (d, J = 12.3 Hz), 117.3 (d, J = 83.9 Hz), and 57.5 (d, J = 65.2 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 17.1 ppm; IR (ATR) 3073, 1439, 1076, 1025, and 998 cm−1. HRMS (TOF-ESI) calcd. for C19H18OP+ [M+] 293.1095, found 293.1085.
- hydroxymethyltriphenylphosphonium triflate (1g) [28]. White crystals (411.4 mg, 93% yield), mp 134–135 °C. 1H NMR (400 MHz, CDCl3) δ 7.89–7.78 (m, 3H, Ph), 7.75–7.63 (m, 12H, Ph), 5.41 (s, 2H, CH2); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.5 (d, J = 3.0 Hz), 134.0 (d, J = 9.5 Hz), 130.6 (d, J = 12.4 Hz), 120.6 (q, J = 319.6 Hz, CF3), 116.7 (d, J = 84.0 Hz), and 58.1 (d, J = 65.1 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 17.2 ppm; IR (ATR) 3313, 1438, 1281, 1249, 1228, 1158, 1113, and 1027 cm−1. HRMS (TOF-ESI) calcd. for C19H18OP+ [M+] 293.1095, found 293.1096.
- hydroxymethyltriphenylphosphonium bromide (1h) [29]. White crystals (354.5 mg, 95% yield), mp 191–193 °C (lit.: mp 203 °C [29]). 1H NMR (400 MHz, CDCl3) δ 7.85–7.61 (m, 15H, 3xPh), 5.47 (s, 2H, CH2); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.4 (d, J = 3.0 Hz), 134.1 (d, J = 9.5 Hz), 130.5 (d, J = 12.3 Hz), 117.3 (d, J = 83.6 Hz), and 57.6 (d, J = 65.1 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 17.1 ppm; IR (ATR) 3087, 1435, 1115, and 1051 cm−1. HRMS (TOF-ESI) calcd. For C19H18OP+ [M+] 293.1095, found 293.1096.
- 1-hydroxyethyltriphenylphosphonium tetrafluoroborate (1i). White crystals (390.3 mg, 99% yield), mp 114–116 °C. 1H NMR (400 MHz, CDCl3) δ 7.90–7.55 (m, 15H, 3xPh), 5.96–5.86 (m, 1H, CαH), 5.29 (dd, J = 13.1, 6.8 Hz, 1H, OH), and 1.70 (dd, J = 18.2, 6.8 Hz, 3H, CH3); 13C{1H} NMR (150 MHz, CDCl3) δ aromatic carbons: 135.3 (br s), 134.2 (d, J = 8.9 Hz), 130.5 (d, J = 12.1 Hz), 116.8 (d, J = 82.1 Hz), 65.4 (d, J = 65.1 Hz, CαH), and 18.5 (br s, CH3); 31P{1H} NMR (161.9 MHz, CDCl3) δ 17.5 ppm; IR (ATR) 3451, 1488, 1439, 1108, 1060, 996, and 976 cm−1. HRMS (TOF-ESI) calcd. for C20H20OP+ [M+] 307.1252, found 307.1251.
- 2-chloro-1-hydroxyethyltriphenylphosphonium tetrafluoroborate (1j). White crystals (369.0 mg, 89% yield), mp 111.5–113.5 °C. 1H NMR (400 MHz, CDCl3) δ 7.90–7.74 (m, 9H, Ph), 7.70–7.59 (m, 6H, Ph), 6.11 (br s, 1H, OH), 5.73 (dd, J = 13.2, 6.2 Hz, 1H, CαH), 4.03 (ddd, J = 19.9, 12.8, 4.3 Hz, 1H, CHH), and 3.88 (ddd, J = 12.9, 8.2, 5.0 Hz, 1H, CHH); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.6 (d, J = 3.1 Hz), 134.3 (d, J = 9.5 Hz), 130.6 (d, J = 12.5 Hz), 116.5 (d, J = 82.7 Hz), 69.8 (d, J = 67.3 Hz, CαH), and 44.4 (d, J = 7.3 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 20.8 ppm; IR (ATR) 3337, 1586, 1483, 1436, 1061, 1023, 995, and 973 cm−1. HRMS (TOF-ESI) calcd. for C20H19ClOP+ [M+] 341.0862, found 341.0861.
- 1-hydroxydecyltriphenylphosphonium tetrafluoroborate (1k). Resin (501.3 mg, 99% yield). 1H NMR (400 MHz, CDCl3) δ 7.87–7.60 (m, 15H, 3xPh), 5.73–5.66 (m, 1H, CαH), 5.29 (br s, 1H, OH), aliphatics (8xCH2): 1.90–1.66 (m, 3H), 1.66–1.46 (m, 1H), 1.35–1.11 (m, 12H), and 0.85 (t, J = 6.9 Hz, 3H, CH3); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.2 (d, J = 3.0 Hz), 134.3 (d, J = 8.9 Hz), 130.5 (d, J = 12.0 Hz), 117.2 (d, J = 80.9 Hz), 68.9 (d, J = 60.1 Hz, CαH), 32.3 (d, J = 4.3 Hz, CH2), 31.9 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 25.7 (d, J = 13.4 Hz, CH2), 22.7 (br s, CH2), and 14.2 (CH3); 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.4 ppm; IR (ATR) 2923, 2854, 1439, 1109, 1056, and 996 cm−1. HRMS (TOF-ESI) calcd. for C28H36OP+ [M+] 419.2504, found 419.2499.
- 1,3-dihydroxypropane-1,3-bis(triphenylphosphonium) bis(tetrafluoroborate) (1l). White crystals (108.1 mg, 14% yield), mp 150–151 °C. 1H NMR (400 MHz, CDCl3) δ 7.90–7.50 (m, 30H, 6xPh), 6.64 (br s, 2H, OH), 5.94 (br s, 2H, CαH), and 2.18–2.05 (m, 2H, CH2); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.4 (br s), 134.7–134.3 (m), 130.9–130.5 (m), 116.1 (d, J = 84.2 Hz), 62.7 (dd, J = 73.2, 13.8 Hz, CαH), and 33.5 (t, J = 8.7 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 22.7 ppm; IR (ATR) 2989, 1586, 1483, 1437, 1109, 1045, and 995 cm−1. HRMS (TOF-ESI) calcd. for C39H36O2P22+ [M2+] 299.1090, found 299.1087.
- 1-hydroxy-3-phenylpropane-1,3-bis(triphenylphosphonium) bis(tetrafluoroborate) (1m). White crystals (815.8 mg, 98% yield), mp 205–207 °C. 1H NMR (400 MHz, CDCl3) δ 7.83–7.71 (m, 6H, Ph), 7.69–7.54 (m, 18H, Ph), 7.47–7.39 (m, 6H, Ph), 7.39–7.30 (m, 1H, Ph), 7.27–7.21 (m, 2H, Ph), 7.06–7.00 (m, 2H, Ph), 6.16 (t, J = 7.1 Hz, 1H, OH), 5.24–5.16 (m, 1H, CH-Ph or CαH), 5.04 (dd, J = 17.1, 10.8 Hz, 1H, CH-Ph or CαH), 2.81–2.69 (m, 1H, CHH), and 2.59–2.50 (m, 1H, CHH); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.6 (d, J = 3.0 Hz), 135.5 (d, J = 3.1 Hz), 134.4 (d, J = 9.4 Hz), 134.1 (d, J = 9.3 Hz), 131.3 (d, J = 5.8 Hz), 130.7 (d, J = 12.4 Hz), 130.6 (d, J = 12.5 Hz), 130.1 (d, J = 3.0 Hz), 129.9 (d, J = 2.2 Hz), 129.4 (d, J = 5.1 Hz), 116.1 (d, J = 83.9 Hz), 115.4 (d, J = 82.6 Hz), 66.5 (dd, J = 66.2, 14.8 Hz, CαH), 40.2 (dd, J = 47.7, 15.6 Hz, CH-Ph), and 35.2 (br d, J = 10.2 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 26.9 (d, J = 9.1 Hz), 21.8 (d, J = 9.1 Hz) ppm; IR (ATR) 3410, 1439, 1108, 1050, and 996 cm−1. HRMS (TOF-ESI) calcd. for C39H36O2P22+ [M2+] 329.1277, found 329.1265.
- 1-hydroxy-3-phenylpropane-1,3-bis(triphenylphosphonium) bis(triflate) (1n). White crystals (108.1 mg, 14% yield), mp 190–192 °C. 1H NMR (400 MHz, CDCl3) δ 7.84–7.70 (m, 6H, Ph), 7.69–7.56 (m, 18H, Ph), 7.49–7.41 (m, 6H, Ph), 7.39–7.33 (m, 1H, Ph), 7.27–7.22 (m, 2H, Ph), 7.05–6.99 (m, 2H, Ph), 5.18–5.02 (m, 2H, CH-Ph and CαH), 2.76–2.64 (m, 1H, CHH), 2.59–and 2.46 (m, 1H, CHH); 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.6 (d, J = 3.0 Hz), 135.4 (d, J = 3.1 Hz), 134.6 (d, J = 9.4 Hz), 134.3 (d, J = 9.3 Hz), 131.4 (d, J = 5.8 Hz), 130.7 (d, J = 12.4 Hz), 130.7 (d, J = 12.4 Hz), 130.2 (d, J = 3.0 Hz), 129.9 (d, J = 2.3 Hz), 129.5 (d, J = 5.1 Hz), 120.6 (q, J = 320.2 Hz, CF3), 116.3 (d, J = 83.9 Hz), 115.8 (d, J = 82.5 Hz), 66.3 (dd, J = 66.8, 14.8 Hz, CαH), 40.0 (dd, J = 47.7, 15.7 Hz, CH-Ph), and 35.2 (dd, J = 9.8, 1.0 Hz, CH2); 31P{1H} NMR (161.9 MHz, CDCl3) δ 26.4 (d, J = 8.7 Hz), 21.4 (d, J = 8.7 Hz) ppm; IR (ATR) 3062, 1439, 1262, 1107, 1025, and 997 cm−1. HRMS (TOF-ESI) calcd. For C39H36O2P22+ [M2+] 329.1277, found 329.1270.
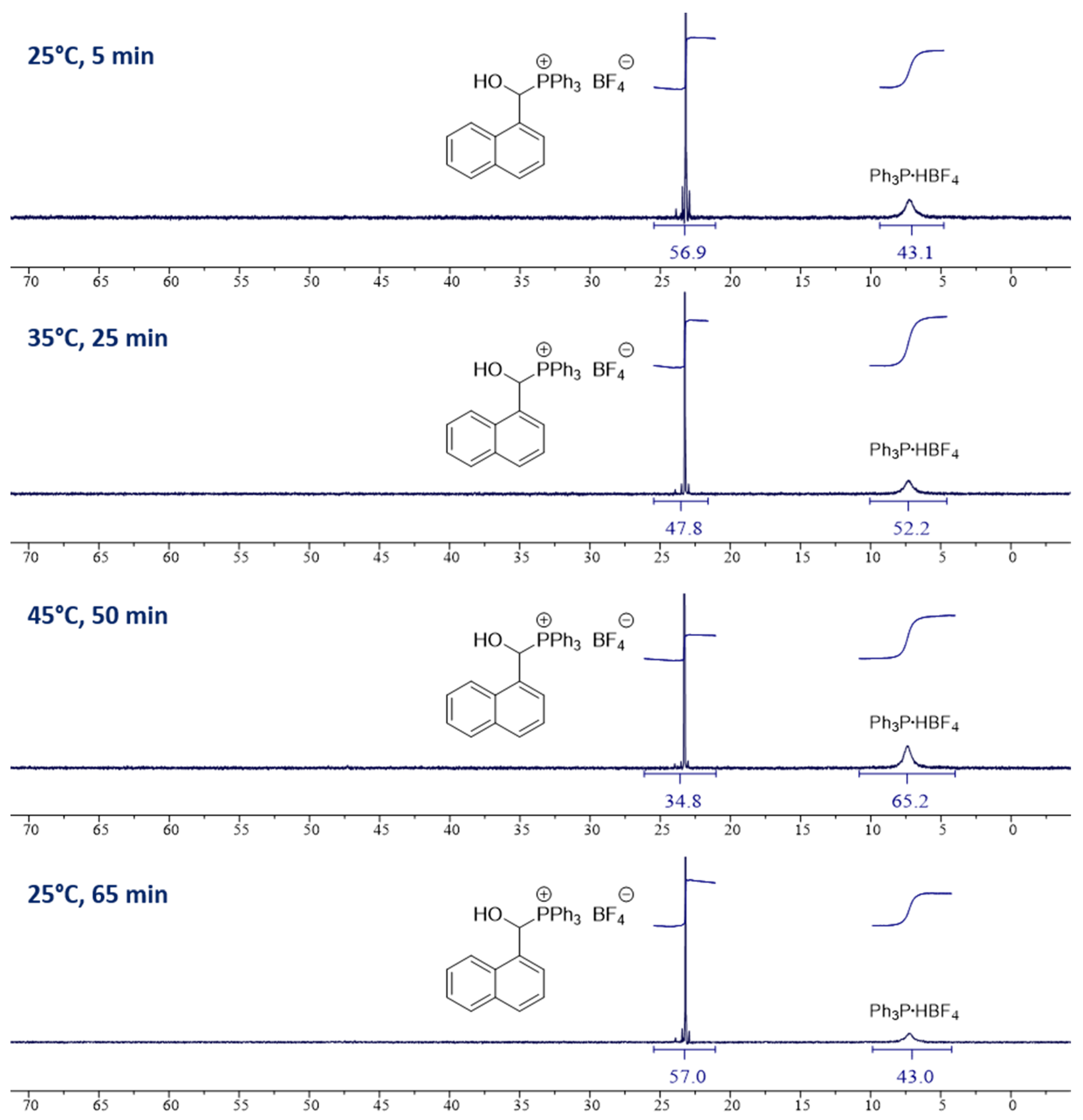
3.5. NMR Experiments (Studies of Equilibria)
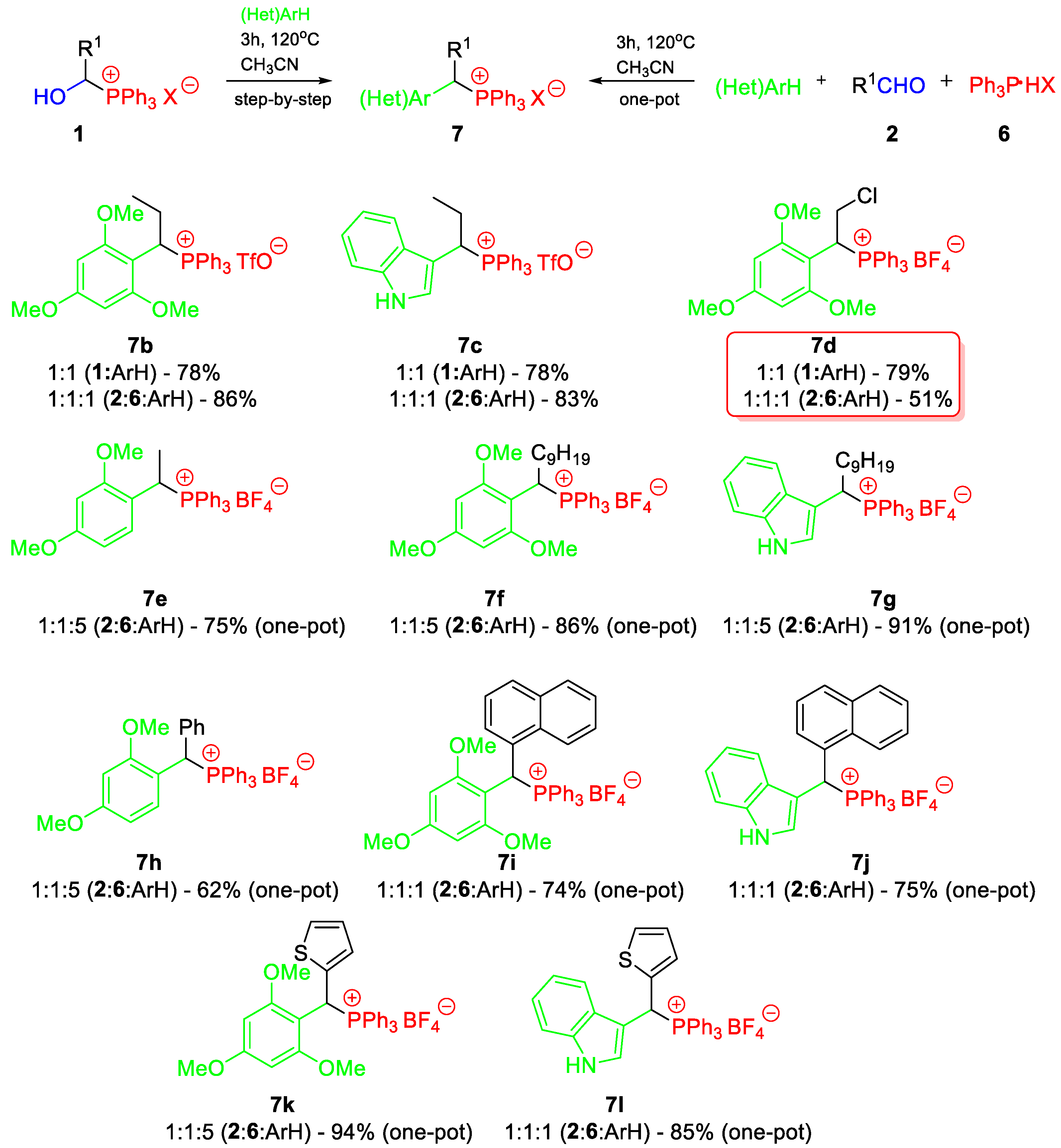
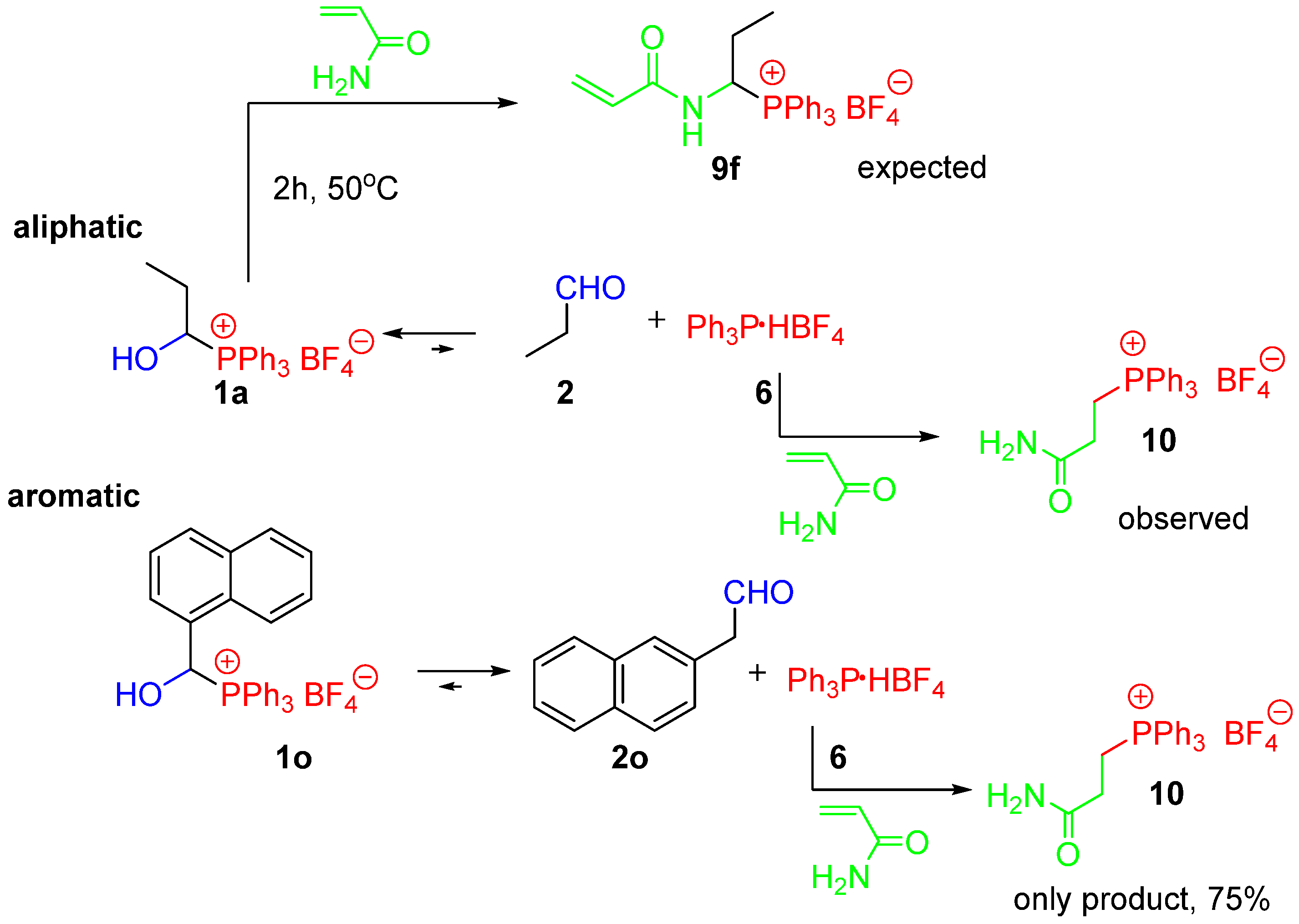
3.6. Reactions of 1-Hydroxyalkylphosphonium Salts with (hetero)arenes (Step-by-Step)
3.7. Reactions of Aldehydes with (hetero)arenes in the Presence of Ar3P∙HX (One-Pot)
3.8. Reactions of Aldehydes with (hetero)arenes in the Presence of PPh3 and HBF4·Et2O (One-Pot)
- 1-(2,4-dimethoxyphenyl)propyltriphenylphosphonium tetrafluoroborate (7a). White crystals (433.2 mg, 82%), mp 174–176 °C. 1H NMR (400 MHz, CDCl3) δ 7.88–7.79 (m, 3H, Ph), 7.73–7.63 (m, 6H, Ph), 7.51–7.40 (m, 6H, Ph), 6.58 (dd, J = 9.3, 2.4 Hz, 1H, aromatic), 6.44–6.33 (m, 2H, aromatic), 4.90 (ddd, J = 15.1, 12.2, 2.6 Hz, 1H, CαH), 3.82 (s, 3H, OCH3), 3.43 (s, 3H, OCH3), 2.35–2.20 (m, 1H, CHH), 2.19–2.08 (m, 1H, CHH), and 0.95 (td, J = 7.1, 1.2 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 161.9 (d, J = 3.0 Hz), 159.4 (d, J = 5.9 Hz), 135.4 (d, J = 3.0 Hz), 134.3 (d, J = 9.0 Hz), 130.5 (d, J = 12.1 Hz), 117.7 (d, J = 82.4 Hz), 110.5 (d, J = 5.2 Hz), 105.8 (d, J = 2.6 Hz), 98.9 (d, J = 2.4 Hz), 55.7 (OCH3), 55.5 (OCH3), 36.7 (d, J = 41.6 Hz, CαH), 24.2 (CH2), and 12.5 (d, J = 15.3 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 23.2 ppm; IR (ATR) 2971, 1608, 1585, 1508, 1438, 1107, 1050, 1027, and 997 cm−1. HRMS (TOF-ESI) calcd. for C29H30O2P+ [M+] 441.1983 found 441.1982.
- 1-(2,4,6-trimethoxyphenyl)propyltriphenylphosphonium triflate (7b). Resin (533.7 mg, 86%). 1H NMR (400 MHz, CDCl3) 7.88–7.75 (m, 3H, Ph), 7.71–7.59 (m, 6H, Ph), 7.44–7.31 (m, 6H, Ph), 6.03 (s, 2H, aromatic), 5.00 (ddd, J = 17.5, 11.9, 3.2 Hz, 1H, CαH), 3.84 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.15 (s, 3H, OCH3), 2.77–2.62 (m, 1H, CHH), 2.13–2.00 (m, 1H, CHH), and 0.91 (td, J = 7.2, 1.1 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 162.7 (d, J = 2.8 Hz), 160.0 (d, J = 7.5 Hz), 135.0 (d, J = 3.0 Hz), 134.0 (d, J = 8.9 Hz), 130.1 (d, J = 12.0 Hz), 121.0 (q, J = 320.7 Hz, CF3), 118.8 (d, J = 82.4 Hz), 98.6 (d, J = 5.3 Hz), 91.0, 56.1 (OCH3), 55.7 (OCH3), 54.7 (OCH3), 36.4 (d, J = 45.5 Hz, CαH), and 21.6 (CH2), 13.0 (d, J = 15.1 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.7 ppm; IR (ATR) 2940, 1609, 1586, 1438, 1266, 1222, 1141, 1119, 1103, and 1030 cm−1. HRMS (TOF-ESI) calcd. for C30H32O3P+ [M+] 471.2089 found 471.2093.
- 1-(H-indol-3-yl)propyltriphenylphosphonium triflate (7c). White crystals (472.8 mg, 83%), mp 191–193 °C. 1H NMR (400 MHz, CDCl3) 10.22 (s, 1H, NH), 7.77–7.69 (m, 3H, Ph), 7.61–7.53 (m, 6H, Ph), 7.51–7.43 (m, 7H, aromatic), 7.07–6.99 (m, 1H, aromatic), 6.97–6.91 (m, 1H, aromatic), 6.81 (br t, J = 7.5 Hz, 1H, aromatic), 6.65 (br t, J = 3.0 Hz, 1H, aromatic), 4.73 (ddd, J = 14.4, 12.2, 2.4 Hz, 1H, CαH), 2.40–2.21 (m, 1H, CHH), 2.15–2.01 (m, 1H, CHH), and 0.91 (td, J = 7.1, 1.3 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 136.2, 135.2 (d, J = 2.0 Hz), 134.3 (d, J = 8.9 Hz), 130.3 (d, J = 12.0 Hz), 127.4 (d, J = 4.6 Hz), 126.3 (d, J = 7.8 Hz), 122.4, 121.0 (q, J = 318.8 Hz, CF3), 119.9, 117.8 (d, J = 80.9 Hz), 117.7, 112.9, 103.0 (d, J = 5.8 Hz), 36.8 (d, J = 45.2 Hz, CαH), 25.6 (CH2), and 12.4 (d, J = 14.9 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.9 ppm; IR (ATR) 3329, 1438, 1277, 1266, 1248, 1225, 1158, 1109, and 1030 cm−1. HRMS (TOF-ESI) calcd. for C29H27NP+ [M+] 420.1881 found 420.1882.
- 2-chloro-1-(2,4,6-trimethoxyphenyl)ethyltriphenylphosphonium tetrafluoroborate (7d). White crystals (457.3 mg, 79%), mp 159–160 °C. 1H NMR (400 MHz, CDCl3) 7.86–7.79 (m, 3H, Ph), 7.72–7.64 (m, 6H, Ph), 7.49–7.41 (m, 6H, Ph), 6.03 (s, 2H, aromatic), 5.50 (ddd, J = 16.4, 7.8, 7.1 Hz, 1H, CαH), 4.45–4.27 (m, 2H, CH2Cl), 3.83 (s, 3H, OCH3), and 3.42 (s, 6H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 163.5 (d, J = 2.7 Hz), 159.5 (br s), 135.4 (d, J = 3.1 Hz), 134.2 (d, J = 9.4 Hz), 130.3 (d, J = 12.4 Hz), 117.8 (d, J = 83.1 Hz), 98.3 (d, J = 5.1 Hz), 91.2, 55.9 (OCH3), 55.5 (OCH3), 41.7 (d, J = 5.0 Hz, CH2Cl), and 38.5 (d, J = 45.6 Hz, CαH) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 22.1 ppm; IR (ATR) 3354, 2843, 1608, 1590, 1438, 1340, 1209, 1157, 1142, 1118, 1103, 1032, and 997 cm−1. HRMS (TOF-ESI) calcd. for C29H29ClO3P+ [M+] 491.1543 found 491.1546.
- 1-(2,4-dimethoxyphenyl)ethyltriphenylphosphonium tetrafluoroborate (7e) [30]. Resin (385.7 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 7.93–7.80 (m, 3H, Ph), 7.73–7.60 (m, 6H, Ph), 7.53–7.42 (m, 6H, Ph), 6.69–6.58 (m, 1H, aromatic), 6.42–6.32 (m, 2H, aromatic), 5.27–5.14 (m, 1H, CαH), 3.81 (s, 3H, OCH3), 3.41 (s, 3H, OCH3), and 1.82 (dd, J = 18.5, 7.4 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 161.8 (d, J = 3.0 Hz), 158.1 (d, J = 5.6 Hz), 135.2 (d, J = 3.0 Hz), 134.2 (d, J = 8.9 Hz), 130.3 (d, J = 12.1 Hz), 130.1 (d, J = 5.0 Hz), 117.3 (d, J = 82.4 Hz), 112.6 (d, J = 5.4 Hz), 105.5 (d, J = 2.6 Hz), 98.7 (d, J = 2.5 Hz), 55.6 (OCH3), 55.2 (OCH3), 29.7 (d, J = 45.7 Hz, CαH), and 16.5 (CH3) ppm; δ 31P{1H} NMR (161.9 MHz, CDCl3) δ 24.2 ppm; IR (ATR) 2936, 1608, 1506, 1486, 1438, 1301, 1209, 1107, 1049, 1023, and 996 cm−1. The spectra reported here are in agreement with previously published data [30].
- 1-(2,4,6-trimethoxyphenyl)decyltriphenylphosphonium tetrafluoroborate (7f). White crystals (564.6 mg, 86%), mp 110–112 °C. 1H NMR (400 MHz, CDCl3) 7.88–7.76 (m, 3H, Ph), 7.70–7.60 (m, 6H, Ph), 7.44–7.31 (m, 6H, Ph), 6.06 (s, 1H, aromatic), 6.03 (s, 1H, aromatic), 5.08 (ddd, J = 17.7, 12.0, 3.0 Hz, 1H, CαH), 3.85 (s, 3H, OCH3), 3.45 (s, 3H, OCH3), 3.16 (s, 3H, OCH3), and aliphatics (8xCH2): 2.81–2.64 (m, 1H), 1.99–1.81 (m, 1H), 1.32–1.09 (m, 14H), and 0.85 (t, J = 7.0 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 162.7 (d, J = 2.9 Hz), 160.1 (d, J = 3.6 Hz), 159.8 (d, J = 6.5 Hz), 135.0 (d, J = 3.0 Hz), 134.1 (d, J = 8.9 Hz), 130.2 (d, J = 12.1 Hz), 118.8 (d, J = 82.3 Hz), 98.8 (d, J = 5.4 Hz), 91.1 (br s), 56.1 (OCH3), 55.8 (OCH3), 54.7 (OCH3), 34.8 (d, J = 45.3 Hz, CαH), 31.9 (CH2), 29.5 (CH2), 29.3 (CH2), 29.3 (CH2), 28.8 (CH2), 28.1 (d, J = 13.6 Hz), 27.7 (CH2), 22.7 (CH2), and 14.2 (CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.7 ppm; IR (ATR) 2925, 2853, 1607, 1589, 1456, 1437, 1206, 1147, 1127, 1101, 1046, 1033, and 996 cm−1. HRMS (TOF-ESI) calcd. for C37H46O3P+ [M+] 569.3185 found 569.3185.
- 1-(H-indol-3-yl)decyltriphenylphosphonium tetrafluoroborate (7g). Creamy crystals (551.0 mg, 91%), mp 195–197 °C. 1H NMR (400 MHz, CDCl3) 9.70 (s, 1H, NH), 7.82–7.70 (m, 3H, Ph), 7.65–7.53 (m, 6H, Ph), 7.52–7.40 (m, 7H, aromatic), 7.12–7.01 (m, 1H, aromatic), 6.91–6.78 (m, 2H, aromatic), 6.77–6.68 (m, 1H, aromatic), 4.81–4.66 (m, 1H, CαH), and aliphatics (8xCH2): 2.27–2.01 (m, 2H), 1.42–0.98 (m, 14H), and 0.82 (t, J = 7.1 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 136.2, 135.3 (d, J = 2.9 Hz), 134.3 (d, J = 8.9 Hz), 130.4 (d, J = 12.0 Hz), 127.2 (d, J = 4.4 Hz), 126.6 (d, J = 6.9 Hz), 122.6, 120.1, 117.9 (d, J = 82.0 Hz), 117.5, 113.0, 103.4 (d, J = 5.9 Hz), 35.4 (d, J = 44.8 Hz, CαH), 32.0 (br s, CH2), 31.9 (CH2), 29.5 (CH2), 29.3 (CH2), 29.2 (CH2), 29.1 (CH2), 27.6 (d, J = 13.6 Hz), 22.7 (CH2), and 14.2 (CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.9 ppm; IR (ATR) 3357, 2927, 2854, 1484, 1459, 1436, 1340, 1106, 1055, 1020, 995, 744, 720, and 691 cm−1. HRMS (TOF-ESI) calcd. for C36H41NP+ [M+] 518.2977 found 518.2982.
- 1-(2,4-dimethoxyphenyl)phenylmethyltriphenylphosphonium tetrafluoroborate (7h) [30]. White crystals (357.4 mg, 62%), mp 209–211 °C (lit.: mp 199.5–200.5 °C [30]). 1H NMR (400 MHz, CDCl3) δ 7.84–7.75 (m, 3H), 7.67–7.58 (m, 8H), 7.51–7.40 (m, 6H), 7.32–7.19 (m, 1H), 7.14–7.07 (m, 2H), 7.02–6.95 (m, 1H), 6.52 (d, J = 18.2 Hz, 1H, CαH), 6.44–6.40 (m, 2H), 3.80 (s, 3H, OCH3), and 3.62 (s, 3H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 161.8 (d, J = 2.1 Hz), 157.7 (d, J = 6.5 Hz), 135.1 (d, J = 3.0 Hz), 134.5 (d, J = 9.0 Hz), 132.5 (d, J = 3.1 Hz), 131.6 (d, J = 6.1 Hz), 130.4 (d, J = 6.2 Hz), 130.1 (d, J = 12.2 Hz), 129.2 (d, J = 1.9 Hz), 129.0 (d, J = 2.8 Hz), 118.3 (d, J = 82.4 Hz), 112.8 (d, J = 3.5 Hz), 105.5 (d, J = 1.6 Hz), 99.1 (d, J = 1.6 Hz), 55.6 (OCH3), 55.5 (OCH3), and 41.8 (d, J = 45.5 Hz, CαH) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 22.0 ppm; IR (ATR) 3058, 1614, 1582, 1505, 1438, 1215, and 1035 cm−1. The spectra reported here are in agreement with previously published data [30].
- 1-(naphthalen-1-yl)-1-(2,4,6-trimethoxyphenyl)methyltriphenylphosphonium tetrafluoroborate (7i). Resin (485.8 mg, 74%). 1H NMR (400 MHz, CDCl3) 7.90–7.76 (m, 3H, aromatic), 7.75–7.62 (m, 4H, aromatic), 7.60–7.39 (m, 13H, aromatic), 7.40–7.27 (m, 2H, aromatic), 7.19 (d, J = 19.4 Hz, 1H, CαH), 6.08 (s, 2H, aromatic), 3.81 (s, 3H, OCH3), and 3.44 (s, 6H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 162.8 (d, J = 1.8 Hz), 158.6 (d, J = 5.5 Hz), 134.7 (d, J = 3.1 Hz), 134.5 (d, J = 8.9 Hz), 134.0, 131.4 (d, J = 8.1 Hz), 130.8 (d, J = 6.4 Hz), 129.9 (d, J = 12.1 Hz), 129.8, 129.3 (d, J = 1.9 Hz), 129.3, 127.1, 126.1, 124.8 (d, J = 2.1 Hz), 122.7, 120.3 (d, J = 82.5 Hz), 102.1 (d, J = 3.6 Hz), 91.5 (d, J = 1.3 Hz), 55.8 (OCH3), 55.4 (OCH3), and 37.7 (d, J = 49.6 Hz, CαH) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 22.4 ppm; IR (ATR) 2940, 1604, 1587, 1458, 1207, 1149, 1097, 1049, 996, and 690 cm−1. HRMS (TOF-ESI) calcd. for C38H34O3P+ [M+] 569.2246 found 569.2252.
- 1-(H-indol-3-yl)-1-(naphthalen-1-yl)methyltriphenylphosphonium tetrafluoroborate (7j). Pink crystals (454.1 mg, 75%), mp 115–117 °C. 1H NMR (400 MHz, CDCl3) 9.81 (s, 1H, NH), 8.07–7.99 (m, 1H, aromatic), 7.87–7.75 (m, 2H, aromatic), 7.73–7.58 (m, 3H, aromatic), 7.54–7.35 (m, 15H, aromatic), 7.33–7.19 (m, 2H, aromatic), 7.07–6.95 (m, 3H, aromatic), 6.96 (d, J = 17.2 Hz, 1H, CαH), and 6.87–6.73 (m, 1H, aromatic) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 136.1, 135.3 (d, J = 3.0 Hz), 134.8 (d, J = 8.9 Hz), 134.2, 131.2 (d, J = 8.1 Hz), 130.3 (d, J = 12.0 Hz), 130.1 (d, J = 2.1 Hz), 130.0 (d, J = 1.8 Hz), 129.5, 128.5 (d, J = 7.1 Hz), 128.4 (d, J = 5.5 Hz), 127.8, 126.7, 126.0 (d, J = 6.4 Hz), 125.1 (d, J = 1.9 Hz), 122.6, 122.2, 120.2, 118.5 (d, J = 80.9 Hz), 117.5, 113.0, 104.9 (d, J = 4.8 Hz), and 38.4 (d, J = 47.1 Hz, CαH) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 19.6 ppm; IR (ATR) 3356, 3059, 1461, 1023, 997, 738, 719, and 690 cm−1. HRMS (TOF-ESI) calcd. for C37H29NP+ [M+] 518.2038 found 518.2036.
- 1-(tiophen-2-yl)-1-(2,4,6-trimethoxyphenyl)methyltriphenylphosphonium (7k). White crystals (575.7 mg, 94%), mp 195–197 °C (decomposition). 1H NMR (400 MHz, CDCl3) 7.83–7.76 (m, 3H, Ph), 7.66–7.58 (m, 6H, Ph), 7.39–7.29 (m, 6H, Ph), 7.24–7.19 (m, 1H, aromatic), 6.90–6.84 (m, 1H, aromatic), 6.84 (d, J = 18.7 Hz, 1H, CαH), 6.70 (br t, J = 3.5 Hz, 1H), 6.13 (s, 2H, aromatic), 3.86 (s, 3H, OCH3), and 3.48 (br s, 6H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 163.2 (d, J = 2.1 Hz), 158.9 (br s), 135.1 (d, J = 3.0 Hz), 134.3 (d, J = 8.8 Hz), 134.1 (d, J = 8.8 Hz), 130.2 (d, J = 12.2 Hz), 129.8 (d, J = 6.7 Hz), 127.4 (d, J = 4.1 Hz), 127.1 (d, J = 3.2 Hz), 119.0 (d, J = 82.5 Hz), 101.8 (d, J = 3.7 Hz), 91.2, 55.9 (OCH3), 55.5 (OCH3), and 37.8 (d, J = 49.2 Hz, CαH) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 21.9 ppm; IR (ATR) 3111, 2947, 1608, 1596, 1435, 1421, 1220, 1158, 1100, 1049, and 997 cm−1. HRMS (TOF-ESI) calcd. for C32H30O3PS+ [M+] 525.1653 found 525.1655.
- 1-(H-indol-3-yl)-1-(tiophen-2-yl)methyltriphenylphosphonium tetrafluoroborate (7l). Pink crystals (477.2 mg, 85%), mp 185–187 °C. 1H NMR (400 MHz, CDCl3) 9.87 (s, 1H, NH), 7.81–7.71 (m, 3H, Ph), 7.60–7.51 (m, 7H, aromatic), 7.40–7.30 (m, 6H, Ph), 7.21–7.15 (m, 2H, aromatic), 7.15–7.08 (m, 1H, aromatic), 6.95–6.87 (m, 2H, aromatic), 6.86–6.82 (m, 1H, aromatic), 6.78–6.72 (m, 1H, aromatic), and 6.45 (d, J = 16.4 Hz, 1H, CαH) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ aromatic carbons: 135.9, 135.6 (d, J = 2.9 Hz), 134.8 (d, J = 8.9 Hz), 130.4 (d, J = 12.1 Hz), 127.8 (d, J = 4.0 Hz), 127.7 (d, J = 3.2 Hz), 127.5 (d, J = 2.9 Hz), 127.4 (d, J = 2.7 Hz), 126.4 (d, J = 6.4 Hz), 123.1, 120.5, 117.5 (d, J = 82.0 Hz), 117.5, 113.2, 104.4 (d, J = 0.9 Hz), 104.4 (d, J = 1.7 Hz), and 38.4 (d, J = 47.5 Hz, CαH) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 19.9 ppm; IR (ATR) 3362, 3060, 1437, 1343, 1107, 1055, 996, and 688 cm−1. HRMS (TOF-ESI) calcd. for C31H25NPS+ [M+] 474.1445 found 474.1437.
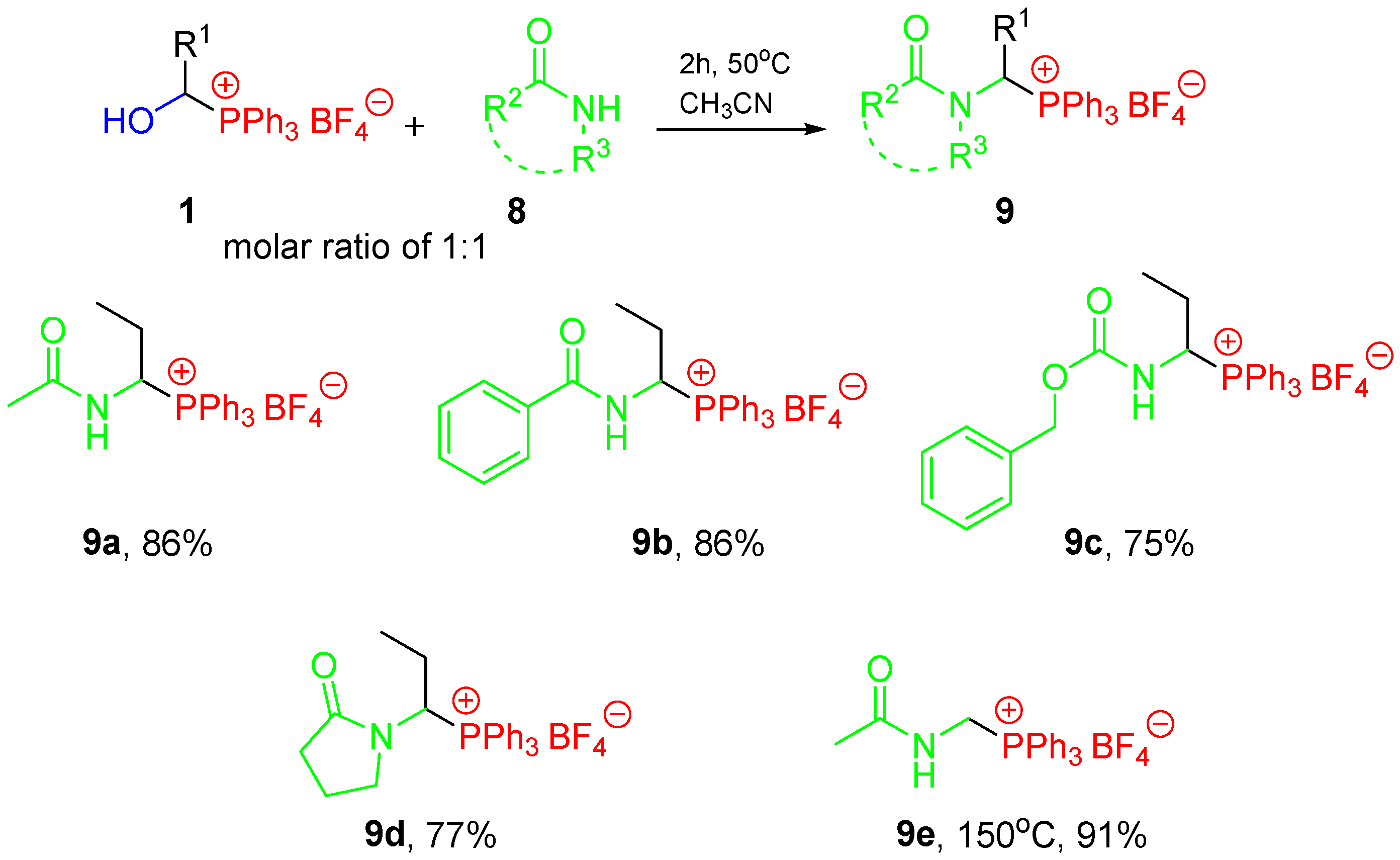
3.9. Reactions of 1-Hydroxyalkylphosphonium Salts with Amide-Type Substrates
- 1-(N-acetylamino)propyltriphenylphosphonium tetrafluoroborate (9a) [24]. White crystals (386.3 mg, 86%), mp 199–201 °C (lit.: mp 185–186 °C [24]). 1H NMR (400 MHz, CDCl3) δ 7.87–7.76 (m, 4H, Ph + NH), 7.74–7.66 (m, 12H, Ph), 5.67 (dddd, J = 12.0, 9.2, 7.7, 2.7 Hz, 1H, CαH), 2.13–1.97 (m, 1H, CHH), 1.91 (d, J = 1.2 Hz, 3H, CH3C=O), 1.86–1.70 (m, 1H, CHH), and 1.12 (td, J = 7.2, 1.3 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 172.6 (d, J = 3.0 Hz, C=O), aromatic carbons: 135.4 (d, J = 3.0 Hz), 134.3 (d, J = 9.3 Hz), 130.6 (d, J = 12.3 Hz), 117.2 (d, J = 81.7 Hz), 49.6 (d, J = 53.4 Hz, CαH), 25.2 (d, J = 5.4 Hz, CH2), 22.3 (CH3C=O), and 11.5 (d, J = 14.1 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 26.3 ppm; IR (ATR) 3330, 1683, 1522, 1440, 1286, 1109, 1062, 1020, and 996 cm−1. The spectra reported here are in agreement with previously published data [24].
- 1-(N-benzoylamino)propyltriphenylphosphonium tetrafluoroborate (9b) [24]. White crystals (383.5 mg, 75%), mp 197–198 °C (lit.: mp 198–199 °C [24]). 1H NMR (400 MHz, CDCl3) δ 8.39 (dd, J = 8.2, 2.5 Hz, 1H, NH), 7.83–7.73 (m, 8H, Ph), 7.72–7.68 (m, 3H, Ph), 7.68–7.59 (m, 6H, Ph), 7.48–7.42 (m, 1H, Ph), 7.39–7.33 (m, 2H, Ph), 5.81 (dtd, J = 11.5, 8.3, 3.0 Hz, 1H, CαH), 2.54–2.37 (m, 1H, CHH), 1.89–1.77 (m, 1H, CHH), and 1.17 (td, J = 7.2, 1.2 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 168.5 (d, J = 2.3 Hz, C=O), aromatic carbons: 134.9 (d, J = 3.0 Hz), 134.4 (d, J = 9.4 Hz), 132.3, 131.7, 130.1 (d, J = 12.3 Hz), 128.6, 127.4, 117.9 (d, J = 81.7 Hz), 51.4 (d, J = 51.7 Hz, CαH), 24.9 (d, J = 5.2 Hz, CH2), and 11.7 (d, J = 13.9 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 27.0 ppm; IR (ATR) 3357, 1670, 1508, 1483, 1438, 1111, 1070, 1030, and 996 cm−1. The spectra reported here are in agreement with previously published data [24].
- 1-(N-benzyloxycarbonylamino)propyltriphenylphosphonium tetrafluoroborate (9c) [24]. White crystals (465.5 mg, 86%), mp 176–177 °C (lit.: mp 161–162 °C [24]). 1H NMR (400 MHz, CDCl3) δ 7.81–7.71 (m, 3H, Ph), 7.70–7.56 (m, 12H, Ph), 7.33–7.24 (m, 3H, Ph), 7.24–7.17 (m, 2H, Ph), 6.93 (d, J = 9.1 Hz, 1H, NH), 5.45–5.33 (m, 1H, CαH), 4.98, 4.89 (ABq, J = 12.5 Hz, 2H, CH2), 2.28–2.14 (m, 1H, CHH), 1.83–1.70 (m, 1H, CHH), and 1.15 (td, J = 7.2, 1.3 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 156.9 (d, J = 2.9 Hz, C=O), aromatic carbons: 136.1, 135.3 (d, J = 2.9 Hz), 134.3 (d, J = 9.3 Hz), 130.5 (d, J = 12.3 Hz), 128.6, 128.1, 128.0, 117.1 (d, J = 81.2 Hz), 67.4 (CH2Ph), 52.7 (d, J = 52.9 Hz, CαH), 25.0 (d, J = 6.2 Hz, CH2), and 11.5 (d, J = 13.9 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 25.6 ppm; IR (ATR) 3334, 1712, 1519, 1439, 1227, 1110, 1063, 1035, 1009, and 995 cm−1. The spectra reported here are in agreement with previously published data [24].
- 1-(2-Oxopyrrolidin-1-yl)propyltriphenylphosphonium tetrafluoroborate (9d) [24]. White crystals (366.0 mg, 77%), mp 189–191 °C (lit.: mp 186–188 °C [24]). 1H NMR (400 MHz, CDCl3) δ 7.91–7.80 (m, 3H, Ph), 7.79–7.65 (m, 12H, Ph), and 5.72 (ddd, J = 12.4, 10.5, 3.1 Hz, 1H, CαH), CH2 groups: 3.57–3.42 (m, 1H), 3.32–3.17 (m, 1H), 2.48–2.28 (m, 1H), 2.27–2.09 (m, 2H), 1.98–1.80 (m, 3H), and 1.07 (td, J = 7.2, 1.3 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 177.0 (d, J = 2.3 Hz, C=O), aromatic carbons: 135.6 (d, J = 3.1 Hz), 134.3 (d, J = 9.7 Hz), 130.8 (d, J = 12.4 Hz), 117.1 (d, J = 81.4 Hz), 53.3 (d, J = 51.4 Hz, CαH), 46.9 (CH2N), 30.3 (CH2), 22.8 (d, J = 5.0 Hz, CH2), 18.6 (CH2), and 11.5 (d, J = 14.1 Hz, CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 24.7 ppm; IR (ATR) 2880, 1692, 1509, 1438, 1405, 1271, 1109, 1047, 1035, and 997 cm−1. The spectra reported here are in agreement with previously published data [24].
- (N-Acetylamino)methyltriphenylphosphonium tetrafluoroborate (9e) [24,31]. White crystals (383.3 mg, 91%), mp 191–193 °C (lit.: mp 191–193 °C [24]). 1H NMR (400 MHz, CDCl3) δ 9.67 (t, J = 6.2 Hz, 1H, NH), 7.86–7.76 (m, 9H, Ph), 7.74–7.63 (m, 6H, Ph), 5.13 (dd, J = 6.3, 2.9 Hz, 2H, CH2), and 1.89 (d, J = 1.4 Hz, 3H, CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 172.2 (d, J = 1.4 Hz, C=O), aromatic carbons: 135.3 (d, J = 3.1 Hz), 134.2 (d, J = 9.8 Hz), 130.3 (d, J = 12.6 Hz), 117.5 (d, J = 83.9 Hz), 37.6 (d, J = 56.8 Hz, CH2), and 22.6 (CH3) ppm; 31P{1H} NMR (161.9 MHz, CDCl3) δ 20.7 ppm; IR (ATR) 3356, 1671, 1508, 1483, 1437, 1111, 1071, 1030, and 996 cm−1. The spectra reported here are in agreement with previously published data [24,31].
- 2-Carbamoyletyhyltriphenylphosphonium tetrafluoroborate (10) [24]. White crystals (315.9 mg, 75%), mp 145–147 °C (lit.: mp 146–148 °C [24]). 1H NMR (400 MHz, CDCl3) δ 7.85–7.64 (m, 15H, Ph), 6.98 (br s, 1H, NH), 5.33 (br s, 1H, NH), 3.49–3.41 (m, 2H, CH2), and 2.85–2.78 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.3 (d, J = 14.2 Hz), 135.4 (d, J = 3.0 Hz), 133.5 (d, J = 10.0 Hz), 130.7 (d, J = 12.7 Hz), 117.5 (d, J = 86.7 Hz), 27.3 (d, J = 2.9 Hz), and 19.1 (d, J = 56.0 Hz); 31P{1H} NMR (161,9 MHz, CDCl3) δ 24.6; IR (ATR) 3422, 3198, 1669, 1441, 1025, and 996 cm−1. The spectra reported here are in agreement with previously published data [24].
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Johnson, A.W. Ylides and Imines of Phosphorus; A Wiley-Interscience Publication: New York, NY, USA, 1993; pp. 221–300. [Google Scholar]
- Kolodiazhnyi, O.I. Phosphorus Ylides; Wiley: Weinheim, Germany, 1999; pp. 359–539. [Google Scholar]
- Hilton, M.C.; Dolewski, R.D.; McNally, A. Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc. 2016, 138, 13806–13809. [Google Scholar] [CrossRef]
- Deng, Z.; Lin, J.-H.; Xiao, J.-C. Nucleophilic arylation with tetraarylphosphonium salts. Nat. Commun. 2016, 7, 10337. [Google Scholar] [CrossRef]
- Babu, K.N.; Massarwe, F.; Shioukhi, I.; Masarwa, A. Sequential Selective C-H and C(sp3)-+P Bond Functionalizations: An Entry to Bioactive Arylated Scaffolds. Angew. Chem. Int. Ed. 2021, 60, 26199–26209. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, M.; Wang, Y.; Man, X.; Lu, X.; Mou, Z.; Luo, Y.; Liang, H. Nickel-Catalyzed Reductive Csp2–Csp3 Cross Coupling Using Phosphonium Salts. Org. Lett. 2021, 23, 8183–8188. [Google Scholar] [CrossRef] [PubMed]
- Adamek, J.; Grymel, M.; Kuźnik, A.; Październiok-Holewa, A. 1-Aminoalkylphosphonium Derivatives: Smart Synthetic Equivalents of N-Acyliminium-Type Cations, and Maybe Something More: A Review. Molecules 2022, 27, 1562. [Google Scholar] [CrossRef] [PubMed]
- Selva, M.; Perosa, A.; Noè, M. Organophosphorous Chemistry; Tebby, J.C., Loakes, D., Allen, D.W., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; pp. 132–169. [Google Scholar]
- Werner, T. Phosphonium Salt Organocatalysis. Adv. Synth. Catal. 2009, 351, 1469–1481. [Google Scholar] [CrossRef]
- He, R.; Ding, C.; Marouka, K. Phosphonium salts as chiral phase-transfer catalysts: Asymmetric Michael and Mannich reactions of 3-aryloxindoles. Angew. Chem. 2009, 48, 4559–4561. [Google Scholar] [CrossRef]
- Bradaric, C.J.; Downard, A.; Kennedy, C.; Robertson, A.J.; Zhou, Y. Industrial preparation of phosphonium ionic liquids. Green Chem. 2003, 5, 143–152. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H. Umsetzung von Triphenylphosphin mit Aldehyden. Angew. Chem. 1960, 72, 77. [Google Scholar] [CrossRef]
- Wittig, G.; Schlosser, M. Über die Herstellung von Vinyläthern, Vinylthioäthern und Vinylhalogeniden auf der Phosphylen-Basis; IV. Mitteil. Über Phosphin-alkylene als olefinbildende Reagenzien. Chem. Ber. 1961, 94, 1373–1383. [Google Scholar] [CrossRef]
- Hellmann, H.; Bader, J.; Birkner, H.; Schumacher, O. Hydroxymethyl-phosphine, Hydroxymethyl-phosphoniumsalze und Chlormethyl-phosphoniumsalze. Liebigs Ann. Chem. 1962, 659, 49–63. [Google Scholar] [CrossRef]
- Henderson, W.; Olsen, G.M. Application of electrospray mass spectrometry to the characterization of hydroxymethylphosphonium salts, -phosphines, and their oxide, sulfide and selenide derivatives. Polyhedron 1996, 15, 2105–2115. [Google Scholar] [CrossRef]
- Allen, D.W.; Millar, I.T. Unusually low PCH coupling constants in the nuclear magnetic resonance spectra of phosphonium salts and phosphine oxides. Tetrahedron Lett. 1968, 9, 745–750. [Google Scholar] [CrossRef]
- Anders, E.; Gaβner, T.; Stankowiak, A. [1-(Aryl-bzw. Alkylcarbonyloxy)alkyl]phosphonium-Salze]—Herstellungsmethoden. Chem. Ber. 1985, 118, 124–131. [Google Scholar] [CrossRef]
- Burton, D.J.; Wiemers, D.M. A practical synthesis of fluoromethyltriphenylphosphonium salts. J. Fluor. Chem. 1985, 27, 85–89. [Google Scholar] [CrossRef]
- Davis, M.C.; Parrish, D.A. Synthesis of 4-(N,N-Dialkylamino)benzyltriphenylphosphonium Iodides from Hydroxymethyltriphenylphosphonium Iodide and N,N-Dialkylaniline. Synth. Commun. 2008, 38, 3909–3918. [Google Scholar] [CrossRef]
- Dal Canto, R.A.; Roskamp, E.J. Addition of triphenylphosphonium salts to aldehydes. Remarkable counterion effects on phosphorus proton couplings. J. Org. Chem. 1992, 57, 406–407. [Google Scholar] [CrossRef]
- Adamek, J.; Zielezny, P.; Erfurt, K. Synthesis of N-Protected 1-Aminoalkylphosphonium Salts from Amides, Carbamates, Lactams, or Imides. J. Org. Chem. 2021, 86, 5852–5862. [Google Scholar] [CrossRef] [PubMed]
- Hazra, G.; Masarwa, A. Synthesis and Functionalization of Thiophosphonium Salts: A Divergent Approach to Access Thioether, Thioester, and Dithioester Derivatives. Org. Lett. 2023, 25, 6396–6400. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.D.; Williams, C.M.; Anders, E.; Bernhardt, P.V. Exploiting the Anders–Gaßner variant on the Wittig reaction: New methodology for the synthesis of 3,3-dimethylacroyl enol esters. Tetrahedron 2008, 64, 6482–6487. [Google Scholar] [CrossRef]
- Huang, W.; Xu, J. In Situ Generation of Formaldehyde and Triphenylphosphine from (Hydroxymethyl)triphenylphosphonium and Its Application in Wittig Olefination. Synth. Commun. 2015, 45, 1777–1782. [Google Scholar] [CrossRef]
- Konrath, R.; Sekine, K.; Jevtovikj, I.; Paciello, R.A.; Hashmi, A.S.K.; Schaub, T. Performance enhancing additives for reusable ruthenium-triphos catalysts in the reduction of CO2 to dimethoxymethane. Green Chem. 2020, 22, 6464–6470. [Google Scholar] [CrossRef]
- Seyferth, D.; Heeren, J.K.; Singh, G.; Grim, S.O.; Hughes, W.B. Studies in phosphinemethylene chemistry: XIII. Routes to triphenylphosphine-halomethylenes and -dihalomethylenes. J. Organomet. Chem. 1966, 5, 267–274. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Krawczyk, M.; Erfurt, K. Catalyst-free Friedel-Crafts reaction of 1-(N-acylamino)alkyltriarylphosphonium salts with electron-rich arenes. Tetrahedron 2018, 74, 2575–2583. [Google Scholar] [CrossRef]
- Adamek, J.; Mrowiec-Białoń, J.; Październiok-Holewa, A.; Mazurkiewicz, R. Thermogravimetrical investigations of the dealkoxycarbonylation of N-acyl-α-triphenylphosphonioglycinates. Thermochim. Acta 2011, 512, 22–27. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Phosphonium Salts 1 | Time, min | Temp., °C | Solvent | Yield, % | ||
| No. | Ar | X | |||||
| 1 | 1a | Ph | BF4 | 30 | r.t. | MeCN | 99 a/92/85 b |
| 2 | 1a | Ph | BF4 | 30 | r.t. | CH2Cl2 | 93 |
| 3 | 1a | Ph | BF4 | 30 | r.t. | THF | c |
| 4 | 1b | Ph | Br | 30 | r.t. | MeCN | 88 |
| 5 | 1c | Ph | TfO | 30 | r.t. | MeCN | 99 a/79 |
| 6 | 1d | 3-C6H4Cl | BF4 | 30 | r.t. | MeCN | d |
| 7 | 1e | 4-C6H4OMe | BF4 | 30 | r.t. | MeCN | 99 a/66 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Phosphonium Salts 1 | Time, min | Temp., °C | Solvent | Yield, % | ||
| No. | R1 | X | |||||
| 1 | 1f | H | BF4 | 30 | r.t. | CHCl3 | n.r. |
| 2 | 1f | H | BF4 | 120 | 50 | CHCl3 | 88 |
| 3 | 1f | H | BF4 | 120 | 50 | MeCN | 95 |
| 4 | 1g | H | TfO | 120 | 50 | MeCN | 93 |
| 5 | 1h | H | Br | 120 | 50 | MeCN | 95/87 a |
| 6 | 1i | Me | BF4 | 30 | r.t. | MeCN | 49 |
| 7 | 1i | Me | BF4 | 30 | r.t. | CH2Cl2 | 99 |
| 8 | 1j | ClCH2 | BF4 | 30 | r.t. | CHCl3 | 89 b |
| 9 | 1k | n-C9H19 | BF4 | 30 | r.t. | CHCl3 | 99 c |
| 10 | 1k | n-C9H19 | BF4 | 30 | r.t. | MeCN | d |
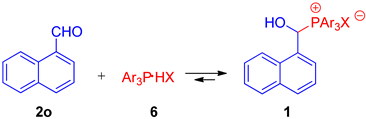 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | No. | Molar Ratio of 2o:6 | Ar | X | Time, min | Temp., °C | Solvent | Molar Ratio of 1:2(6) a |
| 1 | 1o | 1:1 | Ph | BF4 | 5 | 25 | CD3CN | 57:43 |
| 2 | 1o | 1:1 | Ph | BF4 | 15 | 25 | CD3CN | 57:43 |
| 3 | 1o | 1:1 | Ph | BF4 | 30 | 25 | CD3CN | 57:43 |
| 4 | 1o | 1:1 | Ph | BF4 | 5/65 | 25 | CD3CN | 57:43 |
| 5 | 1o | 1:1 | Ph | BF4 | 25 | 35 | CD3CN | 48:52 |
| 6 | 1o | 1:1 | Ph | BF4 | 50 | 45 | CD3CN | 35:65 |
| 7 | 1p | 1:1 | Ph | TfO | 5/65 | 25 | CD3CN | 62:38 |
| 8 | 1p | 1:1 | Ph | TfO | 25 | 35 | CD3CN | 55:45 |
| 9 | 1p | 1:1 | Ph | TfO | 50 | 45 | CD3CN | 44:56 |
| 10 | 1q | 1:1 | 4-C6H4OMe | BF4 | 5/65 | 25 | CD3CN | 59:41 |
| 11 | 1q | 1:1 | 4-C6H4OMe | BF4 | 25 | 35 | CD3CN | 48:52 |
| 12 | 1q | 1:1 | 4-C6H4OMe | BF4 | 50 | 45 | CD3CN | 39:61 |
| 13 | 1o | 1:1 | Ph | BF4 | 5/65 | 25 | CDCl3 | 45:55 |
| 14 | 1o | 1:1 | Ph | BF4 | 25 | 35 | CDCl3 | 35:65 |
| 15 | 1o | 1:1 | Ph | BF4 | 50 | 45 | CDCl3 | 25:75 |
| 16 | 1o | 1:1 | Ph | BF4 | 5/15/30 | 25 | CD3CN | 57:43 |
| 17 | 1o | 2:1 | Ph | BF4 | 5/15/30 | 25 | CD3CN | 75:25 |
| 18 | 1o | 3:1 | Ph | BF4 | 5/15/30 | 25 | CD3CN | 88:12 |
| 19 | 1o | 5:1 | Ph | BF4 | 5/15/30 | 25 | CD3CN | 94:6 |
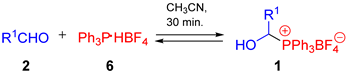 | |||||
|---|---|---|---|---|---|
| Entry | Phosphonium Salts 1 | Temp., °C | Solvent | Molar Ratio of 1:2(6) a | |
| No. | R1 | ||||
| 1 | 1r |  | 25 | CDCl3 | 57:43 |
| CD3CN | 82:18 | ||||
| 2 | 1s | 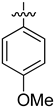 | 25 | CD3CN | 67:33 |
| 3 | 1t | 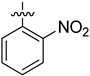 | 25 | CD3CN | 82:18 |
| 4 | 1u | 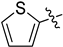 | 25 | CD3CN | 63:37 |
| 5 | 1v | i-Pr | 25 | CD3CN | 88:12 |
| 6 | 1v | i-Pr | 45 | CD3CN | 72:28 |
| 7 | 1w | 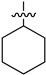 | 25 | CDCl3 | 68:32 |
| 8 | 1x | 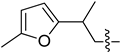 | 25 | CD3CN | 84:16 b |
| 9 | 1y | 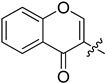 | 25/50 | CD3CN | no reaction |
 | ||||
|---|---|---|---|---|
| Entry | Molar Ratio | Procedure | Solvent | Yield, % |
| 1 | 1:5 (1a:ArH) | step-by-step | CD3CN | 89 a/76 b |
| 2 | 1:2 (1a:ArH) | step-by-step | CD3CN | 83 a/82 b |
| 3 | 1:5 (1a:ArH) | step-by-step | no solvent | 57 b |
| 4 | 1:1:5 (2a:6:ArH) | one-pot | CD3CN | 77 b |
| 5 | 1:1:1:5 (2a:PPh3:HBF4:ArH) c | one-pot | CD3CN | 78 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamek, J.; Kuźnik, A.; Październiok-Holewa, A.; Grymel, M.; Kozicka, D.; Mierzwa, D.; Erfurt, K. 1-Hydroxyalkylphosphonium Salts—Synthesis and Properties. Molecules 2024, 29, 18. https://doi.org/10.3390/molecules29010018
Adamek J, Kuźnik A, Październiok-Holewa A, Grymel M, Kozicka D, Mierzwa D, Erfurt K. 1-Hydroxyalkylphosphonium Salts—Synthesis and Properties. Molecules. 2024; 29(1):18. https://doi.org/10.3390/molecules29010018
Chicago/Turabian StyleAdamek, Jakub, Anna Kuźnik, Agnieszka Październiok-Holewa, Mirosława Grymel, Dominika Kozicka, Dominika Mierzwa, and Karol Erfurt. 2024. "1-Hydroxyalkylphosphonium Salts—Synthesis and Properties" Molecules 29, no. 1: 18. https://doi.org/10.3390/molecules29010018
APA StyleAdamek, J., Kuźnik, A., Październiok-Holewa, A., Grymel, M., Kozicka, D., Mierzwa, D., & Erfurt, K. (2024). 1-Hydroxyalkylphosphonium Salts—Synthesis and Properties. Molecules, 29(1), 18. https://doi.org/10.3390/molecules29010018