
Aptamer-Based Imaging of Polyisoprenoids in the Malaria Parasite

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
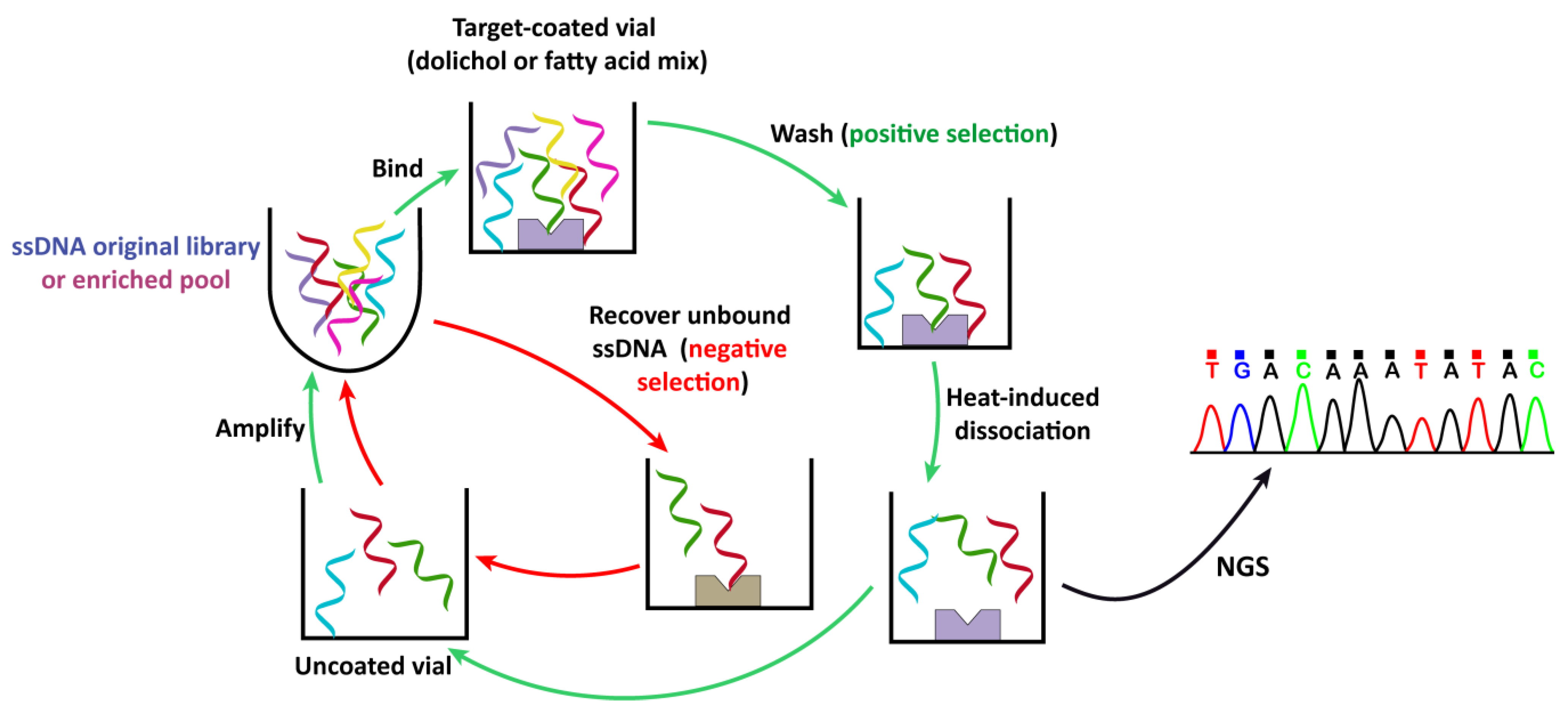
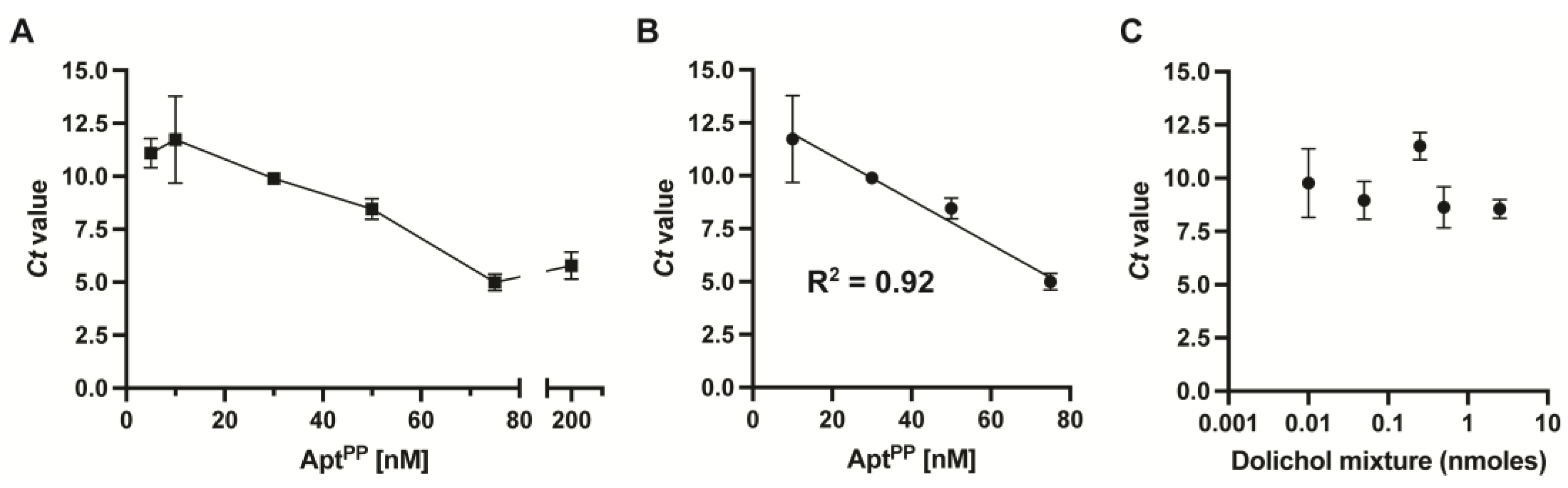
2.1. Selection of Aptamers against Polyisoprenoids
2.2. Families of DNA Aptamers Obtained through the Selection Process
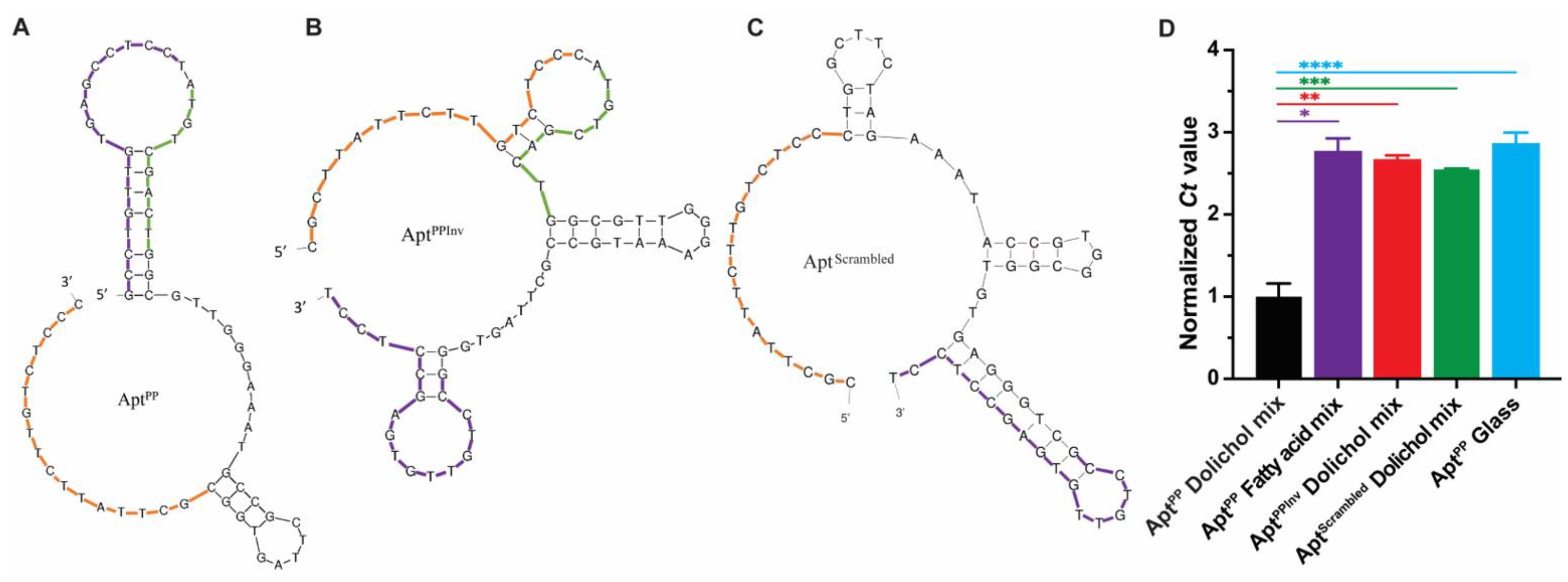
2.3. Secondary Structure Prediction of AptPP and Variant Sequence
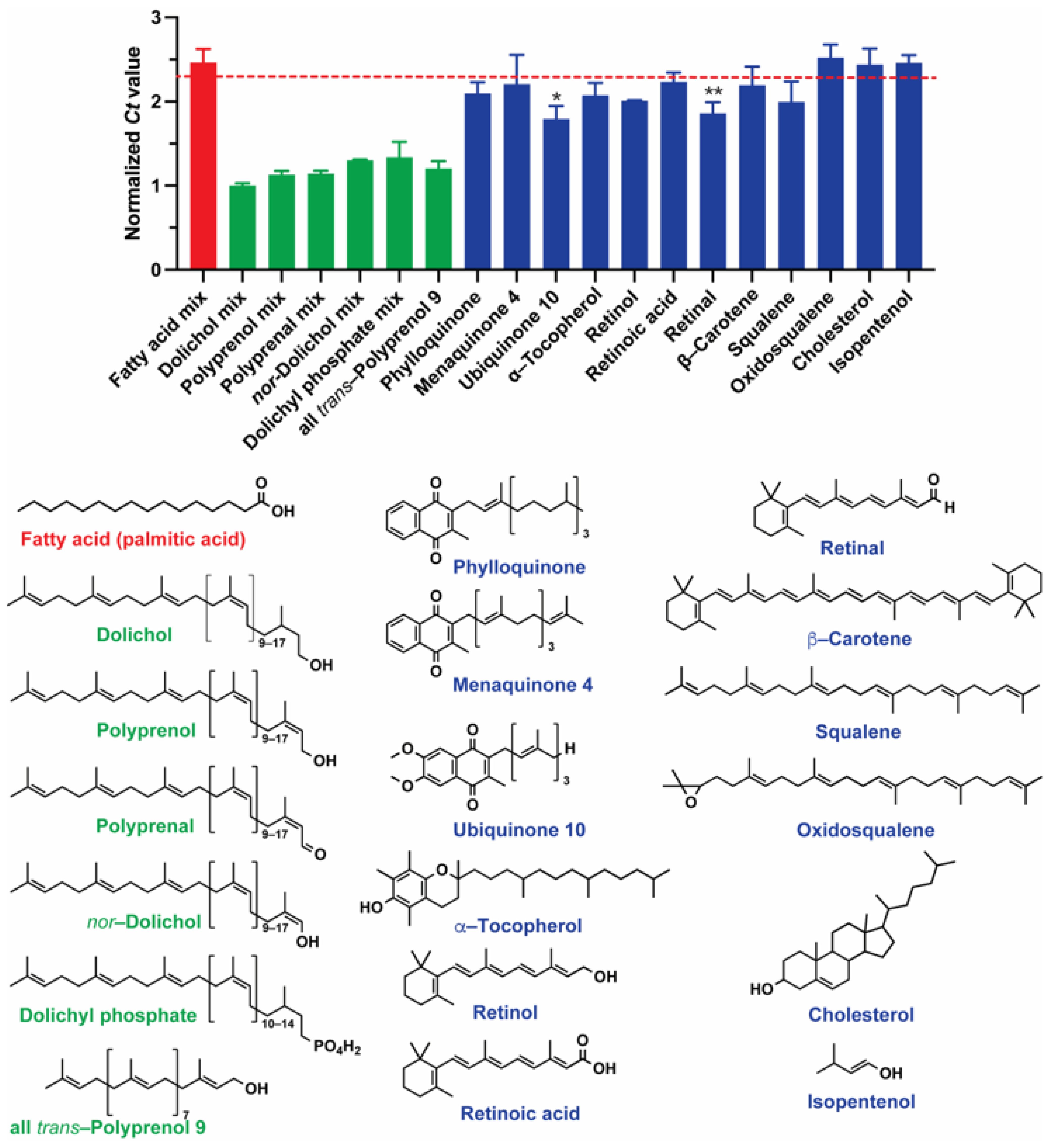
2.4. Specificity of AptPP for Different Isoprenoid Products
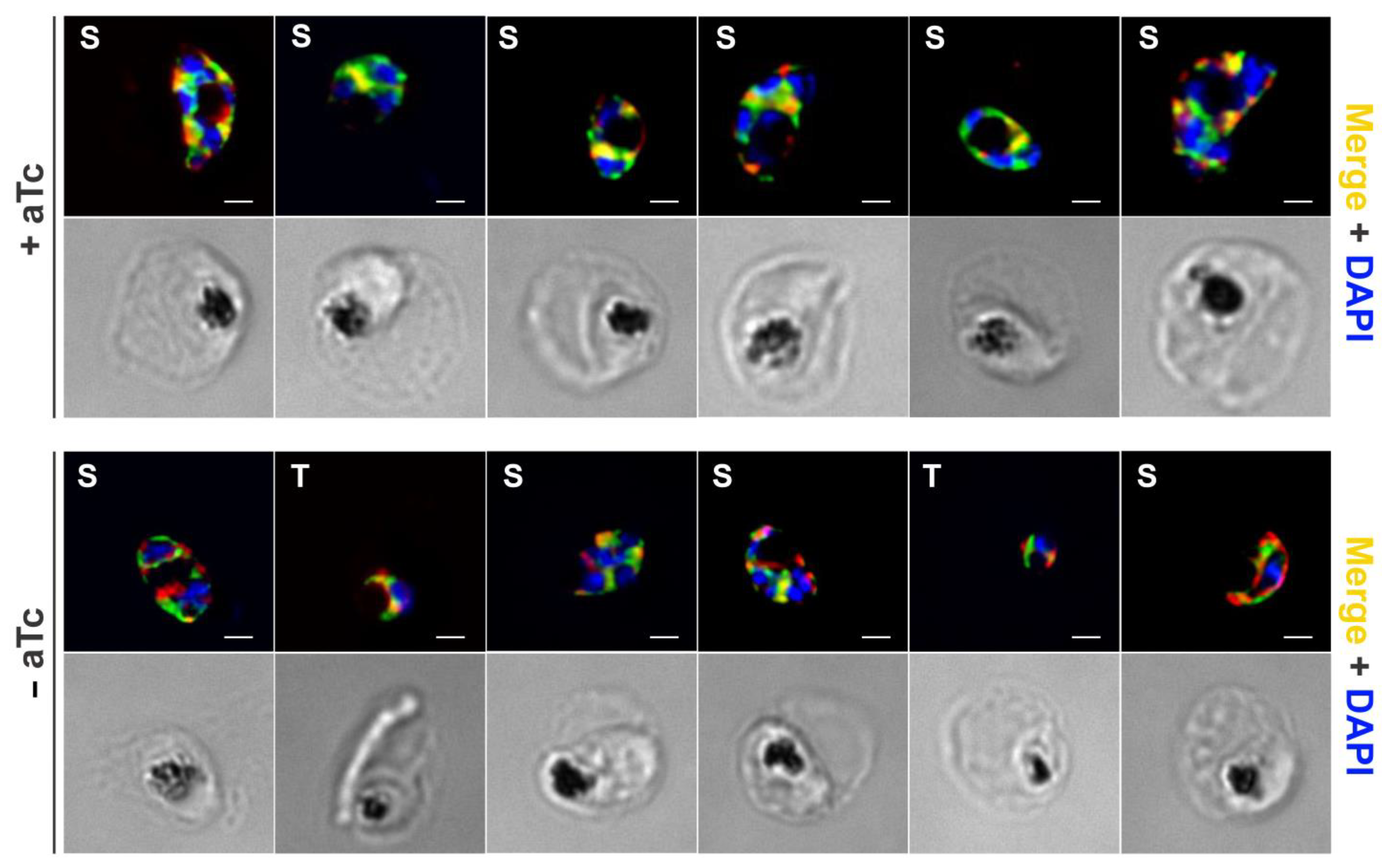
2.5. Subcellular Localization of AptPP in Plasmodium falciparum
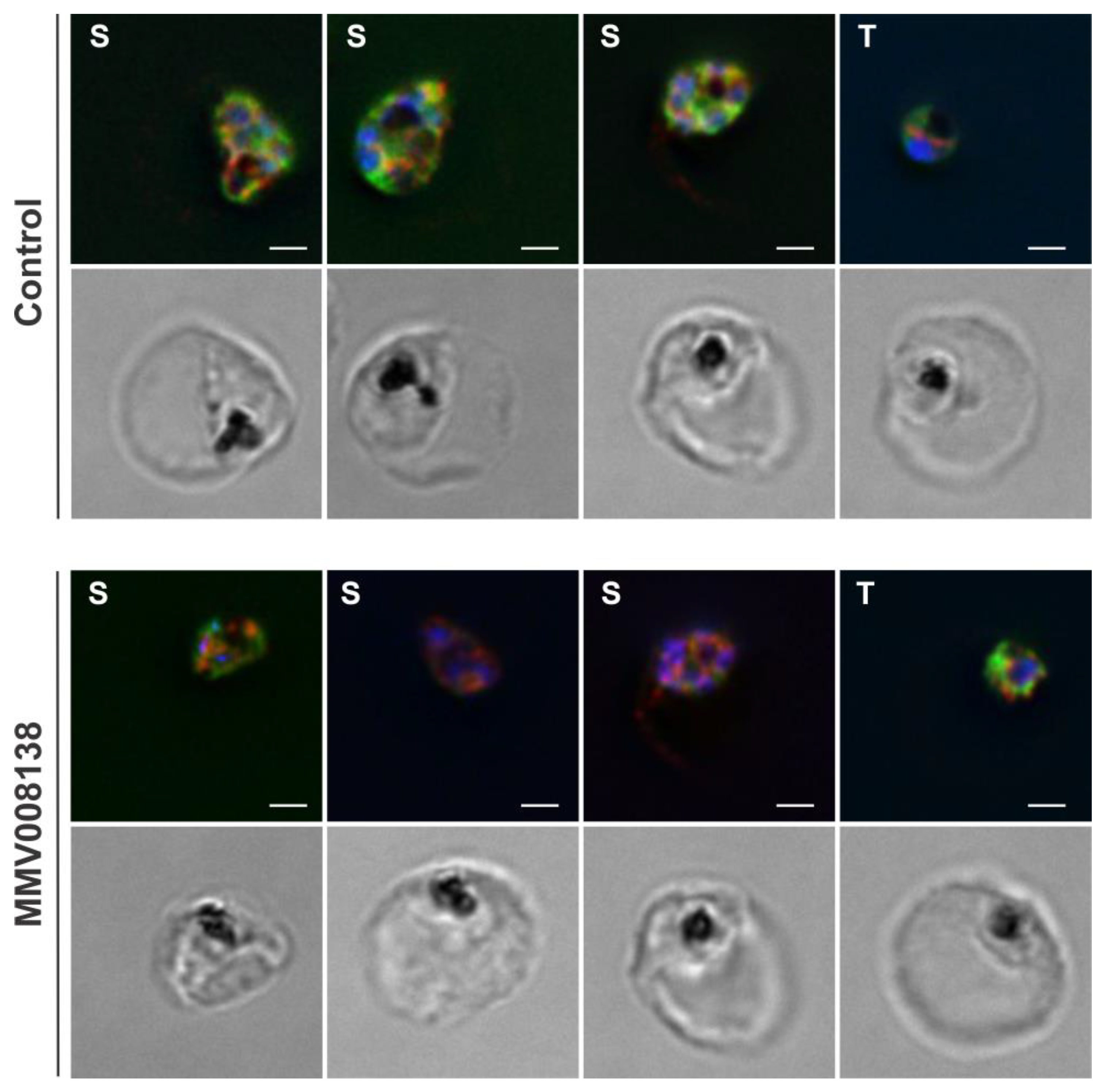
2.6. Validation of the Specificity of AptPP within the Cellular Environment
2.7. Beyond Polyisoprenoid Detection
3. Materials and Methods
3.1. Chemicals
3.2. Systematic Evolution of Ligands by Exponential Enrichment (SELEX)
3.3. Sequencing and Bioinformatics Analysis
3.4. Quantitative Real-Time PCR
3.5. Non-Equilibrium Capillary Electrophoresis of Equilibrium Mixtures (NECEEM)
3.6. Plasmodium falciparum In Vitro Culture
3.7. Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, T.; Ray, J. Aptamer-based molecular imaging. Protein Cell 2012, 3, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Zhuo, Z.; Pan, Y.; Yu, Y.; Li, F.; Liu, J.; Wang, L.; Wu, X.; Li, D.; Wan, Y.; et al. Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications. ACS Appl. Mater. Interfaces 2021, 13, 9500–9519. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Cantagrel, V.; Lefeber, D.J. From glycosylation disorders to dolichol biosynthesis defects: A new class of metabolic diseases. J. Inherit. Metab. Dis. 2011, 34, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, T.; Dallner, G. The biological role of dolichol. Biochem. J. 1988, 251, 1–9. [Google Scholar] [CrossRef]
- Akhtar, T.A.; Surowiecki, P.; Siekierska, H.; Kania, M.; Van Gelder, K.; Rea, K.A.; Virta, L.K.A.; Vatta, M.; Gawarecka, K.; Wojcik, J.; et al. Polyprenols Are Synthesized by a Plastidial cis-Prenyltransferase and Influence Photosynthetic Performance. Plant Cell 2017, 29, 1709–1725. [Google Scholar] [CrossRef] [PubMed]
- Gryz, E.; Perlinska-Lenart, U.; Gawarecka, K.; Jozwiak, A.; Pilsyk, S.; Lipko, A.; Jemiola-Rzeminska, M.; Bernat, P.; Muszewska, A.; Steczkiewicz, K.; et al. Poly-Saturated Dolichols from Filamentous Fungi Modulate Activity of Dolichol-Dependent Glycosyltransferase and Physical Properties of Membranes. Int. J. Mol. Sci. 2019, 20, 3043. [Google Scholar] [CrossRef]
- Hoffmann, R.; Grabinska, K.; Guan, Z.; Sessa, W.C.; Neiman, A.M. Long-Chain Polyprenols Promote Spore Wall Formation in Saccharomyces cerevisiae. Genetics 2017, 207, 1371–1386. [Google Scholar] [CrossRef]
- Cavallini, G.; Sgarbossa, A.; Parentini, I.; Bizzarri, R.; Donati, A.; Lenci, F.; Bergamini, E. Dolichol: A Component of the Cellular Antioxidant Machinery. Lipids 2016, 51, 477–486. [Google Scholar] [CrossRef]
- Bergamini, E.; Bizzarri, R.; Cavallini, G.; Cerbai, B.; Chiellini, E.; Donati, A.; Gori, Z.; Manfrini, A.; Parentini, I.; Signori, F.; et al. Ageing and oxidative stress: A role for dolichol in the antioxidant machinery of cell membranes? J. Alzheimers Dis. 2004, 6, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Komaszylo nee Siedlecka, J.; Kania, M.; Masnyk, M.; Cmoch, P.; Lozinska, I.; Czarnocki, Z.; Skorupinska-Tudek, K.; Danikiewicz, W.; Swiezewska, E. Isoprenoid Alcohols are Susceptible to Oxidation with Singlet Oxygen and Hydroxyl Radicals. Lipids 2016, 51, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Qidwai, T.; Priya, A.; Khan, N.A.; Tripathi, H.; Khan, F.; Darokar, M.P.; Pal, A.; Bawankule, D.U.; Shukla, R.K.; Bhakuni, R.S. Antimalarial drug targets and drugs targeting dolichol metabolic pathway of Plasmodium falciparum. Curr. Drug Targets 2014, 15, 374–409. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.S.; Manjunatha, U.H.; Mikolajczak, S.; Ashigbie, P.G.; Diagana, T.T. Drug discovery for parasitic diseases: Powered by technology, enabled by pharmacology, informed by clinical science. Trends Parasitol. 2023, 39, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Cova, M.; Rodrigues, J.A.; Smith, T.K.; Izquierdo, L. Sugar activation and glycosylation in Plasmodium. Malar. J. 2015, 14, 427. [Google Scholar] [CrossRef] [PubMed]
- Zimbres, F.M.; Valenciano, A.L.; Merino, E.F.; Florentin, A.; Holderman, N.R.; He, G.; Gawarecka, K.; Skorupinska-Tudek, K.; Fernandez-Murga, M.L.; Swiezewska, E.; et al. Metabolomics profiling reveals new aspects of dolichol biosynthesis in Plasmodium falciparum. Sci. Rep. 2020, 10, 13264. [Google Scholar] [CrossRef] [PubMed]
- Janas, T.; Janas, T. The selection of aptamers specific for membrane molecular targets. Cell Mol. Biol. Lett. 2011, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Kara, N.; Ayoub, N.; Ilgu, H.; Fotiadis, D.; Ilgu, M. Aptamers Targeting Membrane Proteins for Sensor and Diagnostic Applications. Molecules 2023, 28, 3728. [Google Scholar] [CrossRef]
- Berezovski, M.V.; Musheev, M.U.; Drabovich, A.P.; Jitkova, J.V.; Krylov, S.N. Non-SELEX: Selection of aptamers without intermediate amplification of candidate oligonucleotides. Nat. Protoc. 2006, 1, 1359–1369. [Google Scholar] [CrossRef]
- Hoinka, J.; Zotenko, E.; Friedman, A.; Sauna, Z.E.; Przytycka, T.M. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 2012, 28, i215–i223. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Liu, Y.; Zeng, J.; Li, X.; Shao, N.; Mao, A.; Wang, L.; Ma, J.; Cen, H.; Wang, Y.; et al. Aptamer-Based Sensitive Detection of Target Molecules via RT-PCR Signal Amplification. Bioconjug Chem. 2010, 21, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.; Cobbold, S.A.; Hanssen, E.; Birnbaum, J.; Spillman, N.J.; McHugh, E.; Brown, H.; Tilley, L.; Spielmann, T.; McConville, M.J.; et al. Delayed death in the malaria parasite Plasmodium falciparum is caused by disruption of prenylation-dependent intracellular trafficking. PLoS Biol. 2019, 17, e3000376. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Rajaram, K.; Swift, R.P.; Mixon, A.; Maschek, J.A.; Prigge, S.T.; Sigala, P.A. Critical role for isoprenoids in apicoplast biogenesis by malaria parasites. Elife 2022, 11, e73208. [Google Scholar] [CrossRef] [PubMed]
- de Macedo, C.S.; Uhrig, M.L.; Kimura, E.A.; Katzin, A.M. Characterization of the isoprenoid chain of coenzyme Q in Plasmodium falciparum. FEMS Microbiol. Lett. 2002, 207, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Tonhosolo, R.; D’Alexandri, F.L.; Genta, F.A.; Wunderlich, G.; Gozzo, F.C.; Eberlin, M.N.; Peres, V.J.; Kimura, E.A.; Katzin, A.M. Identification, molecular cloning and functional characterization of an octaprenyl pyrophosphate synthase in intra-erythrocytic stages of Plasmodium falciparum. Biochem. J. 2005, 392 Pt 1, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Buczkowska, A.; Swiezewska, E.; Lefeber, D.J. Genetic defects in dolichol metabolism. J. Inherit. Metab. Dis. 2015, 38, 157–169. [Google Scholar] [CrossRef]
- Wolf, M.J.; Rush, J.S.; Waechter, C.J. Golgi-enriched membrane fractions from rat brain and liver contain long-chain polyisoprenyl pyrophosphate phosphatase activity. Glycobiology 1991, 1, 405–410. [Google Scholar] [CrossRef]
- Adisa, A.; Frankland, S.; Rug, M.; Jackson, K.; Maier, A.G.; Walsh, P.; Lithgow, T.; Klonis, N.; Gilson, P.R.; Cowman, A.F.; et al. Re-assessing the locations of components of the classical vesicle-mediated trafficking machinery in transfected Plasmodium falciparum. Int. J. Parasitol. 2007, 37, 1127–1141. [Google Scholar] [CrossRef]
- Struck, N.S.; Herrmann, S.; Schmuck-Barkmann, I.; de Souza Dias, S.; Haase, S.; Cabrera, A.L.; Treeck, M.; Bruns, C.; Langer, C.; Cowman, A.F.; et al. Spatial dissection of the cis- and trans-Golgi compartments in the malaria parasite Plasmodium falciparum. Mol. Microbiol. 2008, 67, 1320–1330. [Google Scholar] [CrossRef]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Park. Dis. 2018, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.D.; Merino, E.F.; Brooks, C.F.; Striepen, B.; Carlier, P.R.; Cassera, M.B. Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the malaria box. Antimicrob. Agents Chemother. 2014, 58, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, M.; Merino, E.F.; Yao, Z.K.; Elahi, R.; Simpson, M.E.; Fernandez-Murga, M.L.; Butler, J.H.; Casasanta, M.A.; Krai, P.M.; Totrov, M.M.; et al. Biological Studies and Target Engagement of the 2-C-Methyl-d-Erythritol 4-Phosphate Cytidylyltransferase (IspD)-Targeting Antimalarial Agent (1 R,3 S)-MMV008138 and Analogs. ACS Infect. Dis. 2018, 4, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.A.; Chen, H.; Hicke, B.J.; Swiderek, K.M.; Gold, L. A tenascin-C aptamer identified by tumor cell SELEX: Systematic evolution of ligands by exponential enrichment. Proc. Natl. Acad. Sci. USA 2003, 100, 15416–15421. [Google Scholar] [CrossRef] [PubMed]
- Quang, N.N.; Miodek, A.; Cibiel, A.; Duconge, F. Selection of Aptamers against Whole Living Cells: From Cell-SELEX to Identification of Biomarkers. Methods Mol. Biol. 2017, 1575, 253–272. [Google Scholar] [PubMed]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Cobb, D.W.; Florentin, A.; Fierro, M.A.; Krakowiak, M.; Moore, J.M.; Muralidharan, V. The Exported Chaperone PfHsp70x Is Dispensable for the Plasmodium falciparum Intraerythrocytic Life Cycle. mSphere 2017, 2, e00363-17. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selection Round | ssDNA [nM] | Dolichol Mix (nmol) | Fatty Acid Mix (nmol) | Washing Steps |
|---|---|---|---|---|
| 1 | 15 | 1.25 | - | 1 |
| 2 | 15 | 1.25 | - | 1 |
| 3 | 15 | - | 1.25 | - |
| 4 | 12.5 | 0.5 | - | 2 |
| 5 | 12.5 | - | 1.25 | - |
| 6 | 12.5 | 0.5 | - | 2 |
| 7 | 10 | - | 1.25 | - |
| 8 | 10 | 0.25 | - | 3 |
| 9 | 10 | - | 1.25 | - |
| 10 | 10 | 0.25 | - | 3 |
| Family | Sequence (5′ → 3′) | R00 | R04 | R05 | R06 | R09 | R10 |
|---|---|---|---|---|---|---|---|
| 1 (AptPP) | cst-ATGTCGACTGGCGTTGGGAAATGCCGCTTAGTGG-cst | 0.002 | 0.010 | 0.010 | 2.865 | 5.278 | 13.919 |
| 2 | cst-GTCGAAAGGTTTTGGGAAAGCACCTCAGTTCTTGAG-cst | 0 | 0.023 | 0.002 | 0.001 | 0.010 | 0.040 |
| 3 | cst-AGATCGGAAGAGCACACGTCTGAACTCCAGT-cst | 0 | 0.080 | 0.002 | 0 | 0.028 | 0.025 |
| 4 | cst-GTCACGACGACGTCTCGTATGCCGTCTTCTGCTTG-cst | 0 | 0.055 | 0 | 0 | 0.035 | 0.043 |
| 5 | cst-ACTAACCATGCTCATCATGTGGTTCCGTATTAGG-cst | 0 | 0 | 0 | 0 | 0 | 0.003 |
| 6 | cst-GTGTCGTGTCTTGACTATCAAGCTAGTCTCTTTT-cst | 0 | 0.002 | 0 | 0 | 0 | 0.005 |
| Aptamer | Sequence (5′ → 3′) |
|---|---|
| AptPP | gcctgttgtgagcctcctATGTCGACTGGCGTTGGGAAATGCCGCTTAGTGGcgcttattcttgtctccc |
| AptPPInv | cgcttattcttgtctcccATGTCGACTGGCGTTGGGAAATGCCGCTTAGTGGgcctgttgtgagcctcct |
| AptScrambled | cgcttattcttgtctcccTGGCTTCTAGAAATACCGTGGCGGTGTGAGGGTCgcctgttgtgagcctcct |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimbres, F.M.; Merino, E.F.; Butschek, G.J.; Butler, J.H.; Ducongé, F.; Cassera, M.B. Aptamer-Based Imaging of Polyisoprenoids in the Malaria Parasite. Molecules 2024, 29, 178. https://doi.org/10.3390/molecules29010178
Zimbres FM, Merino EF, Butschek GJ, Butler JH, Ducongé F, Cassera MB. Aptamer-Based Imaging of Polyisoprenoids in the Malaria Parasite. Molecules. 2024; 29(1):178. https://doi.org/10.3390/molecules29010178
Chicago/Turabian StyleZimbres, Flavia M., Emilio F. Merino, Grant J. Butschek, Joshua H. Butler, Frédéric Ducongé, and Maria B. Cassera. 2024. "Aptamer-Based Imaging of Polyisoprenoids in the Malaria Parasite" Molecules 29, no. 1: 178. https://doi.org/10.3390/molecules29010178
APA StyleZimbres, F. M., Merino, E. F., Butschek, G. J., Butler, J. H., Ducongé, F., & Cassera, M. B. (2024). Aptamer-Based Imaging of Polyisoprenoids in the Malaria Parasite. Molecules, 29(1), 178. https://doi.org/10.3390/molecules29010178