


Selenium—More than Just a Fortuitous Sulfur Substitute in Redox Biology


Abstract
:1. Introduction
2. Selenium versus Sulfur
3. Formate Dehydrogenase
3.1. The Current Picture
3.1.1. Enzymatic Machinery
3.1.2. Reaction Mechanism
3.2. How Was the Selenium Locus in Formate Dehydrogenases Established?
3.3. Why Do Some Formate Dehydrogenases Have a Selenocysteine and Not the Less “Expensive” Cysteine Residue?
4. Hydrogenases
4.1. Enzymatic Machineries
4.2. Selenium and the Hydrogenase Reaction Mechanism
4.3. Overview
5. Glutathione Peroxidases
6. Thioredoxin Reductases
7. Iodothyronine Deiodinases
8. Selenoproteins and Human Health
8.1. Cancer
8.2. Diabetes
8.3. Viral Infections
8.3.1. Human Immunodeficiency Virus (HIV)
8.3.2. Coronavirus Disease-2019 (COVID-19)
8.4. Gestational Disorders
8.5. Overview
9. Wrap-Up
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Painter, E.P. The Chemistry and Toxicity of Selenium Compounds, with Special Reference to the Selenium Problem. Chem. Rev. 1941, 28, 179–213. [Google Scholar] [CrossRef]
- Hadrup, N.; Ravn-Haren, G. Acute human toxicity and mortality after selenium ingestion: A review. J. Trace Elem. Med. Biol. 2020, 58, 126435. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; Harris, H.H. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem. Soc. Rev. 2013, 42, 8870–8894. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Foltz, C.M. Selenium as an integral part of factor 3 against dietary necrotic liver degeneration. J. Am. Chem. Soc. 1957, 79, 3292–3293. [Google Scholar] [CrossRef]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M. Selenium–Fascinating Microelement, Properties and Sources in Food. Molecules 2019, 24, 1298. [Google Scholar] [CrossRef]
- Roman, M.; Jitaru, p.; Barbante, C. Selenium biochemistry and its role for human health. Metallomics 2014, 6, 25–54. [Google Scholar] [CrossRef]
- Driscoll, D.M.; Copeland, P.R. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003, 23, 17–40. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef]
- Jacob, C.; Giles, G.I.; Giles, N.M.; Helmut Sies, H. Sulfur and Selenium: The Role of Oxidation State in Protein Structure and Function. Angew. Chem. Int. Ed. 2003, 42, 4742–4758. [Google Scholar] [CrossRef] [PubMed]
- Wessjohann, L.A.; Schneider, A.; Abbas, M.; Brandt, W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol. Chem. 2007, 388, 997–1006. [Google Scholar] [CrossRef]
- Boyd, R. Selenium stories. Nat. Chem. 2011, 3, 570. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K. Cross-talk Between (Hydrogen)Sulfite and Metalloproteins: Impact on Human Health. Chem.–A Eur. J. 2022, 28, e202104342. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Yukio Sugiura, Y.; Hojo, Y.; Tamai, Y.; Hisashi Tanaka, H. Selenium protection against mercury toxicity. Binding of methylmercury by the selenohydryl-containing ligand. J. Am. Chem. Soc. 1976, 98, 2339–2341. [Google Scholar] [CrossRef]
- Huber, R.E.; Criddle, R.S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar] [CrossRef]
- Bell, I.M.; Fisher, M.L.; Wu, Z.P.; Hilvert, D. Kinetic studies on the peroxidase activity of selenosubtilisin. Biochemistry 1993, 32, 3754–3762. [Google Scholar] [CrossRef]
- Ruggles, E.L.; Snider, G.W.; Hondal, R.J. Chemical basis for the use of selenocysteine. In Selenium: Its Molecular Biology and Role in Human Health, 3rd ed.; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer: New York, NY, USA, 2012; pp. 73–83. [Google Scholar]
- Abdo, M.; Knapp, S. Biomimetic Seleninates and Selenonates. J. Am. Chem. Soc. 2008, 130, 9234–9235. [Google Scholar] [CrossRef]
- Nauser, T.; Steinmann, D.; Grassi, G.; Koppenol, W.H. Why selenocysteine replaces cysteine in thioredoxin reductase: A radical hypothesis. Biochemistry 2014, 53, 5017–5022. [Google Scholar] [CrossRef] [PubMed]
- Nauser, T.; Dockheer, S.; Kissner, R.; Koppenol, W.H. Catalysis of Electron Transfer by Selenocysteine. Biochemistry 2006, 45, 6038–6043. [Google Scholar] [CrossRef] [PubMed]
- Nauser, T.; Steinmann, D.; Koppenol, W.H. Why do proteins use selenocysteine instead of cysteine? Amino Acids 2012, 42, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Joan, C.; Pleasants, J.C.; Guo, W.; Rabenstein, D.L. A comparative study of the kinetics of selenol/diselenide and thiol/disulfide exchange reactions. J. Am. Chem. Soc. 1989, 111, 6553–6558. [Google Scholar]
- Hondal, R.J.; Marino, S.M.; Gladyshev, V.N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid Redox Signal. 2013, 18, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Andreesen, J.R.; Ljungdahl, L.G. Formate dehydrogenase of Clostridium thermoaceticum: Incorporation of selenium-75, and the effects of selenite, molybdate, and tungstate on the enzyme. J. Bacteriol. 1973, 116, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Zinoni, F.; Birkmann, A.; Stadtman, T.C.; Böck, A. Nucleotide sequence and expression of the selenocysteine-containing polypeptide of formate dehydrogenase (formate-hydrogen-lyase-linked) from Escherichia coli. Proc. Natl. Acad. Sci. USA 1986, 83, 4650–4654. [Google Scholar] [CrossRef]
- Zhang, Y.; Romero, H.; Salinas, G.; Gladyshev, V.N. Dynamic evolution of selenocysteine utilization in bacteria: A balance between selenoprotein loss and evolution of selenocysteine from redox active cysteine residues. Genome Biol. 2006, 7, R94. [Google Scholar]
- Thauer, R.K.; Fuchs, G.; Jungermann, K. Role of iron-sulfur proteins in formate metabolism. In Iron–Sulfur Proteins; Lovenber, W., Ed.; Academic: New York, NY, USA, 1977; pp. 121–156. [Google Scholar]
- Maden, B.; Edward, H. Tetrahydrofolate and tetrahydromethanopterin compared: Functionally distinct carriers in C1 metabolism. Biochem. J. 2000, 350, 609–629. [Google Scholar] [CrossRef]
- Adams, M.W.W.; Mortenson, L.E. Mo reductases: Nitrate reductase and formate dehydrogenase. In Molybdenum Enzymes; Spiro, T.G., Ed.; Wiley Interscience: New York, NY, USA, 1985; pp. 519–593. [Google Scholar]
- Ferry, J.G. Formate dehydrogenase. FEMS Microbiol. Rev. 1990, 7, 377–382. [Google Scholar] [CrossRef]
- Unden, G.; Bongaerts, J. Alternative respiratory pathways of Escherichia coli: Energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1997, 1320, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.J. Bacterial respiration: A flexible process for a changing environment. Microbiology 2000, 146, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.; Sawers, G. Structural biology—PMF through the redox loop. Science 2002, 295, 1842–1843. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A.; Thauer, R.K. Molybdenum and tungsten enzymes in C1 metabolism. Met. Biol. Sys. 2002, 39, 571–619. [Google Scholar]
- Sawers, R.G. Formate and its role in hydrogen production in Escherichia coli. Biochem. Soc. Trans. 2005, 33, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, S.; Schoepp-Cothenet, B.; Ceccaldi, P.; Guigliarelli, B.; Magalon, A. The prokaryotic Mo/W-bisPGD enzymes family: A catalytic workhorse in bioenergetic. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1048–1085. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.; Moura, I. Molybdenum and tungsten-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 2015, 20, 287–309. [Google Scholar] [CrossRef]
- Hartmann, T.; Schwanhold, N.; Leimkühler, S. Assembly and catalysis of molybdenum or tungsten-containing formate dehydrogenases from bacteria. Biochim. Biophys. Acta 2015, 1854, 1090–1100. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, I.; Moura, J.J. Molybdenum and tungsten-containing formate dehydrogenases: Aiming to inspire a catalyst for carbon dioxide utilization. Inorg. Chim. Acta 2017, 455, 350–363. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, I.; Moura, J.J. Molybdenum and tungsten-containing enzymes: An overview. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–80. [Google Scholar]
- Niks, D.; Hille, R. Reductive activation of CO2 by formate dehydrogenases. Methods Enzymol. 2018, 613, 277–295. [Google Scholar] [PubMed]
- Niks, D.; Hille, R. Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion. Prot. Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.F.; Lange, L.; Meyer, A.S. Classification and enzyme kinetics of formate dehydrogenases for biomanufacturing via CO2 utilization. Biotechnol. Adv. 2019, 37, 107408. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Carbon Dioxide Utilisation—The Formate Route. In Enzymes for Solving Humankind’s Problems; Moura, J.J.G., Moura, I., Maia, L.B., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 29–81. [Google Scholar]
- Kato, N. Formate dehydrogenase from methylotrophic yeasts. Methods Enzymol. 1990, 188, 459–462. [Google Scholar] [PubMed]
- Vinals, C.; Depiereux, E.; Feytmans, E. Prediction of structurally conserved regions of D-specific hydroxy acid dehydrogenases by multiple alignment with formate dehydrogenase. Biochem. Biophys. Res. Commun. 1993, 192, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.O.; Lamzin, V.S. NAD (+)-dependent formate dehydrogenase. Biochem. J. 1994, 301, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Filippova, E.V.; Polyakov, K.M.; Tikhonova, T.V.; Stekhanova, T.N.; Booeko, K.M.; Popov, V.O. Structure of a new crystal modification of the bacterial NAD-dependent formate dehydrogenase with a resolution of 2.1 Å. Crystallogr. Rep. 2005, 50, 796–800. [Google Scholar] [CrossRef]
- Shabalin, I.G.; Polyakov, K.M.; Tishkov, V.I.; Popov, V.O. Atomic resolution crystal structure of nad+ dependent formate dehydrogenase from bacterium Moraxella sp. C-1. Acta Nat. 2009, 1, 89–93. [Google Scholar] [CrossRef]
- Alekseeva, A.A.; Savin, S.S.; Tishkov, V.I. NAD+-dependent formate dehydrogenase from plants. Acta Nat. 2011, 3, 38–54. [Google Scholar] [CrossRef]
- Hille, R. The mononuclear molybdenum enzymes. Chem. Rev. 1996, 96, 2757–2816. [Google Scholar] [CrossRef]
- Johnson, M.K.; Rees, D.C.; Adams, M.W.W. Tungstoenzymes. Chem. Rev. 1996, 96, 2817–2839. [Google Scholar] [CrossRef] [PubMed]
- Bertram, P.A.; Karrasch, M.; Schmitz, R.A.; Böcher, R.; Albracht, S.P.J.; Thauer, R.K. Formylmethanofuran dehydrogenases from methanogenic Archaea. Substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron-sulfur proteins. Eur. J. Biochem. 1994, 220, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Hochheimer, A.; Hedderich, R.; Thauer, R.K. The formylmethanofuran dehydrogenase isozymes in Methanobacterium wolfeii and Methanobacterium terhmoautotrophicum: Induction of the molybdenum isozyme by molybdate and constitutive synthesis of the tungsten isozyme. Arch. Microbiol. 1998, 170, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Ermler, U.; Shima, S. The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 2016, 354, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Hemmann, J.L.; Wagner, T.; Shima, S.; Vorholt, J.A. Methylofuran is a prosthetic group of the formyltransferase/hydrolase complex and shuttles one-carbon units between two active sites. Proc. Natl. Acad. Sci. USA 2019, 116, 25583–25590. [Google Scholar] [CrossRef] [PubMed]
- Niks, D.; Duvvuru, J.; Escalona, M.; Hille, R. Spectroscopic and Kinetic Properties of the Molybdenum-containing, NAD+-dependent Formate Dehydrogenase from Ralstonia eutropha. J. Biol. Chem. 2016, 291, 1162–1174. [Google Scholar] [CrossRef]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J. Reduction of carbon dioxide by a molybdenum-containing formate dehydrogenase: A kinetic and mechanistic study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Niks, D.; Mulchandani, A.; Hille, R. Efficient reduction of CO2 by the molybdenum-containing formate dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J. Biol. Chem. 2017, 292, 16872–16879. [Google Scholar] [CrossRef]
- Meneghello, M.; Oliveira, A.R.; Jacq-Bailly, A.; Pereira, I.A.C.; Léger, C.; Fourmond, V. Formate Dehydrogenases Reduce CO2 Rather than HCO3−: An Electrochemical Demonstration. Angew. Chem. 2021, 60, 9964–9967. [Google Scholar] [CrossRef]
- Meneghello, M.; Uzel, A.; Broc, M.; Manuel, R.R.; Magalon, A.; Léger, C.; Pereira, I.A.C.; Walburger, A.; Fourmond, V. Electrochemical Kinetics Support a Second Coordination Sphere Mechanism in Metal-Based Formate Dehydrogenase. Angew. Chem. 2023, 62, e202212224. [Google Scholar] [CrossRef]
- Harmer, J.R.; Hakopian, S.; Niks, D.; Hille, R.; Bernhardt, P.V. Redox Characterization of the Complex Molybdenum Enzyme Formate Dehydrogenase from Cupriavidus necator. J. Am. Chem. Soc. 2023, 145, 25850–25863. [Google Scholar] [CrossRef] [PubMed]
- Leimkühler, S. Metal-Containing Formate Dehydrogenases, a Personal View. Molecules 2023, 28, 5338. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, E.I. Proposed molecular mechanism for the action of molybedenum in enzymes: Coupled proton and electron transfer. Proc. Natl. Acad. Sci. USA 1973, 70, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, E.I. The coordination and bioinorganic chemistry of molybdenum. Prog. Inorg. Chem. 1977, 22, 1–223. [Google Scholar]
- Rajapakshe, A.; Snyder, R.A.; Astashkin, A.V.; Bernardson, P.; Evans, D.J.; Young, C.G.; Evans, D.H.; Enemark, J.H. Insights into the nature of Mo(V) species in solution: Modeling catalytic cycles for molybdenum enzymes. Inorg. Chim. Acta 2009, 362, 4603–4608. [Google Scholar] [CrossRef]
- Maia, L.; Moura, I.; Moura, J.J.G. EPR spectroscopy on mononuclear molybdenum-containing enzymes. In Future Directions in Metalloprotein and Metalloenzyme Research, Biological Magnetic Resonance; Hanson, G., Berliner, L.J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 33, pp. 55–101. [Google Scholar]
- Kirk, M.L.; Hille, R. Spectroscopic Studies of Mononuclear Molybdenum Enzyme Centers. Molecules 2022, 27, 4802. [Google Scholar] [CrossRef]
- Hille, R.; Niks, D. Application of EPR and related methods to molybdenum-containing enzymes. Methods Enzymol. 2022, 666, 373–412. [Google Scholar]
- Gladyshev, V.N.; Khangulov, S.V.; Stadtman, T.C. Nicotinic acid hydroxylase from Clostridium barkeri: Electron paramagnetic resonance studies show that selenium is coordinated with molybdenum in the catalytically active selenium-dependent enzyme. Proc. Natl. Acad. Sci. USA 1994, 91, 232–236. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Khangulov, S.V.; Axley, M.J.; Stadtman, T.C. Coordination of selenium to molybdenum in formate dehydrogenase H from Escherichia coli. Proc. Natl. Acad. Sci. USA 1994, 91, 7708–7711. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Boyington, J.C.; Khangulov, S.V.; Grahame, D.A.; Stadtman, T.C.; Sun, P.D. Characterization of crystalline formate dehydrogenase H from Escherichia coli: Stabilization, EPR spectroscopy, and preliminary crystallographic analysis. J. Biol. Chem. 1996, 271, 8095–8100. [Google Scholar] [CrossRef]
- Khangulov, S.V.; Gladyshev, V.N.; Dismukes, G.C.; Stadtman, T.C. Selenium-containing formate dehydrogenase H from Escherichia coli: A molybdopterin enzyme that catalyzes formate oxidation without oxygen transfer. Biochemistry 1998, 37, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.R.; Wilson, G.L.; Bailey, T.D.; Pilbrow, J.R.; Wedd, A.G. Multifrequency electron spin resonance of molybdenum (V) and tungsten (V) compounds. J. Am. Chem. Soc. 1987, 109, 2609–2616. [Google Scholar] [CrossRef]
- Rivas, M.G.; González, P.J.; Brondino, C.D.; Moura, J.J.; Moura, I. EPR characterization of the molybdenum (V) forms of formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774 upon formate reduction. J. Inorg. Biochem. 2007, 101, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.R.; Mota, C.; Klymanska, K.; Biaso, F.; Romão, M.J.; Guigliarelli, B.; Pereira, I.C. Spectroscopic and Structural Characterization of Reduced Desulfovibrio vulgaris Hildenborough W-FdhAB Reveals Stable Metal Coordination during Catalysis. ACS Chem. Biol. 2022, 17, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.R.; Mota, C.; Mourato, C.; Domingos, R.M.; Santos, M.F.; Gesto, D.; Pereira, I.A. Toward the mechanistic understanding of enzymatic CO2 reduction. ACS Catal. 2020, 10, 3844–3856. [Google Scholar] [CrossRef]
- Graham, J.E.; Niks, D.; Zane, G.M.; Gui, Q.; Hom, K.; Hille, R.; Raman, C.S. How a formate dehydrogenase responds to oxygen: Unexpected O2 insensitivity of an enzyme harboring tungstopterin, selenocysteine, and [4Fe–4S] clusters. ACS Catal. 2022, 12, 10449–10471. [Google Scholar] [CrossRef]
- Raman, C.S. Reply to Comment on ‘How a Formate Dehydrogenase Responds to Oxygen: Unexpected O2 Insensitivity of an Enzyme Harboring Tungstopterin, Selenocysteine, and [4Fe–4S] Clusters’. ACS Catal. 2023, 13, 9629–9632. [Google Scholar] [CrossRef]
- Almendra, M.J.; Brondino, C.D.; Gavel, O.; Pereira, A.S.; Tavares, P.; Bursakov, S.; Moura, I. Purification and characterization of a tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Biochemistry 1999, 38, 16366–16372. [Google Scholar] [CrossRef]
- Raaijmakers, H.; Teixeira, S.; Dias, J.M.; Almendra, M.J.; Brondino, C.D.; Moura, I.; Romão, M.J. Tungsten-containing formate dehydrogenase from Desulfovibrio gigas: Metal identification and preliminary structural data by multi-wavelength crystallography. J. Biol. Inorg. Chem. 2001, 6, 398–404. [Google Scholar] [CrossRef]
- Jollie, D.R.; Lipscomb, J.D. Formate dehydrogenase from Methylosinus trichosporium OB3b. Purification and spectroscopic characterization of the cofactors. J. Biol. Chem. 1991, 266, 21853–21863. [Google Scholar] [CrossRef]
- Cramer, S.P.; Liu, C.L.; Mortenson, L.E.; Spence, J.T.; Liu, S.M.; Yamamoto, I.; Ljungdahl, L.G. Formate dehydrogenase molybdenum and tungsten sites—Observation by EXAFS of structural differences. J. Inorg. Biochem. 1985, 23, 119–124. [Google Scholar] [CrossRef]
- George, G.N.; Colangelo, C.M.; Dong, J.; Scott, R.A.; Khangulov, S.V.; Gladyshev, V.N.; Stadtman, T.C. X-ray absorption spectroscopy of the molybdenum site of Escherichia coli formate dehydrogenase. J. Am. Chem. Soc. 1998, 120, 1267–1273. [Google Scholar] [CrossRef]
- George, G.N.; Costa, C.; Moura, J.J.G.; Moura, I. Observation of ligand-based redox chemistry at the active site of a molybdenum enzyme. J. Am. Chem. Soc. 1999, 121, 2625–2626. [Google Scholar] [CrossRef]
- Schrapers, P.; Hartmann, T.; Kositzki, R.; Dau, H.; Reschke, S.; Schulzke, C.; Haumann, M. Sulfido and cysteine ligation changes at the molybdenum cofactor during substrate conversion by formate dehydrogenase (FDH) from Rhodobacter capsulatus. Inorg. Chem. 2015, 54, 3260–3271. [Google Scholar] [CrossRef] [PubMed]
- Duffus, B.R.; Schrapers, P.; Schuth, N.; Mebs, S.; Dau, H.; Leimkühler, S.; Haumann, M. Anion binding and oxidative modification at the molybdenum cofactor of formate dehydrogenase from Rhodobacter capsulatus studied by X-ray absorption spectroscopy. Inorg. Chem. 2020, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Boyington, J.C.; Gladyshev, V.N.; Khangulov, S.V.; Stadtman, T.C.; Sun, P.D. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 1997, 275, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Jormakka, M.; Tornroth, S.; Byrne, B.; Iwata, S. Molecular basis of proton motive force generation: Structure of formate dehydrogenase-N. Science 2002, 295, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, H.; Macieira, S.; Dias, J.M.; Teixeira, S.; Bursakov, S.; Huber, R.; Moura, I.; Moura, M.J.; Romão, M.J. Gene sequence and the 1.8 Å crystal structure of the tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Structure 2002, 10, 1261–1272. [Google Scholar] [CrossRef]
- Radon, C.; Mittelstädt, G.; Duffus, B.R.; Burger, J.; Hartmann, T.; Mielke, T.; Teutloff, C.; Leimkuhler, S.; Wendler, P. Cryo-EM structures reveal intricate Fe-S cluster arrangement and charging in Rhodobacter capsulatus formate dehydrogenase. Nat. Commun. 2020, 11, 1912. [Google Scholar] [CrossRef]
- Young, T.; Niks, D.; Hakopian, S.; Tam, T.K.; Yu, X.; Hille, R.; Blaha, G.M. Crystallographic and kinetic analyses of the FdsBG subcomplex of the cytosolic formate dehydrogenase FdsABG from Cupriavidus necator. J. Biol. Chem. 2020, 295, 6570–6585. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Righetto, R.D.; Kumar, A.; Wietrzynski, W.; Trischler, R.; Schuller, S.K.; Schuller, J.M. Membrane-anchored HDCR nanowires drive hydrogen-powered CO2 fixation. Nature 2022, 607, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Makino, F.; Miyata, T.; Suzuki, Y.; Tanaka, H.; Namba, K.; Kano, K.; Sowa, K.; Kitazumi, Y.; Shirai, O. Multiple electron transfer pathways of tungsten-containing formate dehydrogenase in direct electron transfer-type bioelectrocatalysis. Chem. Commun. 2022, 58, 6478–6481. [Google Scholar] [CrossRef] [PubMed]
- Vilela-Alves, G.; Manuel, R.R.; Oliveira, A.R.; Pereira, I.C.; Romão, M.J.; Mota, C. Tracking W-Formate Dehydrogenase Structural Changes during Catalysis and Enzyme Reoxidation. Int. J. Mol. Sci. 2022, 24, 476. [Google Scholar] [CrossRef] [PubMed]
- Zinoni, F.; Birkmann, A.; Leinfelder, W.; Bock, A. Cotranslational insertion of selenocysteine into formate dehydrogenase from Escherichia coli directed by a UGA codon. Proc. Natl. Acad. Sci. USA 1987, 84, 3156–3160. [Google Scholar] [CrossRef] [PubMed]
- Axley, M.J.; Böck, A.; Stadtman, T.C. Catalytic properties of an Escherichia coli formate dehydrogenase mutant in which sulfur replaces selenium. Proc. Natl. Acad. Sci. USA 1991, 88, 8450–8454. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.J.; Martin, G.W.; Tujebajeva, R.; Grundner-Culemann, E.; Mansell, J.B.; Morozova, N.; Harney, J.W. Selenocysteine Insertion Sequence Element Characterization and Selenoprotein Expression. Methods Enzymol. 2002, 347, 17–24. [Google Scholar]
- Hatfield, D.L.; Carlson, B.A.; Xu, X.M.; Mix, H.; Gladyshev, V.N. Selenocysteine Incorporation Machinery and the Role of Selenoproteins in Development and Health. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 97–142. [Google Scholar]
- Allmang, C.; Wurth, L.; Krol, A. The Selenium to Selenoprotein Pathway in Eukaryotes: More Molecular Partners than Anticipated. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1415–1423. [Google Scholar] [CrossRef]
- Yoshizawa, S.; Böck, A. The Many Levels of Control on Bacterial Selenoprotein Synthesis. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1404–1414. [Google Scholar] [CrossRef]
- Bulteau, A.-L.; Chavatte, L. Update on Selenoprotein Biosynthesis. Antioxid. Redox Signal. 2015, 23, 775–794. [Google Scholar] [CrossRef]
- Arnér, E.S. Selenoproteins—What unique properties can arise with selenocysteine in place of cysteine? Exp. Cell Res. 2010, 316, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R. The Evolving Versatility of Selenium in Biology. Antioxid. Redox Signal. 2015, 23, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Bortoli, M.; Torsello, M.; Bickelhaupt, F.M.; Orian, L. Role of the Chalcogen (S, Se, Te) in the Oxidation Mechanism of the Glutathione Peroxidase Active Site. ChemPhysChem 2017, 18, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Maroney, M.J.; Hondal, R.J. Selenium Versus Sulfur: Reversibility of Chemical Reactions and Resistance to Permanent Oxidation in Proteins and Nucleic Acids. Free Radic. Biol. Med. 2018, 127, 228–237. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPx4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef]
- Fauque, G.; Peck, H.D., Jr.; Moura, J.J.G.; Huynh, B.H.; Berlier, Y.; DerVartanian, D.V.; Teixeira, M.; Przybyla, A.E.; Lespinat, P.A.; Moura, I. The three classes of hydrogenases from sulfate-reducing bacteria of the genus Desulfovibrio. FEMS Microbiol. Rev. 1988, 4, 299–344. [Google Scholar] [CrossRef]
- Pereira, A.S.; Tavares, P.; Moura, I.; Moura, J.J.; Huynh, B.H. Mössbauer characterization of the iron-sulfur clusters in Desulfovibrio vulgaris hydrogenase. J. Am. Chem. Soc. 2001, 123, 2771–2782. [Google Scholar] [CrossRef]
- Patil, D.S.; Moura, J.J.; He, S.H.; Teixeira, M.; Prickril, B.C.; DerVartanian, D.V.; Peck, H.D., Jr.; LeGall, J.; Huynh, B.H. EPR-detectable redox centers of the periplasmic hydrogenase from Desulfovibrio vulgaris. J. Biol. Chem. 1988, 263, 18732–18738. [Google Scholar] [CrossRef]
- Moura, J.J.G.; Teixeira, M.; Moura, I. The role of nickel and iron-sulfur centers in the bioproduction of hydrogen. Pure Appl. Chem. 1989, 61, 915–921. [Google Scholar] [CrossRef]
- Wombwell, C.; Caputo, C.A.; Reisner, E. [NiFeSe]-Hydrogenase Chemistry. Acc. Chem. Res. 2015, 48, 2858–2865. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, T.M.; Baltazar, C.S.A.; Cruz, D.R.; Lousa, D.; Soares, C.M. Studying O2 pathways in [NiFe]- and [NiFeSe]-hydrogenases. Sci. Rep. 2020, 10, 10540. [Google Scholar] [CrossRef] [PubMed]
- Happe, R.P.; Roseboom, W.; Pierik, A.J.; Albracht, S.P.; Bagley, K.A. Biological Activation of Hydrogen. Nature 1997, 385, 126. [Google Scholar] [CrossRef] [PubMed]
- Bleijlevens, B.; van Broekhuizen, F.A.; De Lacey, A.L.; Roseboom, W.; Fernandez, V.M.; Albracht, S.P. The Activation of the [NiFe]-Hydrogenase from Allochromatium Vinosum. An Infrared Spectro-Electrochemical Study. J. Biol. Inorg. Chem. 2004, 9, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, C.; Laurich, C.; Bothe, E.; Lubitz, W. Spectroelectrochemical Characterization of the [NiFe] Hydrogenase of Desulfovibrio vulgaris Miyazaki F. Biochemistry 2006, 5, 9706–9716. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.; Fontecilla-Camps, J.C.; Volbeda, A. Nickel–Iron Hydrogenases. In Handbook of Metalloproteins; Messerschmidt, A., Huber, R., Poulos, T., Wieghardt, K., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2001; p. 880. [Google Scholar]
- LeGall, J.; Ljungdahl, P.O.; Moura, I.; Peck, H.D., Jr.; Xavier, A.V.; Moura, J.J.G.; Teixera, M.; Huynh, B.H. DerVartanian DV. The presence of redox-sensitive nickel in the periplasmic hydrogenase from Desulfovibrio gigas. Biochem. Biophys. Res. Commun. 1982, 106, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Yagi, T.; Yasuoka, N. Unusual Ligand Structure in Ni-Fe Active Center and an Additional Mg Site in Hydrogenase Revealed by High Resolution X-Ray Structure Analysis. Structure 1997, 5, 1671–1680. [Google Scholar] [CrossRef]
- Volbeda, A.; Garcin, E.; Piras, C.; deLacey, A.L.; Fernandez, V.M.; Hatchikian, E.C.; Frey, M.; FontecillaCamps, J.C. Structure of the [NiFe] Hydrogenase Active Site: Evidence for Biologically Uncommon Fe Ligands. J. Am. Chem. Soc. 1996, 118, 12989–12996. [Google Scholar] [CrossRef]
- Carepo, M.; Tierney, D.L.; Brondino, C.D.; Yang, T.C.; Pamplona, A.; Telser, J.; Moura, I.; Moura, J.J.; Hoffman, B.M. 17O ENDOR detection of a solvent-derived Ni-(OH(x))-Fe bridge that is lost upon activation of the hydrogenase from Desulfovibrio gigas. J. Am. Chem. Soc. 2002, 124, 281–286. [Google Scholar] [CrossRef]
- Teixeira, M.; Moura, I.; Xavier, A.V.; Huynh, B.H.; DerVartanian, D.V.; Peck, H.D., Jr.; LeGall, J.; Moura, J.J. Electron paramagnetic resonance studies on the mechanism of activation and the catalytic cycle of the nickel-containing hydrogenase from Desulfovibrio gigas. J. Biol. Chem. 1985, 260, 8942–8950. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.; Moura, I.; Xavier, A.V.; Moura, J.J.; LeGall, J.; DerVartanian, D.V.; Peck, H.D., Jr.; Huynh, B.H. Redox intermediates of Desulfovibrio gigas [NiFe] hydrogenase generated under hydrogen. Mössbauer and EPR characterization of the metal centers. J. Biol. Chem. 1989, 264, 16435–16450. [Google Scholar] [CrossRef] [PubMed]
- Foerster, S.; van Gastel, M.; Brecht, M.; Lubitz, W. An Orientation-Selected ENDOR and HYSCORE Study of the Ni-C Active State of Desulfovibrio vulgaris Miyazaki F Hydrogenase. J. Biol. Inorg. Chem. 2005, 10, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Darensbourg, M.Y. The roles of chalcogenides in O2 protection of H2ase active sites. Chem. Sci. 2020, 11, 9366–9377. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.C.; Coelho, R.; De Lacey, A.L.; Pereira, I.A.; Matias, P.M. The Three-Dimensional Structure of [NiFeSe] Hydrogenase from Desulfovibrio vulgaris Hildenborough: A Hydrogenase without a Bridging Ligand in the Active Site in its Oxidised, “As-Isolated” State. J. Mol. Biol. 2010, 396, 893–907. [Google Scholar] [CrossRef]
- Marques, M.C.; Tapia, C.; Gutierrez-Sanz, O.; Ramos, A.R.; Keller, K.L.; Wall, J.D.; De Lacey, A.L.; Matias, P.M.; Pereira, I.A.C. The Direct Role of Selenocysteine in [NiFeSe] Hydrogenase Maturation and Catalysis. Nat. Chem. Biol. 2017, 13, 544–550. [Google Scholar] [CrossRef]
- Baltazar, C.S.A.; Marques, M.C.; Soares, C.M.; DeLacey, A.M.; Pereira, I.A.C.; Matias, P.M. Nickel–iron–selenium hydrogenases—An overview. Eur. J. Inorg. Chem. 2011, 2011, 948–962. [Google Scholar] [CrossRef]
- Teixeira, M.; Moura, I.; Fauque, G.; Dervartanian, D.V.; Legall, J.; Peck, H.D., Jr.; Moura, J.J.; Huynh, B.H. The iron-sulfur centers of the soluble [NiFeSe] hydrogenase, from Desulfovibrio baculatus (DSM 1743). EPR and Mossbauer Characterization. Eur. J. Biochem. 1990, 189, 381–386. [Google Scholar] [CrossRef]
- Parkin, A.; Goldet, G.; Cavazza, C.; Fontecilla-Camps, J.C.; Armstrong, F.A. The Difference a se Makes? Oxygen-Tolerant Hydrogen Production by the [NiFeSe]-Hydrogenase from Desulfomicrobium baculatum. J. Am. Chem. Soc. 2008, 130, 13410–13416. [Google Scholar] [CrossRef]
- Rüdiger, O.; Gutiérrez-Sánchez, C.; Olea, D.; Pereira, I.A.C.; Vélez, M.; Fernández, V.M.; De Lacey, A.L. Enzymatic Anodes for Hydrogen Fuel Cells Based on Covalent Attachment of Ni-Fe Hydrogenases and Direct Electron Transfer to SAM-Modified Gold Electrodes. Electroanalysis 2010, 22, 776–783. [Google Scholar] [CrossRef]
- Valente, F.M.A.; Oliveira, A.S.F.; Gnadt, N.; Pacheco, I.; Coelho, A.V.; Xavier, A.V.; Teixeira, M.; Soares, C.M.; Pereira, I.A.C. Hydrogenases in Desulfovibrio vulgaris Hildenborough: Structural and Physiologic Characterisation of the Membrane-Bound [NiFeSe] Hydrogenase. J. Biol. Inorg. Chem. 2005, 10, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Claude Hatchikian, E.; Cammack, R. Studies of Light-Induced Nickel EPR Signals in Hydrogenase: Comparison of Enzymes with and without Selenium. Biochim. Biophys. Acta 1996, 1275, 227–236. [Google Scholar] [CrossRef]
- Teixeira, M.; Fauque, G.; Moura, I.; Lespinat, P.A.; Berlier, Y.; Prickril, B.; Peck, H.D., Jr.; Xavier, A.V.; LeGall, J.; Moura, J.J.G. Nickel-[iron-sulfur]-selenium-containing hydrogenases from Desulfovibrio baculatus (DSM 1743). Redox centers and catalytic properties. Eur. J. Biochem. 1987, 167, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.; Moura, I.; Fauque, G.; Czechowski, M.; Berlier, Y.; Lespinat, P.A.; Le Gall, J.; Xavier, A.V.; Moura, J.J.G. Redox properties and activity studies on a nickel-containing hydrogenase isolated from a halophilic sulfate reducer Desulfovibrio salexigens. Biochimie 1986, 68, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lespinat, P.A.; Berlier, Y.; Fauque, G.; Czechowski, M.H.; Dimon, B.; LeGall, J. The pH dependence of proton-deuterium exchange, hydrogen production and uptake catalyzed by hydrogenases from sulfate-reducing bacteria. Biochimie 1986, 68, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Moura, J.J.G.; Moura, I.; Huynh, B.H.; Krüger, H.J.; Teixeira, M.; DuVarney, R.C.; DerVartanian, D.V.; Xavier, A.V.; Peck, H.D., Jr.; LeGall, J. Unambiguous identification of the nickel EPR signal in 61Ni-enriched Desulfovibrio gigas hydrogenase. J. Biochem. Biophys. Res. Commun. 1982, 108, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sanz, O.; Marques, M.C.; Baltazar, C.S.; Fernandez, V.M.; Soares, C.M.; Pereira, I.A.; De Lacey, A.L. Influence of the Protein Structure Surrounding the Active Site on the Catalytic Activity of [NiFeSe] Hydrogenases. J. Biol. Inorg. Chem. 2013, 18, 419–427. [Google Scholar] [CrossRef] [PubMed]
- He, S.H.; Teixeira, M.; LeGall, J.; Patil, D.S.; Moura, I.; Moura, J.J.; DerVartanian, D.V.; Huynh, B.H.; Peck, H.D., Jr. EPR studies with 77Se-enriched (NiFeSe) hydrogenase of Desulfovibrio baculatus. Evidence for a selenium ligand to the active site nickel. J. Biol. Chem. 1989, 264, 2678–2682. [Google Scholar] [CrossRef]
- Eidsness, M.K.; Scott, R.A.; Prickril, B.C.; DerVartanian, D.V.; LeGall, J.; Moura, I.; Moura, J.J.; Peck, H.D., Jr. Evidence for selenocysteine coordination to the active site nickel in the [NiFeSe] hydrogenases from Desulfovibrio baculatus. Proc. Natl. Acad. Sci. USA 1989, 86, 147–151. [Google Scholar] [CrossRef]
- Moura, J.J.G.; Teixeira, M.; Moura, I.; LeGall, J. [Ni-Fe] hydrogenases from sulfate reducing bacteria: Nickel catalytic and regulatory roles. In Nickel in Biochemistry; Lancaster, J.R., Ed.; VCH Publishers: New York, NY, USA, 1988; Chapter 9; pp. 191–224. [Google Scholar]
- van der Zwaan, J.W.; Albracht, S.P.J.; Fontijn, R.D.; Slater, E.C. Monovalent nickel in hydrogenase from Chromatium vinosum. FEBS Lett. 1985, 179, 271–277. [Google Scholar] [CrossRef]
- Fauque, G.D.; Barton, L.L.; LeGall, J. Oxidative Phosphorylation Linked to the Dissimilatory Reduction of Elemental Sulphur by Desulfovibrio. Ciba Found. Symp. 1980, 72, 71–86. [Google Scholar]
- Fischer, H.F.; Krasna, A.I.; Rittenberg, D. The interaction of hydrogenase with oxygen. J. Biol. Chem. 1954, 209, 569–578. [Google Scholar] [CrossRef]
- Olive, H.; Olive, S. Hydrogenation catalysts: A synthetic hydrogenase model. J. Mol. Catal. 1976, 1, 121–125. [Google Scholar] [CrossRef]
- Moura, I.; Cordas, C.; Moura, J.J.G. Direct electrochemistry study of the multiple redox centers of hydrogenase from Desulfovibrio gigas. Bioelectrochemistry 2008, 74, 83–89. [Google Scholar]
- Zacarias, S.; Vélez, M.; Pita, M.; De Lacey, A.L.; Matias, P.M.; Pereira, I.A.C. Characterization of the [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough. Methods Enzymol. 2018, 613, 169–201. [Google Scholar] [PubMed]
- Stolwijk, J.M.; Garje, R.; Sieren, J.C.; Buettner, G.R.; Zakharia, Y. Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies. Antioxidants 2020, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.C. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [CrossRef]
- Little, C.; Olinescu, R.; Reid, K.G.; O’Brien, P.J. Properties and Regulation of Glutathione peroxidase. J. Biol. Chem. 1970, 245, 3632–3636. [Google Scholar] [CrossRef]
- Flohe, L.; Günzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical role as a component of glutathione peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Forstrom, J.W.; Zakowski, J.J.; Tappel, A.L. Identification of the catalytic site of rat liver glutathione peroxidase as selenocysteine. Biochemistry 1978, 17, 2639–2644. [Google Scholar] [CrossRef] [PubMed]
- Trenz, T.S.; Delaix, C.L.; Turchetto-Zolet, A.C.; Zamocky, M.; Lazzarotto, F.; Margis-Pinheiro, M. Going Forward and Back: The Complex Evolutionary History of the GPx. Biology 2021, 10, 1165. [Google Scholar] [CrossRef] [PubMed]
- Herbette, S.; Roeckel-Drevet, P.; Drevet, J.R. Seleno-independent glutathione peroxidases. More than simple antioxidant scavengers. FEBS J. 2007, 274, 2163–2180. [Google Scholar] [CrossRef]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef]
- Toppo, S.; Vanin, S.; Bosello, V.; Tosatto, S.C.E. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid. Redox Signal. 2008, 10, 1501–1514. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Janowski, R.; Scanu, S.; Niessing, D.; Madl, T. Crystal and solution structural studies of mouse phospholipid hydroperoxide glutathione peroxidase 4. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 743–749. [Google Scholar] [CrossRef]
- Tosatto, S.C.E.; Bosello, V.; Fogolari, F.; Mauri, P.; Roveri, A.; Toppo, S.; Flohe, L.; Ursini, F.; Maiorino, M. The Catalytic Site of Glutathione Peroxidases. Antioxid. Redox Signal. 2008, 10, 1515–1526. [Google Scholar] [CrossRef]
- Borchert, A.; Kalms, J.; Roth, S.R.; Rademacher, M.; Schmidt, A.; Holzhutter, H.-G.; Hartmut Kuhn, H.; Scheerer, P. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1095–1107. [Google Scholar] [CrossRef]
- Takahashi, K.; Avissar, N.; Whitin, J.; Cohen, H. Purification and characterization of human plasma glutathione peroxidase: A selenoglycoprotein distinct from the known cellular enzyme. Arch. Biochem. Biophys. 1987, 256, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Epp, O.; Ladenstein, R.; Wendel, A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur. J. Biochem. 1983, 133, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, C.L.; Fuglestad, B. Electrostatic Drivers of GPx4 Interactions with Membrane, Lipids, and DNA. Biochemistry 2021, 60, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Foster, S.J.; Ganther, H.E. Identification of selenocysteine in glutathione peroxidase by mass spectroscopy. Biochemistry 1983, 22, 5853–5858. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Factor, V.M.; Housseau, F.; Hatfield, D.L. Contrasting patterns of regulation of the antioxidant selenoproteins, thioredoxin reductase, and glutathione peroxidase, in cancer cells. Biochem. Biophys. Res. Commun. 1998, 251, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Masuda, R.; Kimura, R.; Karasaki, T.; Sase, S.; Goto, K. Modeling the Catalytic Cycle of Glutathione Peroxidase by Nuclear Magnetic Resonance Spectroscopic Analysis of Selenocysteine Selenenic Acids. J. Am. Chem. Soc. 2021, 143, 6345–6350. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1–8. [Google Scholar] [CrossRef]
- Williams, C.H., Jr. Chemistry and Biochemistry of Flavoenzymes; Müller, F., Ed.; CRC: Boca Raton, FL, USA, 1992; Volume III, pp. 121–211. [Google Scholar]
- Arscott, L.D.; Gromer, S.; Schirmer, R.H.; Williams, C.H., Jr. The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 3621–3626. [Google Scholar] [CrossRef]
- Williams, C.H.; Arscott, L.D.; Müller, S.; Lennon, B.W.; Ludwig, M.L.; Wang, P.F.; Veine, D.M.; Becker, K.; Schirmer, R.H. Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 2000, 267, 6110–6117. [Google Scholar] [CrossRef]
- Williams, C.H., Jr. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995, 9, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Arnér, E.S.; Ljung, J.; Aslund, F.; Holmgren, A. Rat and calf thioredoxin reductase are homologous to glutathione reductase with a carboxyl-terminal elongation containing a conserved catalytically active penultimate selenocysteine residue. J. Biol. Chem. 1998, 273, 8581–8591. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Jeang, K.T.; Stadtman, T.C. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. USA 1996, 93, 6146–6151. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Vizuete, A.M.; Damdimopoulos, A.E.; Pedrajas, J.R.; Gustafsson, J.-Å.; Spyrou, G. Human mitochondrial thioredoxin reductase. Eur. J. Biochem. 1999, 261, 405–412. [Google Scholar] [CrossRef]
- Lee, S.R.; Bar-Noy, S.; Kwon, J.; Levine, R.L.; Stadtman, T.C.; Rhee, S.G. Mammalian thioredoxin reductase: Oxidation of the C-terminal cysteine/selenocysteine active site forms a thioselenide, and replacement of selenium with sulfur markedly reduces catalytic activity. Proc. Natl Acad. Sci. USA 2000, 97, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Björnstedt, M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995, 252, 199–208. [Google Scholar]
- Lee, S.R.; Kim, J.R.; Kwon, K.S.; Yoon, H.W.; Levine, R.L.; Ginsburg, A.; Rhee, S.G. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J. Biol. Chem. 1999, 274, 4722–4734. [Google Scholar] [CrossRef]
- Turanov, A.A.; Su, D.; Gladyshev, V.N. Characterization of alternative cytosolic forms and cellular targets of mouse mitochondrial thioredoxin reductase. J. Biol. Chem. 2006, 281, 22953–22963. [Google Scholar] [CrossRef]
- Arnér, E.S.J. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Sandalova, T.; Zhong, L.; Lindqvist, Y.; Holmgren, A.; Schneider, G. Three-dimensional structure of a mammalian thioredoxin reductase: Implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc. Natl Acad. Sci. USA 2001, 98, 9533–9538. [Google Scholar] [CrossRef]
- Biterova, E.I.; Turanov, A.A.; Gladyshev, V.N.; Barycki, J.J. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 15018–15023. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Schulz, G.E. Refined structure of glutathione reductase at 1.54 A resolution. J. Mol. Biol. 1987, 195, 701–729. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Urig, S.; Becker, K. The structure of human thioredoxin reductase 1 provides insights into C-terminal rearrangements during catalysis. J. Mol. Biol. 2007, 370, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Eckenroth, B.; Harris, K.; Turanov, A.A.; Gladyshev, V.N.; Raines, R.T.; Hondal, R.J. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry 2006, 45, 5158–5170. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Sandalova, T.; Lindqvist, Y.; Arnér, E.S.J. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 2009, 284, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Kehr, S.; Stumpf, M.; Rahlfs, S.; Becker, K. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat. Commun. 2011, 2, 383. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Besse, D.; Siedler, F.; Diercks, T.; Kessler, H.; Moroder, L. The redox potentials of selenocystine in unconstrained cyclic peptides. Angew. Chem. Int. Ed. Engl. 1997, 36, 883–885. [Google Scholar] [CrossRef]
- Marsan, E.S.; Bayse, C.A. A Halogen Bonding Perspective on Iodothyronine Deiodinase Activity. Molecules 2020, 25, 1328. [Google Scholar] [CrossRef]
- Behne, D.; Kyriakopoulos, A.; Meinhold, H.; Köhrle, J. Identification of type I iodothyronine 5′-deiodinase as a selenoenzyme. Biochem. Biophys. Res. Commun. 1990, 173, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.J.; Banu, L.; Larsen, P.R. Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature 1991, 349, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.R.; Berry, M.J. Nutritional and hormonal regulation of thyroid hormone deiodinases. Annu. Rev. Nutr. 1995, 15, 323–352. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, Cellular and Molecular Biology, and Physiological Roles of the Iodothyronine Selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. Iodothyronine deiodinases. Methods Enzymol. 2002, 347, 125–167. [Google Scholar] [PubMed]
- Koehrle, J.; Auf’mkolk, M.; Rokos, H.; Hesch, R.D.; Cody, V. Rat liver iodothyronine monodeiodinase. Evaluation of the iodothyronine ligand-binding site. J. Biol. Chem. 1986, 261, 11613–11622. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.J.M.; Kester, M.H.A.; Peeters, R.P.; Visser, T.J. Biochemical mechanisms of thyroid hormone deiodination. Thyroid 2005, 15, 787–798. [Google Scholar] [CrossRef]
- Visser, T.J.; Schoenmakers, C.H. Characteristics of type III iodothyronine deiodinase. Acta Med. Austriaca 1992, 19, 18–21. [Google Scholar]
- Köhrle, J. Local activation and inactivation of thyroid hormones: The deiodinase family. Mol. Cell Endocrinol. 1999, 151, 103–119. [Google Scholar] [CrossRef]
- Köhrle, J.; Jakob, F.; Contempré, B.; Dumont, J.E. Selenium, the thyroid, and the endocrine system. Endocr. Rev. 2005, 26, 944–984. [Google Scholar] [CrossRef]
- Gentile, F.; DiLauro, R.; Salvatore, G. Biosynthesis and Secretion of Thyroid Hormones. In Endocrinology, 3rd ed.; DeGroot, L.J., Ed.; WB Saunders Company: Philadelphia, PA, USA, 1995; pp. 517–542. [Google Scholar]
- Taurog, A. Hormone synthesis. In The Thyroid; Braverman, L.E., Utiger, R., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2000; pp. 61–85. [Google Scholar]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Debasish Manna, D.; Mugesh, G. Regioselective Deiodination of Thyroxine by Iodothyronine Deiodinase Mimics: An Unusual Mechanistic Pathway Involving Cooperative Chalcogen and Halogen Bonding. J. Am. Chem. Soc. 2012, 134, 4269–4279. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Hu, Y.J.; Diamond, A.M. Role of glutathione peroxidase 1 in breast cancer: Loss of heterozygosity and allelic differences in the response to selenium. Cancer Res. 2003, 63, 3347–3351. [Google Scholar]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Dagnell, M.; Schmidt, E.E.; Arnér, E.S.J. The A to Z of modulated cell patterning by mammalian thioredoxin reductases. Free Radic. Biol. Med. 2018, 115, 484–496. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Zhang, M.-L.; Wu, H.-T.; Chen, W.-J.; Xu, Y.; Ye, Q.-Q.; Shen, J.-X.; Liu, J. Involvement of glutathione peroxidases in the occurrence and development of breast cancers. J. Transl. Med. 2020, 18, 247. [Google Scholar] [CrossRef]
- Baliga, M.S.; Wang, H.; Zhuo, P.; Schwartz, J.L.; Diamond, A.M. Selenium and GPx-1 overexpression protect mammalian cells against UV-induced DNA damage. Biol. Trace. Elem. Res. 2007, 115, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, J.; Zhang, Y.; Sun, W.; Smith, B.J.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in pancreatic cancer by overexpression of phospholipid hydroperoxide glutathione peroxidase. Hum. Gene Ther. 2006, 17, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Metere, A.; Frezzotti, F.; Graves, C.E.; Vergine, M.; De Luca, A.; Pietraforte, D.P.; Giacomelli, L. A possible role for selenoprotein glutathione peroxidase (GPx1) and thioredoxin reductases (TrxR1) in thyroid cancer: Our experience in thyroid surgery. Cancer Cell Int. 2018, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Min, S.Y.; Kim, H.S.; Jung, E.J.; Jung, E.J.; Jee, C.D.; Kim, W.H. Prognostic significance of glutathione peroxidase 1 (GPX1) down-regulation and correlation with aberrant promoter methylation in human gastric cancer. Anticancer Res. 2012, 32, 3169–3175. [Google Scholar] [PubMed]
- Nalkiran, I.; Turan, S.; Arikan, S.; Kahraman, Ö.T.; Acar, L.; Yaylim, I.; Ergen, A. Determination of Gene Expression and Serum Levels of MnSOD and GPX1 in Colorectal Cancer. Anticancer Res. 2015, 35, 255–259. [Google Scholar] [PubMed]
- Cheng, Y.; Xu, T.; Li, S.; Ruan, H. GPX1, a biomarker for the diagnosis and prognosis of kidney cancer, promotes the progression of kidney cancer. Aging 2019, 11, 12165–12176. [Google Scholar] [CrossRef]
- Meng, Q.; Xu, J.; Liang, C.; Liu, J.; Hua, J.; Zhang, Y.; Ni, Q.; Shi, S.; Yu, X. GPx1 is involved in the induction of protective autophagy in pancreatic cancer cells in response to glucose deprivation. Cell Death Dis. 2018, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Al-Taie, O.H.; Uceyler, N.; Eubner, U.; Jakob, F.; Mork, H.; Scheurlen, M.; Brigelius-Flohe, R.; Schottker, K.; Abel, J.; Thalheimer, A.; et al. Expression Profiling and Genetic Alterations of the Selenoproteins GI-GPx and SePP in Colorectal Carcinogenesis. Nutr. Cancer 2004, 48, 6–14. [Google Scholar] [CrossRef]
- Woenckhaus, M.; Klein-Hitpass, L.; Grepmeier, U.; Merk, J.; Pfeifer, M.; Wild, P.; Bettstetter, M.; Wuensch, P.; Blaszyk, H.; Hartmann, A.; et al. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J. Pathol. 2006, 210, 192–204. [Google Scholar] [CrossRef]
- Banning, A.; Kipp, A.; Schmitmeier, S.; Löwinger, M.; Florian, S.; Krehl, S.; Thalmann, S.; Thierbach, R.; Steinberg, P.; Brigelius-Flohé, R. Glutathione Peroxidase 2 Inhibits Cyclooxygenase-2–Mediated Migration and Invasion of HT-29 Adenocarcinoma Cells but Supports Their Growth as Tumors in Nude Mice. Cancer Res. 2008, 68, 9746–9753. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Guo, S.; Wang, G. Glutathione peroxidases as oncotargets. Oncotarget 2017, 8, 80093–80102. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Worley, B.L.; Phaëton, R.; Hempel, N. Extracellular Glutathione Peroxidase GPx3 and Its Role in Cancer. Cancers 2020, 12, 2197. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.-J.; Schneider-Stock, R.; McChesney, P.A.; Kuester, D.; Roessner, A.; Vieth, M.; Moskaluk, C.A.; El-Rifai, W. Hypermethylation and Loss of Expression of Glutathione Peroxidase-3 in Barrett’s Tumorigenesis1. Neoplasia 2005, 7, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Yu, G.; Tseng, G.; Cieply, K.; Nelson, J.; Defrances, M.; Zarnegar, R.; Michalopoulos, G.; Luo, J.-H. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007, 67, 8043–8050. [Google Scholar] [CrossRef] [PubMed]
- Eva Falck, E.; Karlsson, S.; Carlsson, J.; Helenius, G.; Karlsson, M.; Klinga-Levan, K. Loss of glutathione peroxidase 3 expression is correlated with epigenetic mechanisms in endometrial adenocarcinoma. Cancer Cell Int. 2010, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Cejas, P.; García-Cabezas, M.A.; Casado, E.; Belda-Iniesta, C.; De Castro, J.; Fresno, J.A.; Sereno, M.; Barriuso, J.; Espinosa, E.; Zamora, P.; et al. Phospholipid hydroperoxide glutathione peroxidase (PHGPx) expression is downregulated in poorly differentiated breast invasive ductal carcinoma. Free Radic. Res. 2007, 41, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Heirman, I.; Ginneberge, D.; Brigelius-Flohé, R.; Hendrickx, N.; Agostinis, P.; Brouckaert, P.; Rottiers, P.; Grooten, J. Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic. Biol. Med. 2006, 40, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.-Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.-J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef]
- Kim, J.-W.; Gao, P.; Dang, C.V. Effects of hypoxia on tumor metabolism. Cancer Metastasis Rev. 2007, 26, 291–298. [Google Scholar] [CrossRef]
- Nyengaard, J.R.; Ido, Y.; Kilo, C.; Williamson, J.R. Interactions Between Hyperglycemia and Hypoxia: Implications for Diabetic Retinopathy. Diabetes 2004, 53, 2931–2938. [Google Scholar] [CrossRef] [PubMed]
- Tilton, R.G. Diabetic vascular dysfunction: Links to glucose-induced reductive stress and VEGF. Microsc. Res. Technol. 2002, 57, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.C.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA 1996, 276, 1957–1963. [Google Scholar] [CrossRef] [PubMed]
- Short, S.P.; Williams, C.S. Selenoproteins in Tumorigenesis and Cancer Progression. Adv. Cancer Res. 2017, 136, 49–83. [Google Scholar] [PubMed]
- Combs, G.F., Jr. Status of selenium in prostate cancer prevention. Br. J. Cancer 2004, 91, 195–199. [Google Scholar] [CrossRef]
- Imyanitov, E.N.; Togo, A.V.; Hanson, K.P. Searching for cancer-associated gene polymorphisms: Promises and obstacles. Cancer Lett. 2004, 204, 3–14. [Google Scholar] [CrossRef]
- Jablonska, E.; Gromadzinska, J.; Peplonska, B.; Fendler, W.; Reszka, E.; Krol, M.B.; Wieczorek, E.; Bukowska, A.; Gresner, P.; Galicki, M.; et al. Lipid peroxidation and glutathione peroxidase activity relationship in breast cancer depends on functional polymorphism of GPX1. BMC Cancer 2015, 15, 657. [Google Scholar] [CrossRef]
- Hu, J.; Zhou, G.-W.; Wang, N.; Wang, Y.-J. GPX1 Pro198Leu polymorphism and breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2010, 124, 425–431. [Google Scholar] [CrossRef]
- Arsova-Sarafinovska, Z.; Matevska, N.; Eken, A.; Petrovski, D.; Banev, S.; Dzikova, S.; Georgiev, V.; Sikole, A.; Erdem, O.; Sayal, A.; et al. Glutathione peroxidase 1 (GPX1) genetic polymorphism, erythrocyte GPX activity, and prostate cancer risk. Int. Urol. Nephrol. 2009, 41, 63–70. [Google Scholar] [CrossRef]
- Raaschou-Nielsen, O.; Sørensen, M.; Hansen, R.D.; Frederiksen, K.; Tjønneland, A.; Overvad, K.; Vogel, U. GPX1 Pro198Leu polymorphism, interactions with smoking and alcohol consumption, and risk for lung cancer. Cancer Lett. 2007, 247, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Men, T.; Zhang, X.; Yang, J.; Shen, B.; Li, X.; Chen, D.; Wang, J. The rs1050450 C > T polymorphism of GPX1 is associated with the risk of bladder but not prostate cancer: Evidence from a meta-analysis. Tumor Biol. 2014, 35, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Bănescu, C.; Trifa, A.P.; Voidăzan, S.; Moldovan, V.G.; Macarie, I.; Lazar, E.B.; Dima, D.; Duicu, C.; Dobreanu, M. CAT, GPX1, MnSOD, GSTM1, GSTT1, and GSTP1 Genetic Polymorphisms in Chronic Myeloid Leukemia: A Case-Control Study. Oxid. Med. Cell. Longev. 2014, 2014, 875861. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Saebø, M.; Skjelbred, C.F.; Nexø, B.A.; Hagen, P.C.; Bock, G.; Lothe, I.M.B.; Johnson, E.; Aase, S.; Hansteen, I.-L.; et al. GPX Pro198Leu and OGG1 Ser326Cys polymorphisms and risk of development of colorectal adenomas and colorectal cancer. Cancer Lett. 2005, 229, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Weïwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef]
- Moosmayer, D.; Hilpmann, A.; Hoffmann, J.; Schnirch, L.; Zimmermann, K.; Badock, V.; Furst, L.; Eaton, J.K.; Viswanathan, V.S.; Schreiber, S.L.; et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr. D Struct. Biol. 2021, 77, 237–248. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Emadi, E.M.A.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Smart, D.K.; Ortiz, K.L.; Mattson, D.; Bradbury, C.M.; Bisht, K.S.; Sieck, L.K.; Brechbiel, M.W.; Gius, D. Thioredoxin Reductase as a Potential Molecular Target for Anticancer Agents That Induce Oxidative Stress. Cancer Res. 2004, 64, 6716–6724. [Google Scholar] [CrossRef] [PubMed]
- di Bernardo, D.; Thompson, M.J.; Gardner, T.S.; Chobot, S.E.; Eastwood, E.L.; Wojtovich, A.P.; Elliott, S.J.; Schaus, S.E.; Collins, J.J. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat. Biotechnol. 2005, 23, 377–383. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Chen, T.; You, Y.; Hu, H.; Zheng, W.; Kwong, W.-L.; Zou, T.; Che, C.-M. A cancer-targeted nanosystem for delivery of gold(III) complexes: Enhanced selectivity and apoptosis-inducing efficacy of a gold(III) porphyrin complex. Angew. Chem. Int. Ed. Engl. 2014, 53, 12532–12536. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hou, Y.; Meng, X.; Ge, C.; Ma, H.; Li, J.; Fang, J. Selective Activation of a Prodrug by Thioredoxin Reductase Providing a Strategy to Target Cancer Cells. Angew. Chem. Int. Ed. Engl. 2018, 57, 6141–6145. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, C.; He, Y.; Zhenqin Zhang, Z.; Zhou, W.; Muhammad, N.; Guo, Y.; Wang, X.; Guo, Z. Restraining Cancer Cells by Dual Metabolic Inhibition with a Mitochondrion-Targeted Platinum(II) Complex. Angew. Chem. Int. Ed. Engl. 2019, 58, 4638–4643. [Google Scholar] [CrossRef]
- Gromer, S.; Arscott, L.D.; Williams, C.H., Jr.; Schirmer, R.H.; Becker, K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998, 273, 20096–20101. [Google Scholar] [CrossRef]
- Sasada, T.; Nakamura, H.; Ueda, S.; Sato, N.; Kitaoka, Y.; Gon, Y.; Takabayashi, A.; Spyrou, G.; Holmgren, A.; Yodoi, J. Possible involvement of thioredoxin reductase as well as thioredoxin in cellular sensitivity to cis-diamminedichloroplatinum (II). Free Radic. Biol. Med. 1999, 27, 504–514. [Google Scholar] [CrossRef]
- Tibodeau, J.D.; Benson, L.M.; Isham, C.R.; Owen, W.G.; Bible, K.C. The Anticancer Agent Chaetocin Is a Competitive Substrate and Inhibitor of Thioredoxin Reductase. Antioxid. Redox Signal. 2008, 11, 1097–1106. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, B.; Duan, D.; Wu, J.; Fang, J. Curcumin targeting the thioredoxin system elevates oxidative stress in HeLa cells. Toxicol. Appl. Pharmacol. 2012, 262, 341–348. [Google Scholar] [CrossRef]
- Jin-Jing Jia, J.-J.; Geng, W.-S.; Wang, Z.-Q.; Chen, L.; Zeng, X.-S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar]
- Liang, Y.-W.; Zheng, J.; Li, X.; Zheng, W.; Chen, T. Selenadiazole derivatives as potent thioredoxin reductase inhibitors that enhance the radiosensitivity of cancer cells. Eur. J. Med. Chem. 2014, 84, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, J.; Peng, S.; Liu, R.; Li, X.; Hou, Y.; Han, X.; Fang, J. Thioredoxin reductase inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zachary Bloomgarden, Z. Evolution of type 2 diabetes mellitus treatment approaches: 2. J. Diabetes Res. 2019, 11, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Jetton, T.L.; Lausier, J.; LaRock, K.; Trotman, W.E.; Larmie, B.; Habibovic, A.; Peshavaria, M.; Leahy, J.L. Mechanisms of compensatory beta-cell growth in insulin-resistant rats: Roles of Akt kinase. Diabetes 2005, 54, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef]
- Evren Okur, M.E.; Karantas, I.D.; Siafaka, P.I. Diabetes Mellitus: A Review on Pathophysiology, Current Status of Oral Pathophysiology, Current Status of Oral Medications and Future Perspectives. ACTA Pharm. Sci. 2017, 55, 61–82. [Google Scholar]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Abad-Jiménez, Z.; de Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef]
- Karunakaran, U.; Park, K.-G. A systematic review of oxidative stress and safety of antioxidants in diabetes: Focus on islets and their defense. Diabetes Metab. J. 2013, 37, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in beta-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Speckmann, B.; Pinto, A.; Sies, H. High selenium intake and increased diabetes risk: Experimental evidence for interplay between selenium and carbohydrate metabolism. J. Clin. Biochem. Nutr. 2011, 48, 40–45. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; de Haën, C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 1979, 254, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Vatamaniuk, M.Z.; Wang, S.K.; Roneker, C.A.; Simmons, R.A.; Lei, X.G. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia 2008, 51, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, W.C. Association of Glutathione Peroxidase Activity with Insulin Resistance and Dietary Fat Intake during Normal Pregnancy. J. Clin. Endocrinol. Metab. 2004, 89, 4772–4773. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Agostini, M.; Mitchell, C.; Schoenmakers, N.; Papp, L.; Rajanayagam, O.; Padidela, R.; Ceron-Gutierrez, L.; Doffinger, R.; Prevosto, C.; et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Investig. 2010, 120, 4220–4235. [Google Scholar] [CrossRef]
- Molteni, C.G.; Principi, N.; Esposito, S. Reactive oxygen and nitrogen species during viral infections. Free Radic. Res. 2014, 48, 1163–1169. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.S.; Lee, C.-Y.J.; Lim, E.C.H.; Quek, A.M.L.; Yeo, L.L.L.; Huang, S.-H.; Halliwell, B. Oxidative damage in dengue fever. Free Radic. Biol. Med. 2009, 47, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.R.; Rojo, D.R.; von Lindern, J.J.; O’Brien, W.A. HIV-1 replication cycle. Clin. Lab. Med. 2002, 22, 611–635. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Martin, M.A. HIVs and their replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2107–2185. [Google Scholar]
- Ryser, H.J.-P.; Flückiger, R. Progress in targeting HIV-1 entry. Drug Discov. Today 2005, 10, 1085–1194. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Kim, P.S. HIV Entry and Its Inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Barbouche, R.; Miquelis, R.; Jones, I.M.; Fenouillet, E. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J. Biol. Chem. 2003, 278, 3131–3136. [Google Scholar] [CrossRef]
- Gallina, A.; Hanley, T.M.; Mandel, R.; Trahey, M.; Broder, C.C.; Viglianti, G.A.; Ryser, H.J.-P. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J. Biol. Chem. 2002, 277, 50579–50588. [Google Scholar] [CrossRef]
- Markovic, I.; Stantchev, T.S.; Fields, K.H.; Tiffany, L.J.; Tomiç, M.; Weiss, C.D.; Broder, C.C.; Strebel, K.; Clouse, K.A. Thiol/disulfide exchange is a prerequisite for CXCR4-tropic HIV-1 envelope-mediated T-cell fusion during viral entry. Blood 2004, 103, 1586–1594. [Google Scholar] [CrossRef]
- Cerutti, N.; Killick, M.; Jugnarain, V.; Papathanasopoulos, M.; Capovilla, A. Disulfide reduction in CD4 domain 1 or 2 is essential for interaction with HIV glycoprotein 120 (gp120), which impairs thioredoxin-driven CD4 dimerization. J. Biol. Chem. 2014, 289, 10455–10465. [Google Scholar] [CrossRef]
- Matthias, L.J.; Azimi, I.; Tabrett, C.A.; Hogg, P.J. Reduced monomeric CD4 is the preferred receptor for HIV. J. Biol. Chem. 2010, 285, 40793–40799. [Google Scholar] [CrossRef] [PubMed]
- Matthias, L.J.; Yam, P.T.W.; Jiang, X.-M.; Vandegraaff, N.; Li, P.; Poumbourios, P.; Donoghue, N.; Hogg, P.J. Disulfide exchange in domain 2 of CD4 is required for entry of HIV-1. Nat. Immunol. 2002, 3, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Moolla, N.; Killick, M.; Papathanasopoulos, M.; Capovilla, A. Thioredoxin (Trx1) regulates CD4 membrane domain localization and is required for efficient CD4-dependent HIV-1 entry. Biochim. Biophys. Acta 2016, 1860, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J.; Isacsson, O.; Söderlund, J.; Balzarini, J.; Johansson, M.; Lundberg, M. Human glutaredoxin-1 catalyzes the reduction of HIV-1 gp120 and CD4 disulfides and its inhibition reduces HIV-1 replication. Int. J. Biochem. Cell Biol. 2009, 41, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Ryser, H.J.; Levy, E.M.; Mandel, R.; DiSciullo, G.J. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. USA 1994, 91, 4559–4563. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Kuppuswamy, M.; Subramanian, T.; Srinivasan, A.; Chinnadurai, G. Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res. 1989, 17, 3551–3561. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Koken, S.E.; Greijer, A.E.; Verhoef, K.; van Wamel, J.; Bukrinskaya, A.G.; Berkhout, B. Intracellular analysis of in vitro modified HIV Tat protein. J. Biol. Chem. 1994, 269, 8366–8375. [Google Scholar] [CrossRef]
- Price, T.O.; Ercal, N.; Nakaoke, R.; Banks, W.A. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005, 1045, 57–63. [Google Scholar] [CrossRef]
- Banerjee, A.; Zhang, X.; Manda, K.R.; Banks, W.A.; Nuran Ercal, N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: Potential role of the thiol antioxidant N-acetylcysteine amide. Free Radic. Biol. Med. 2010, 48, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Samikkannu, T.; Ranjith, D.; Rao, K.V.K.; Atluri, V.S.R.; Pimentel, E.; El-Hage, N.; Nair, M.P.N. HIV-1 gp120 and morphine induced oxidative stress: Role in cell cycle regulation. Front. Microbiol. 2015, 6, 614. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.-J.; Guiraud, P.; Didier, C.; Seve, M.; Flores, S.C.; Favier, A. Impairs Selenoglutathione Peroxidase Expression and Activity by a Mechanism Independent of Cellular Selenium Uptake: Consequences on Cellular Resistance to UV-A Radiation. Arch. Biochem. Biophys. 2001, 386, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Stadtman, T.C.; Hatfield, D.L.; Jeang, K.T. Levels of major selenoproteins in T cells decrease during HIV infection and low molecular mass selenium compounds increase. Proc. Natl. Acad. Sci. USA 1999, 96, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.E.; Klaus, J.R.; Llabre, M.M.; Gonzalez, A.; Lawrence, P.J.; Maher, K.J.; Greeson, J.M.; Baum, M.K.; Shor-Posner, G.; Skyler, J.S.; et al. Suppression of human immunodeficiency virus type 1 viral load with selenium supplementation: A randomized controlled trial. Arch. Intern. Med. 2007, 167, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Mattsson, Å.; Reiser, K.; Holmgren, A.; Curbo, S. Inhibition of the thioredoxin system by PX-12 (1-methylpropyl 2-imidazolyl disulfide) impedes HIV-1 infection in TZM-bl cells. Sci. Rep. 2019, 9, 5656. [Google Scholar] [CrossRef] [PubMed]
- Reiser, K.; Mathys, L.; Curbo, S.; Pannecouque, C.; Noppen, S.; Liekens, S.; Engman, L.; Lundberg, M.; Balzarini, J.; Karlsson, A. The Cellular Thioredoxin-1/Thioredoxin Reductase-1 Driven Oxidoreduction Represents a Chemotherapeutic Target for HIV-1 Entry Inhibition. PLoS ONE 2016, 11, e0147773. [Google Scholar] [CrossRef]
- Reiser, K.; François, K.O.; Schols, D.; Bergman, T.; Jörnvall, H.; Balzarini, J.; Karlsson, A.; Lundberg, M. Thioredoxin-1 and protein disulfide isomerase catalyze the reduction of similar disulfides in HIV gp120. Int. J. Biochem. Cell Biol. 2012, 44, 556–562. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.; Lau, E.H.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Maiti, B.K. Can Papain-like Protease Inhibitors Halt SARS-CoV-2 Replication? ACS Pharmacol. Transl. Sci. 2020, 3, 1017–1019. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, I.A.; Todd, D.A.; Lima, S.T.; Jonathan, R.; Chekan, J.R.; Chiu, N.H.; Taylor, E.W. SARS-CoV-2 Main Protease Targets Host Selenoproteins and Glutathione Biosynthesis for Knockdown via Proteolysis, Potentially Disrupting the Thioredoxin and Glutaredoxin Redox Cycles. Antioxidants 2023, 12, 559. [Google Scholar] [CrossRef] [PubMed]
- Tomo, S.; Saikiran, G.; Banerjee, M.; Paul, S. Selenium to selenoproteins—Role in COVID-19. EXCLI J. 2021, 20, 781–791. [Google Scholar] [PubMed]
- Moghaddam, A.; Heller, R.A.; Sun, Q.; Seelig, J.; Cherkezov, A.; Seibert, L.; Hackler, J.; Seemann, P.; Diegmann, J.; Pilz, M.; et al. Selenium Deficiency Is Associated with Mortality Risk from COVID-19. Nutrients 2020, 12, 2098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Taylor, E.W.; Bennett, K.; Saad, R.; Rayman, M.P. Association between regional selenium status and reported outcome of COVID-19 cases in China. Am. J. Clin. Nutr. 2020, 111, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Saad, R.; Taylor, E.W.; Rayman, M.P. Selenium and selenoproteins in viral infection with potential relevance to COVID-19. Redox Biol. 2020, 37, 101715. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.A.; Kolbeck, P.C.; Rohr, L.H.; Shi, Q.; Morris, V.C.; Levander, O.A. Benign human enterovirus becomes virulent in selenium-deficient mice. J. Med. Virol. 1994, 43, 166–170. [Google Scholar] [CrossRef]
- Avery, J.C.; Hoffmann, P.R. Selenium, Selenoproteins, and Immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef]
- Barchielli, G.; Capperucci, A.; Tanini, D. The Role of Selenium in Pathologies: An Updated Review. Antioxidants 2022, 11, 251. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Maiti, B.K. Potential Role of Peptide-Based Antiviral Therapy against SARS-CoV-2 Infection. ACS Pharmacol. Transl. Sci. 2020, 3, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zeida, A.; Edwards, C.E.; Mallory, M.L.; Sastre, S.; Machado, M.R.; Pickles, R.J.; Fu, L.; Liu, K.; Yang, J.; et al. Thiol-based chemical probes exhibit antiviral activity against SARS-CoV-2 via allosteric disulfide disruption in the spike glycoprotein. Proc. Natl. Acad. Sci. USA 2022, 119, e2120419119. [Google Scholar] [CrossRef] [PubMed]
- Hati, S.; Sudeep Bhattacharyya, S. Impact of Thiol-Disulfide Balance on the Binding of COVID-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS Omega 2020, 5, 16292–16298. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Santucci, A.; Bartolini, D.; Galli, F.; Rossi, R. The age-dependent decline of the extracellular thiol-disulfide balance and its role in SARS-CoV-2 infection. Redox Biol. 2021, 41, 101902. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Soria-Castro, E.; Soto, M.E.; Guarner-Lans, V.; Rojas, G.; Perezpeña-Diazconti, M.; Críales-Vera, S.A.; Manzano Pech, L.; Pérez-Torres, I. The kidnapping of mitochondrial function associated with the SARS-CoV-2 infection. Histol. Histopathol. 2021, 36, 947–965. [Google Scholar]
- Yang, M.; Lai, C.L. SARS-CoV-2 infection: Can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Taylor, E.W.; Radding, W. Understanding Selenium and Glutathione as Antiviral Factors in COVID-19: Does the Viral Mpro Protease Target Host Selenoproteins and Glutathione Synthesis? Front. Nutr. 2020, 7, 143. [Google Scholar] [CrossRef]
- Li, F.; Leier, A.; Liu, Q.; Wang, Y.; Xiang, D.; Akutsu, T.; Webb, G.I.; Smith, A.I.; Marquez-Lago, T.; Li, J.; et al. Procleave: Predicting Protease-specific Substrate Cleavage Sites by Combining Sequence and Structural Information. Genom. Proteom. Bioinform. 2020, 18, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Hogan, C.; Perkins, A.V. Selenoproteins in the Human Placenta: How Essential Is Selenium to a Healthy Start to Life? Nutrients 2022, 14, 628. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Murtaza, G.; Metwally, E.; Kalhoro, D.H.; Kalhoro, M.S.; Rahu, B.A.; Sahito, R.G.A.; Yin, Y.; Yang, H.; Chughtai, M.I.; et al. The Role of Oxidative Stress and Antioxidant Balance in Pregnancy. Mediat. Inflamm. 2021, 2021, 9962860. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Vázquez, C.M.; Mate, A.; Sobrevia, L.; Marín, R. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165354. [Google Scholar] [CrossRef] [PubMed]
- Scaife, P.J.; Simpson, A.; Kurlak, L.O.; Briggs, L.V.; Gardner, D.S.; Broughton Pipkin, F.; Jones, C.J.P.; Mistry, H.D. Increased Placental Cell Senescence and Oxidative Stress in Women with Pre-Eclampsia and Normotensive Post-Term Pregnancies. Int. J. Mol. Sci. 2021, 22, 7295. [Google Scholar] [CrossRef] [PubMed]
- Farzin, L.; Sajadi, F. Comparison of serum trace element levels in patients with or without pre-eclampsia. J. Res. Med. Sci. 2012, 17, 938–941. [Google Scholar]
- Khera, A.; Vanderlelie, J.J.; Perkins, A.V. Selenium supplementation protects trophoblast cells from mitochondrial oxidative stress. Placenta 2013, 34, 594–598. [Google Scholar] [CrossRef]
- Biswas, K.; McLay, J.; Campbell, F.M. Selenium Supplementation in Pregnancy-Maternal and Newborn Outcomes. J. Nutr. Metab. 2022, 2022, 4715965. [Google Scholar] [CrossRef]
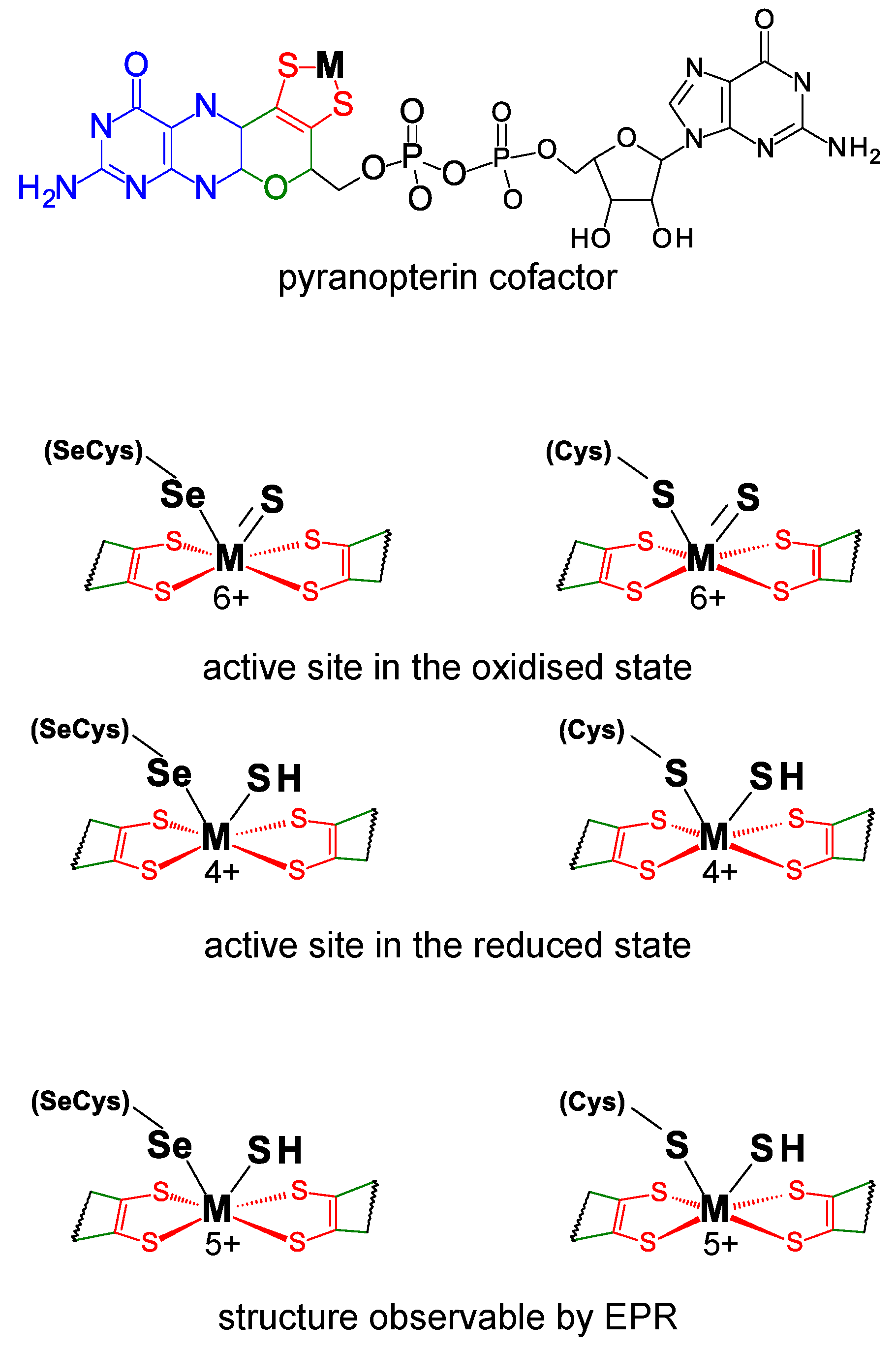


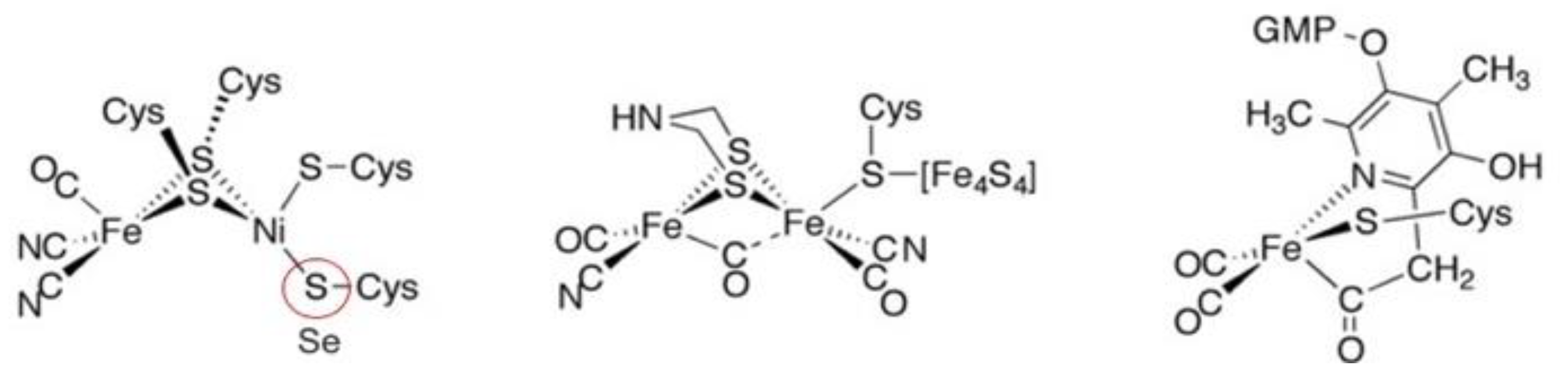
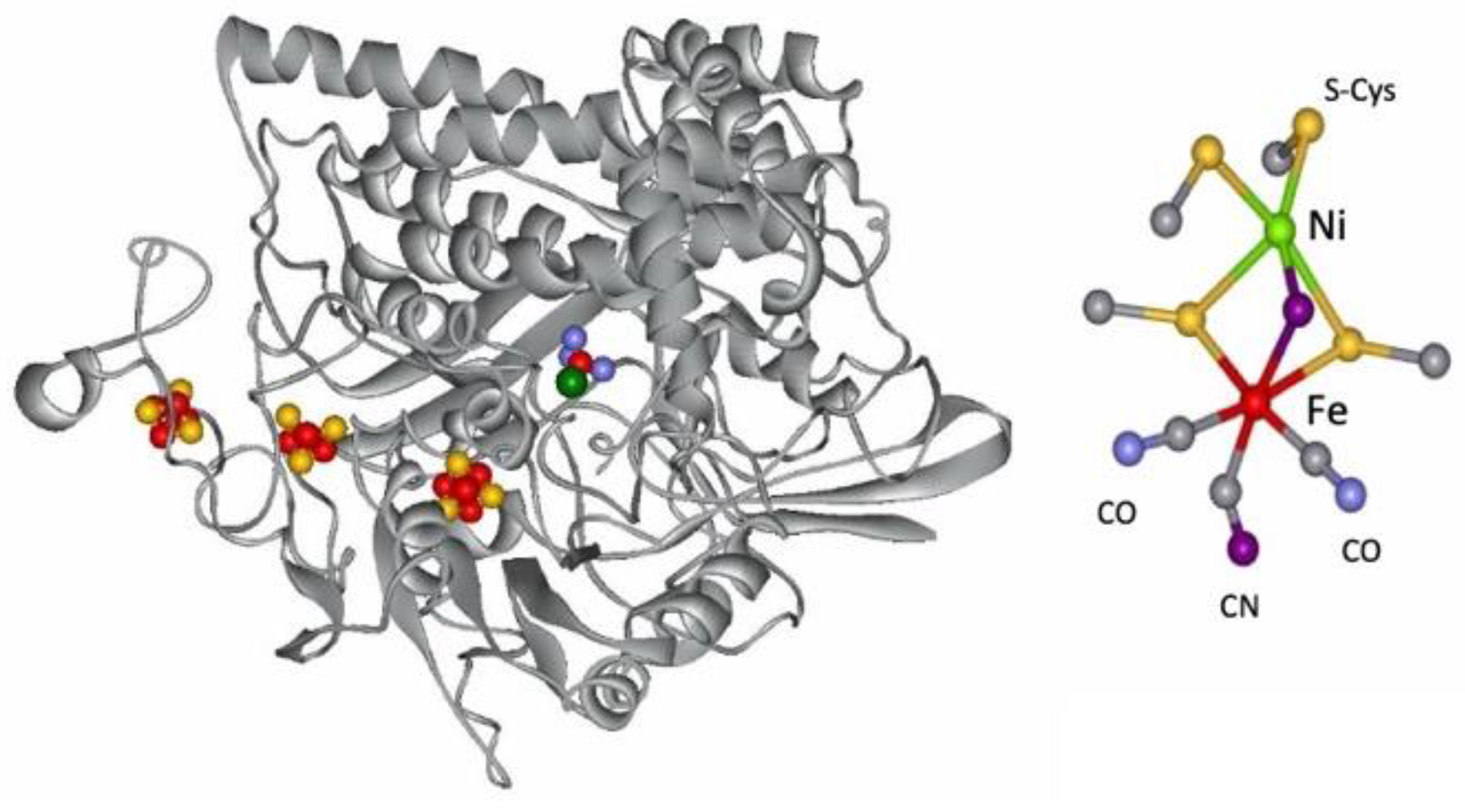
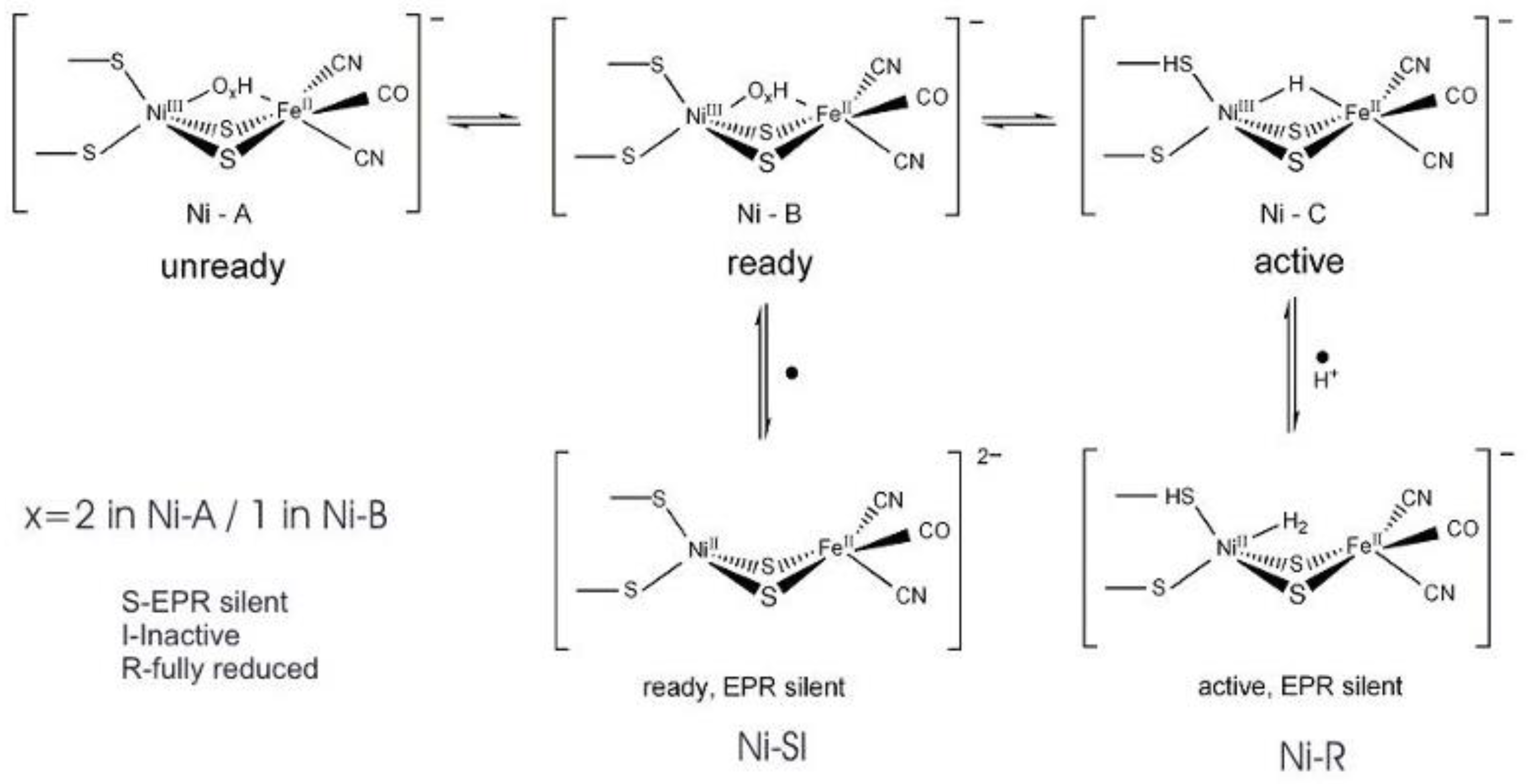
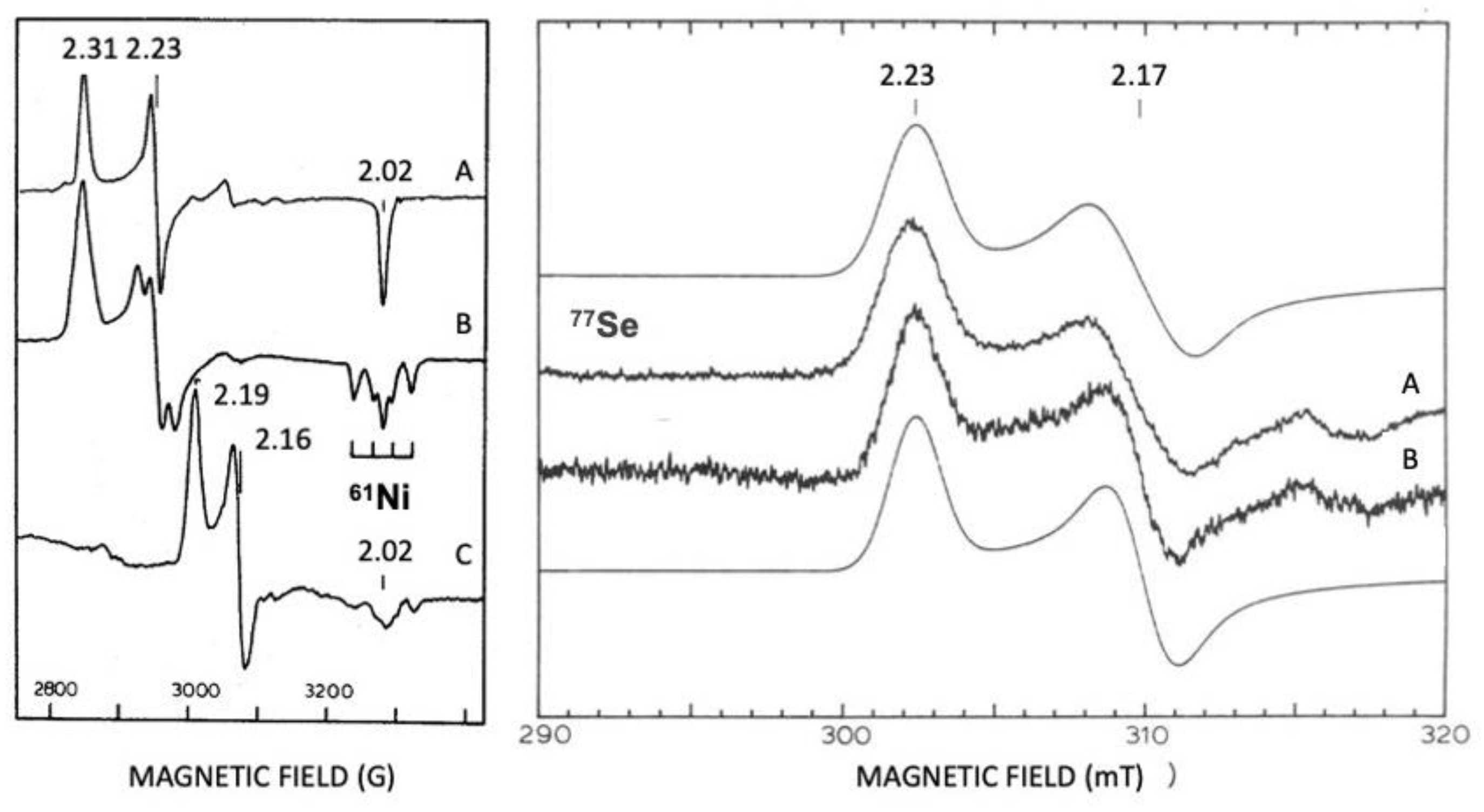
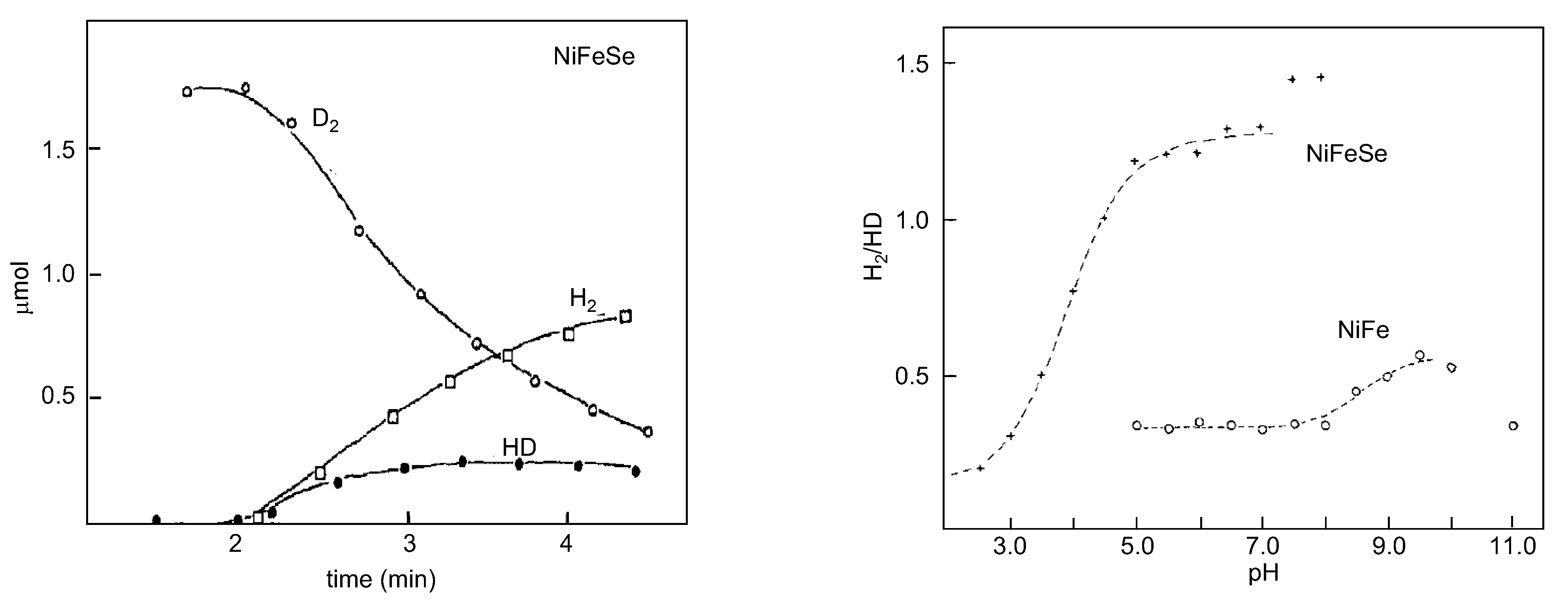

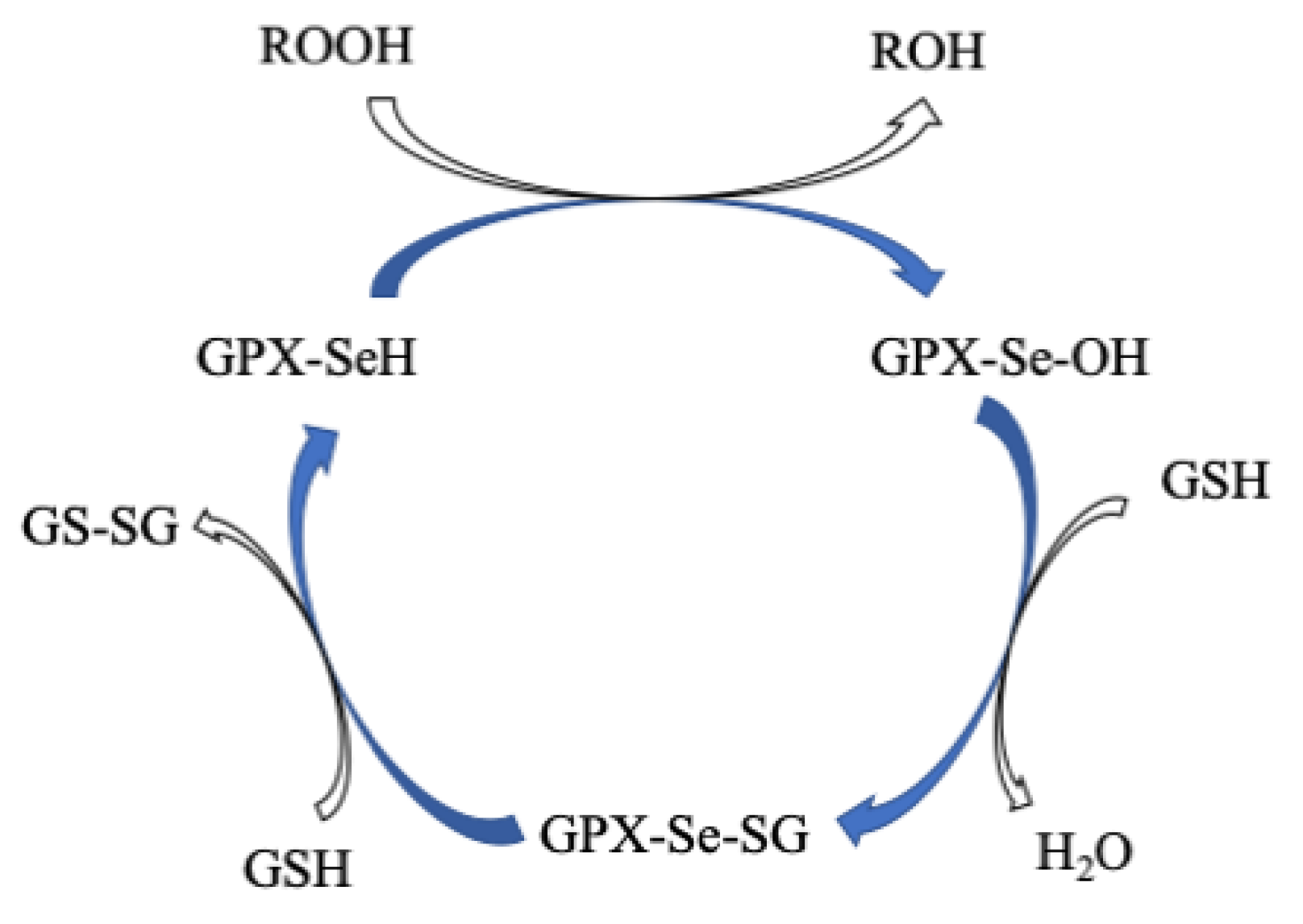
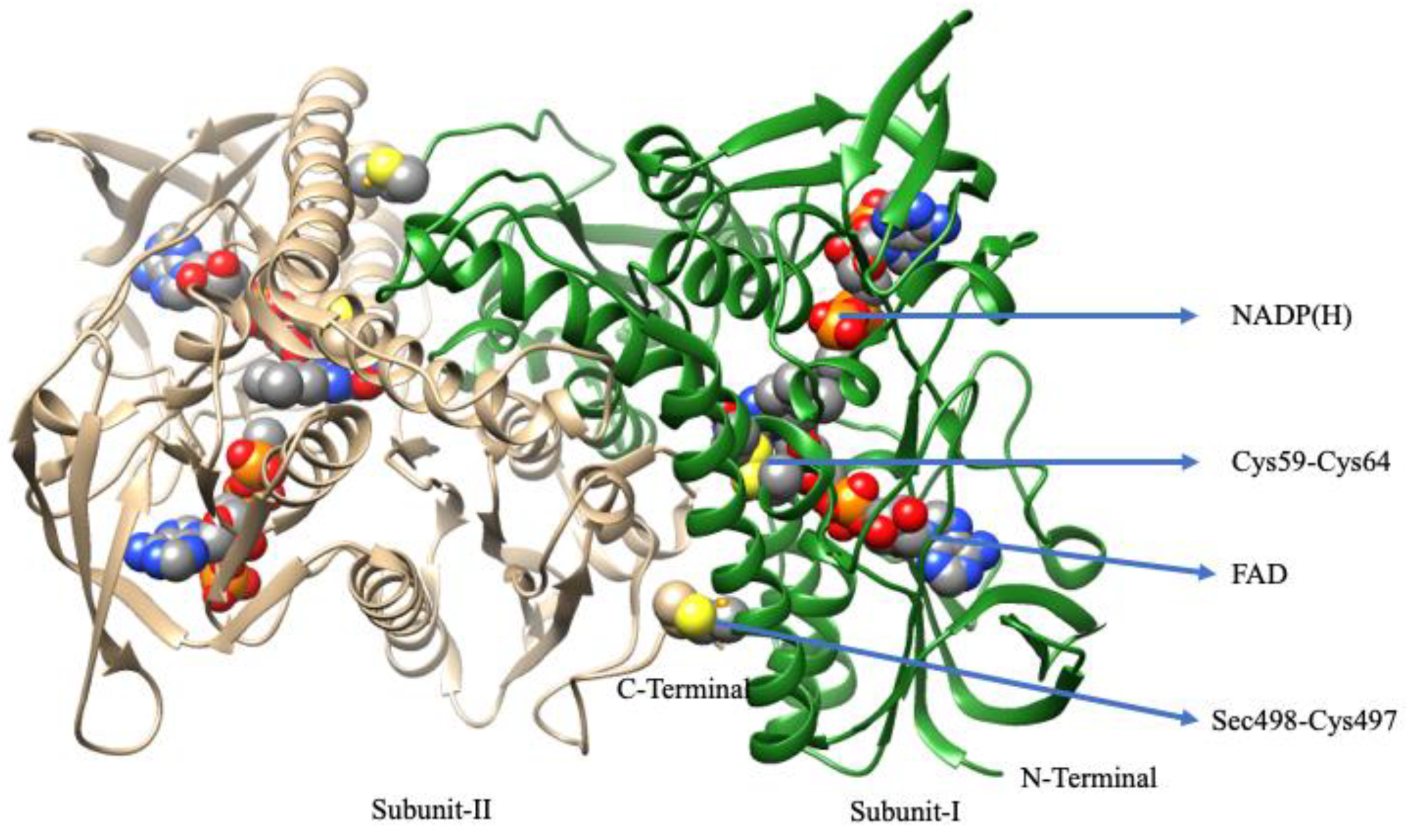

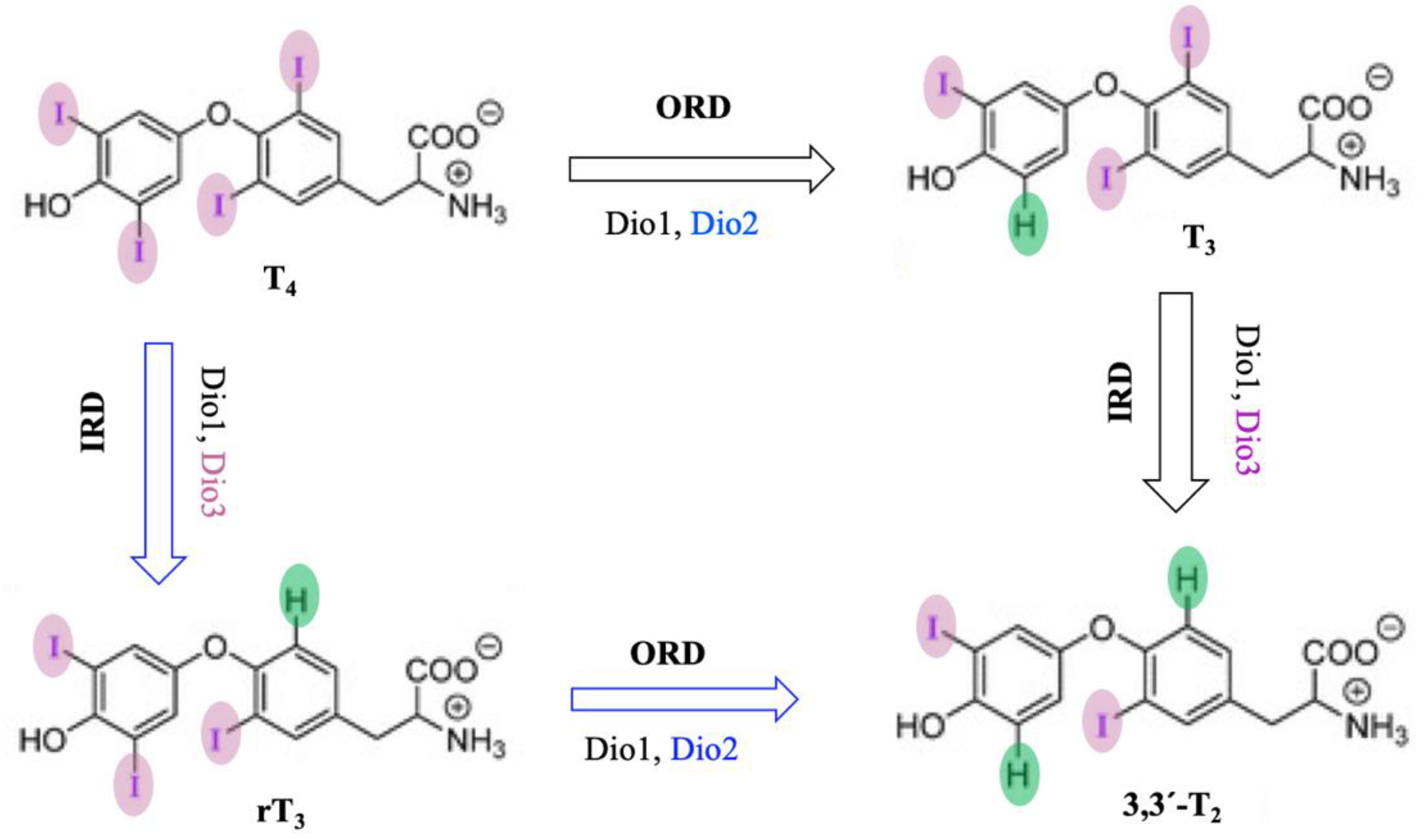
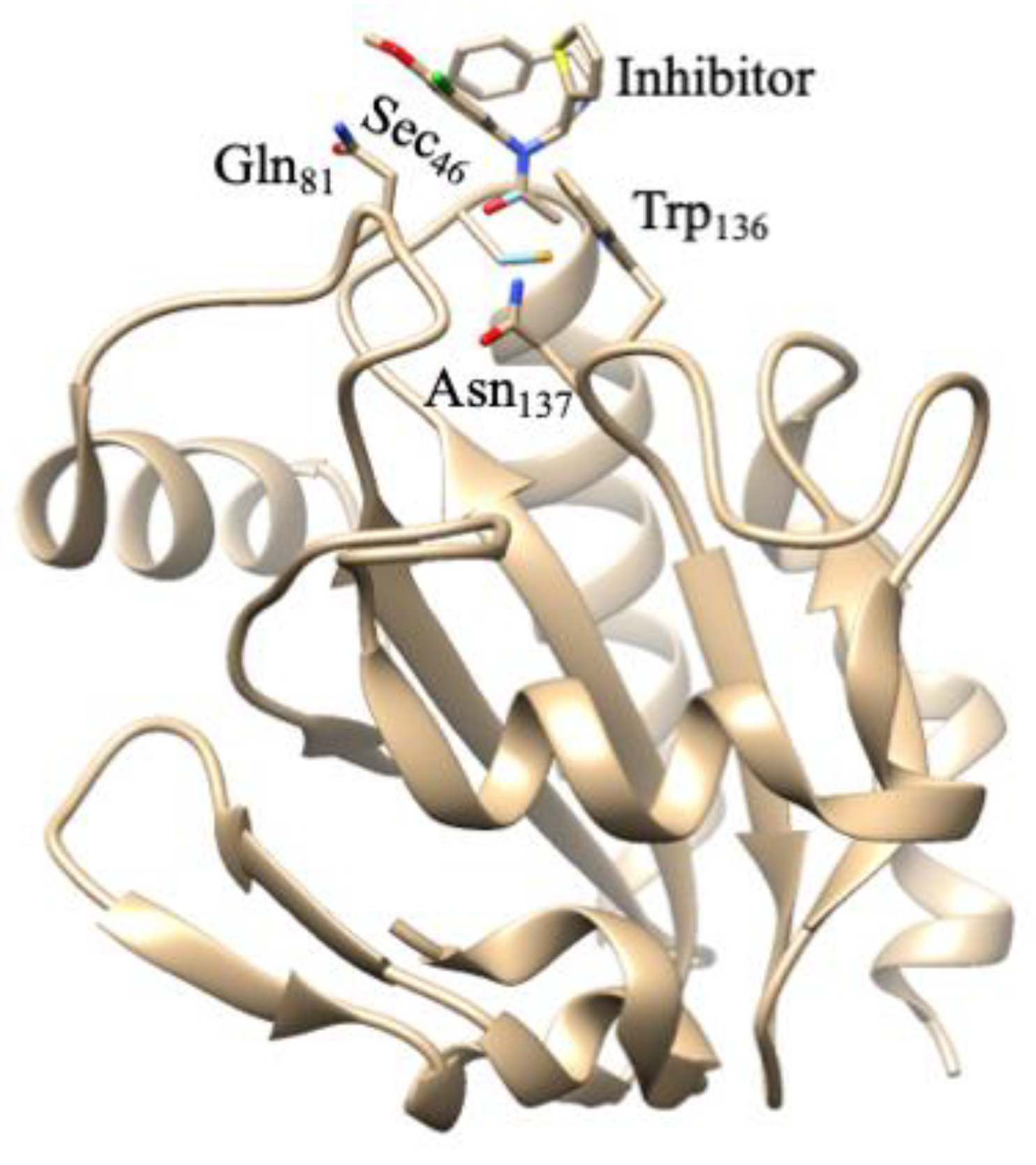
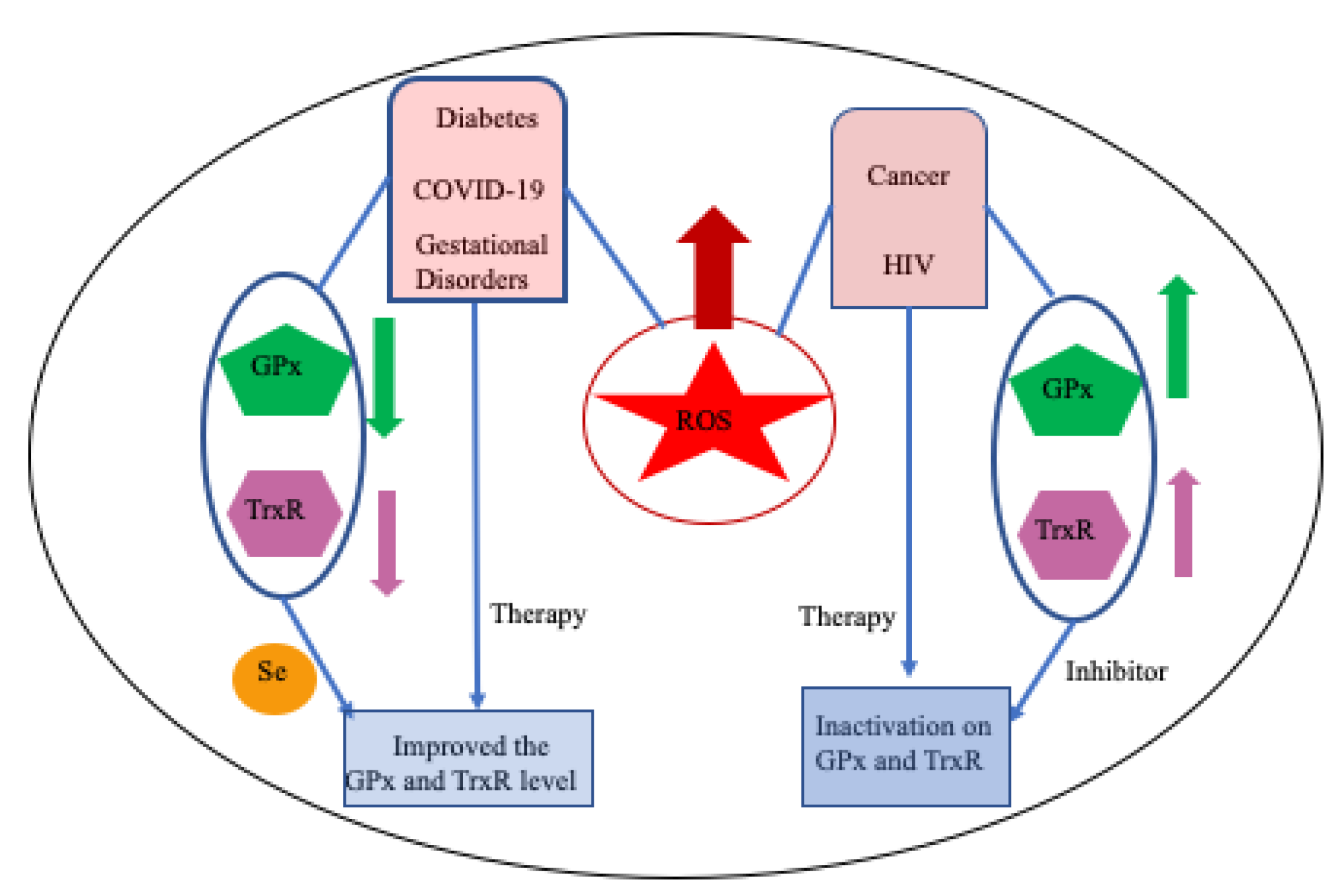

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Site a | Subunit Composition | Examples | Notes |
|---|---|---|---|
| no metal | α2 no redox-active cofactors | Candida boidinii FDH | ● NAD-dependent |
| W SeCys | α W, [4Fe-4S] | Clostridium carboxidivorans FDH | ● cytoplasmic? ● NAD-dependent |
| Thermoanaerobacter kivui FDH | ● hydrogen-dependent CO2 reductase | ||
| αβ α: W, [4Fe-4S] β: 3 [4Fe-4S] | Desulfovibrio gigas, Desulfovibrio alaskensis, Desulfovibrio vulgaris FDHs | ● periplasmic | |
| (αβ)2 α: W, [4Fe-4S] β: 3 [4Fe-4S] | Moorella thermoacetica FDH | ● cytoplasmic ● NADP-dependent | |
| (αβγ)2 W, Fe | Synthrobacter fumaroxidans FDH | ● periplasmic? | |
| W Cys | αβ α: W, ≥ 1 Fe/S β: [4Fe-4S], FMN | Methylobacterium extorquens FDH | ● cytoplasmic ● NAD-dependent |
| Mo SeCys | α Mo, [4Fe-4S] | Escherichia coli FDH H | ● cytoplasmic ● formate–hydrogen lyase system |
| Acetobacterium woodii FDH | ● hydrogen-dependent CO2 reductase | ||
| αβγ α: Mo, [4Fe-4S] β: 3 [4Fe-4S] γ: 4 c haems | Desulfovibrio desulfuricans FDH | ● periplasmic | |
| Desulfovibrio vulgaris FDH | |||
| (αβγ)3 α: Mo, [4Fe-4S] β: 4 [4Fe-4S] γ: 2 b haems | E. coli FDH N | ● membrane-bound periplasm-faced ● partner anaerobic nitrate–formate respir. system | |
| E. coli FDH O | ● membrane-bound periplasm-faced ● partner microaerobic nitrate–formate respir. syst. | ||
| Mo Cys | α Mo, [4Fe-4S] | Pectobacterium atrosepticum, Corynebacterium glutamicum FDHs | ● cytoplasmic |
| αβ Mo, several Fe/S | Clostridium pasteurianum FDH | ● cytoplasmic | |
| αβ Mo, FAD, several Fe/S, Zn | Methanobacterium formicicum FDH | ● cytoplasmic ● F420-dependent | |
| αβγ α: Mo, [4Fe-4S] β: 4 [4Fe-4S] γ: 4 b haems | Wolinella succinogenes FDH | ● membrane-bound | |
| (αβγ)2 α: Mo, [2Fe-2S], 4 [4Fe-4S] β: [4Fe-4S], FMN γ: [2Fe-2S] | Cupriavidus necator, Rhodobacter capsulatus, Methylosinus trichosporium, Pseudomonas oxalatus FDHs | ● cytoplasmic ● NAD-dependent | |
| (αβγδ)2 Mo, ≥1 [2Fe–2S], ≥1 [4Fe–4S], FMN | Methylosinus trichosporium FDH | ● cytoplasmic ● NAD-dependent | |
| (αβγδεω)4 α: 2 Zn β: Mo, [4Fe-4S] γ: 2 [4Fe-4S] γ: 4 b haems δ ε: 8 [4Fe-4S] ω | Methanothermobacter wolfeiir FMFDH | ● cytoplasmic |
| Hase | SRB | Localization | EPR g-Values Native | EPR g-Values H2 Red—Ni-C |
|---|---|---|---|---|
| [NiFe] | D. gigas | periplasm | 2.31 2.23 2.02 | 2.19 2.14 2.02 |
| [NiFe] | D. desulfuricans (ATCC 2774) | periplasm | 2.32 2.16 2.02 | 2.19 2.14 2.02 |
| [NiFeSe] | D. desulfuricans (Norway 4) | soluble | weak Ni(III) signals | 2.20 2.15~2.0 |
| [NiFeSe] | D. salexigens | periplasm | EPR silent | 2.22 2.14 2.01 |
| [NiFeSe] | D. africanus | soluble | EPR silent | 2.21 2.17 2.01 |
| [NiFeSe] | D. baculatus | soluble | weak Ni(III) signals | 2.20 2.16 2.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia, L.B.; Maiti, B.K.; Moura, I.; Moura, J.J.G. Selenium—More than Just a Fortuitous Sulfur Substitute in Redox Biology. Molecules 2024, 29, 120. https://doi.org/10.3390/molecules29010120
Maia LB, Maiti BK, Moura I, Moura JJG. Selenium—More than Just a Fortuitous Sulfur Substitute in Redox Biology. Molecules. 2024; 29(1):120. https://doi.org/10.3390/molecules29010120
Chicago/Turabian StyleMaia, Luisa B., Biplab K. Maiti, Isabel Moura, and José J. G. Moura. 2024. "Selenium—More than Just a Fortuitous Sulfur Substitute in Redox Biology" Molecules 29, no. 1: 120. https://doi.org/10.3390/molecules29010120
APA StyleMaia, L. B., Maiti, B. K., Moura, I., & Moura, J. J. G. (2024). Selenium—More than Just a Fortuitous Sulfur Substitute in Redox Biology. Molecules, 29(1), 120. https://doi.org/10.3390/molecules29010120