
Asymmetric Monomethine Cyanine Dyes with Hydrophobic Functionalities for Fluorescent Intercalator Displacement Assay


 , and
, and
Abstract
:1. Introduction
2. Results
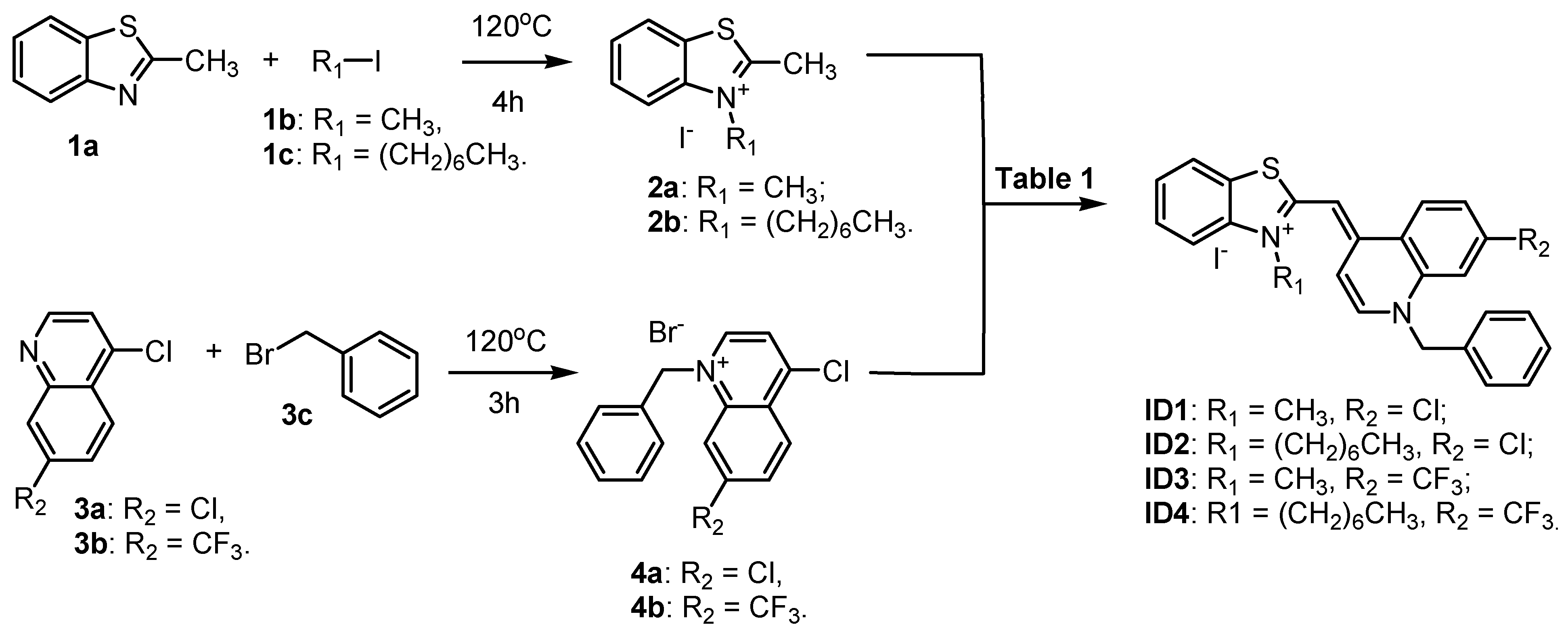
2.1. Synthesis of Intermediates and Dyes
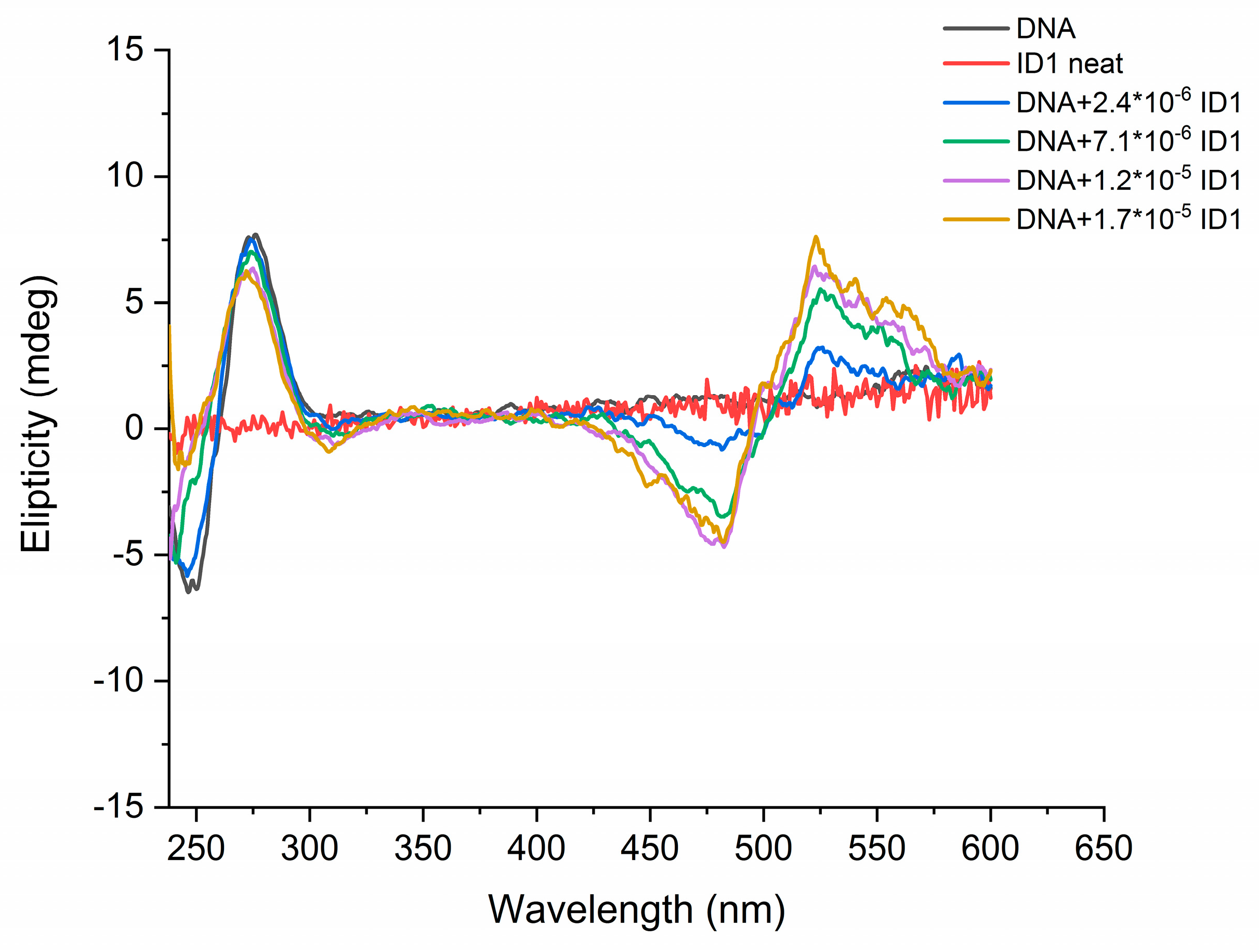
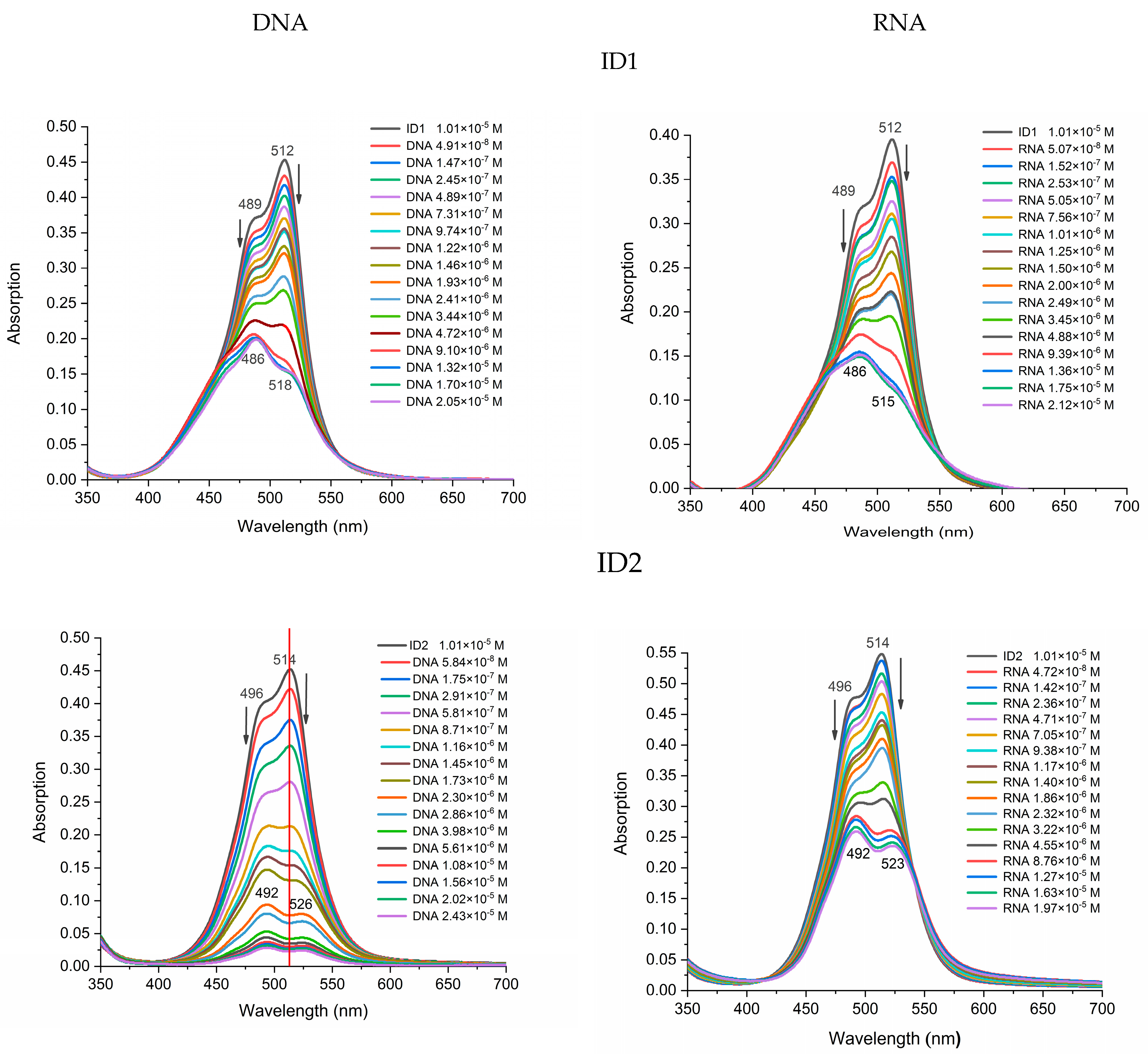
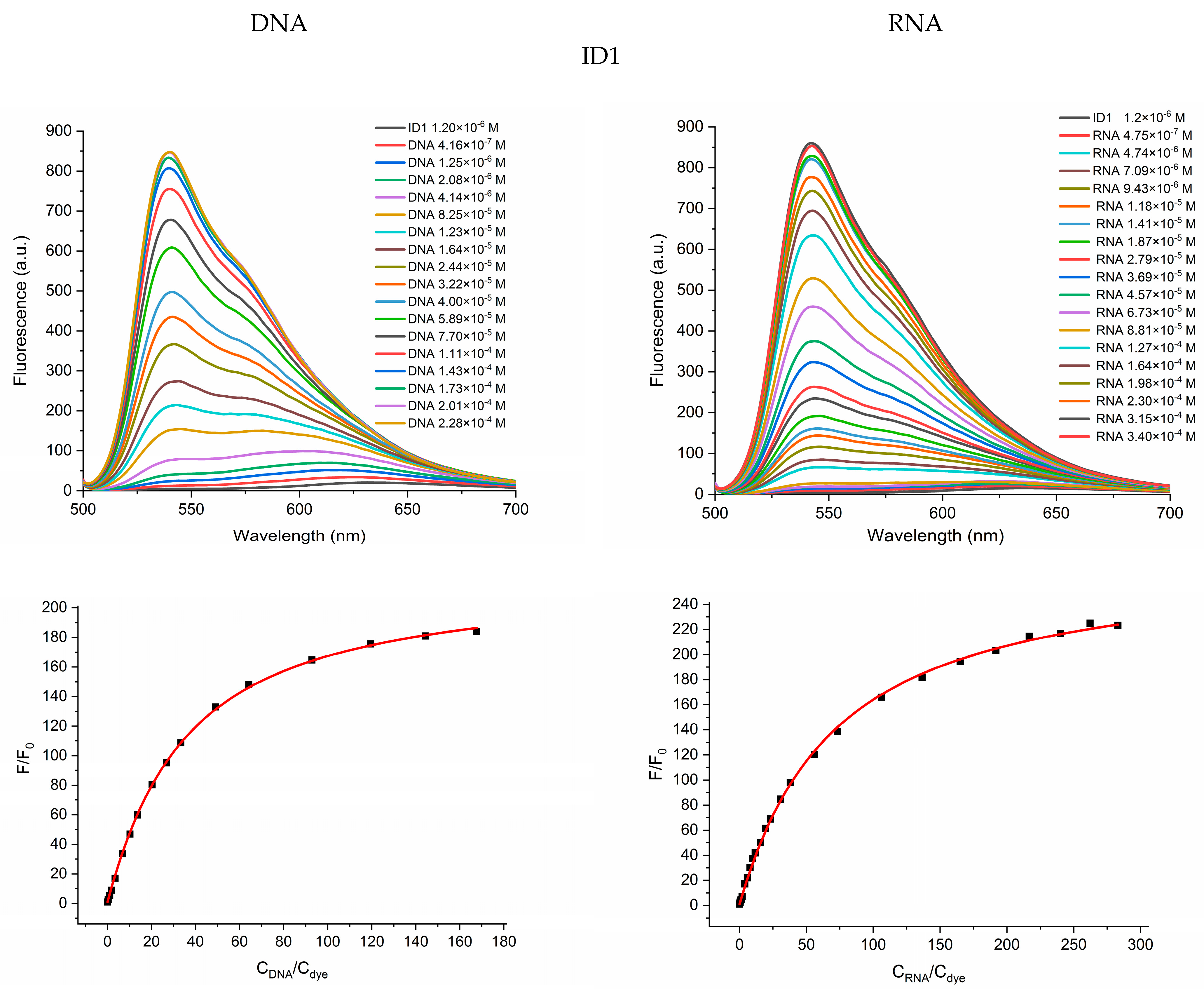
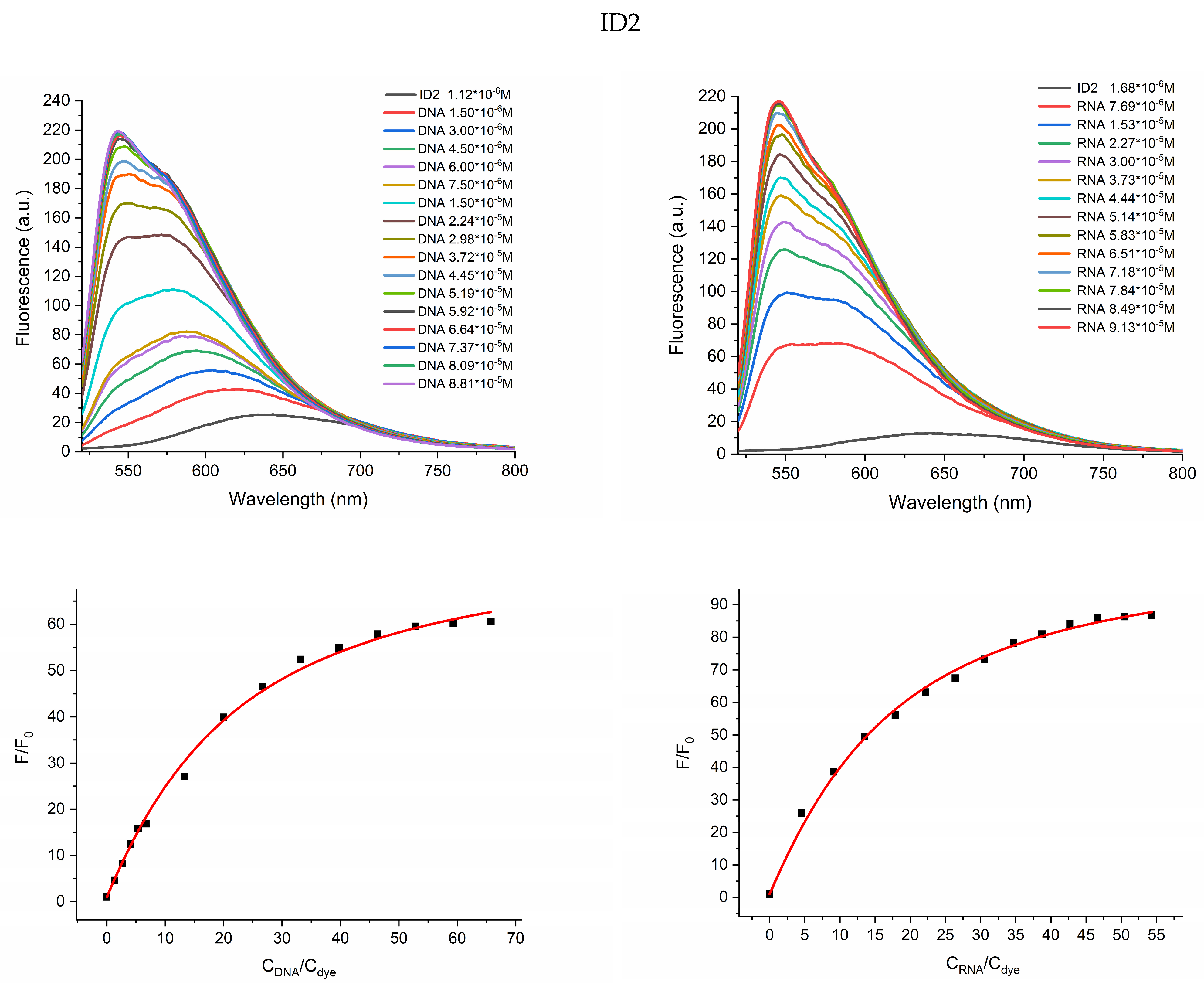
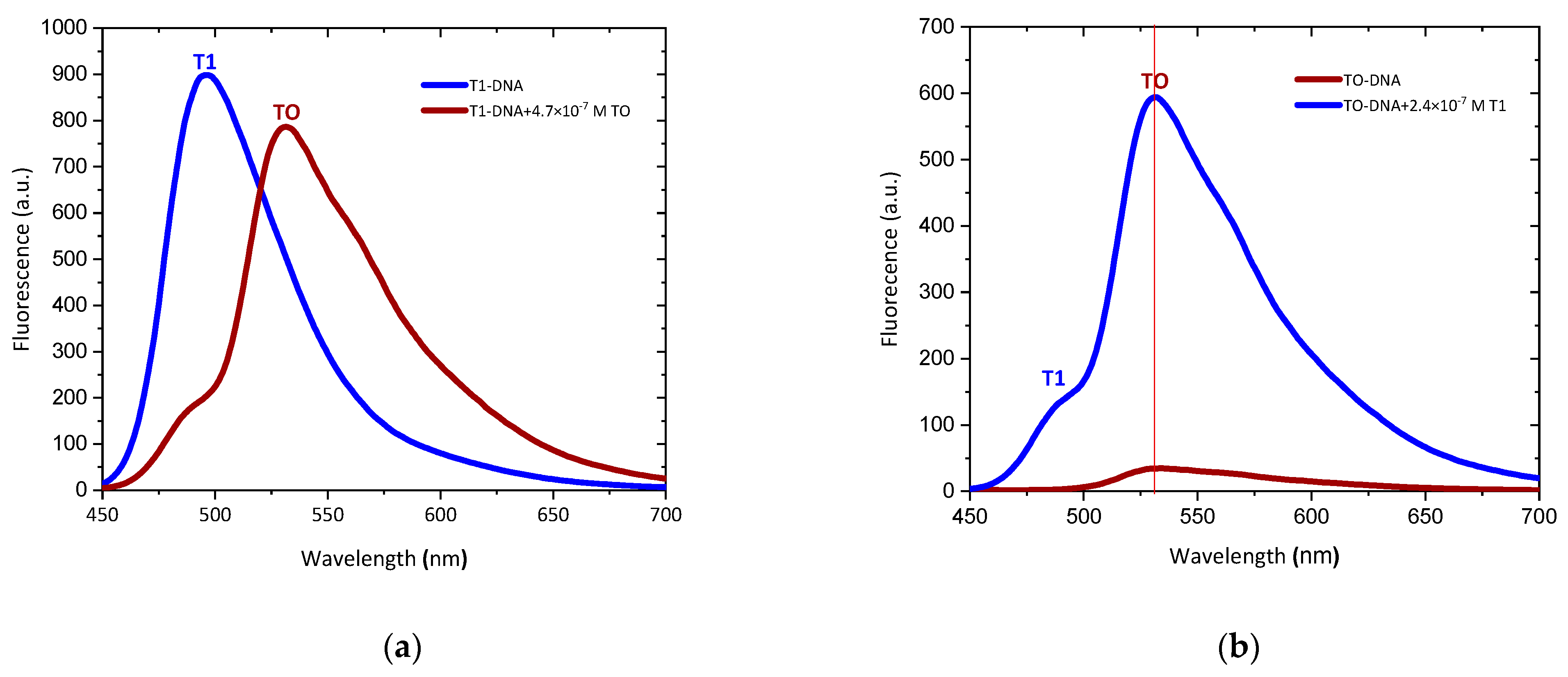
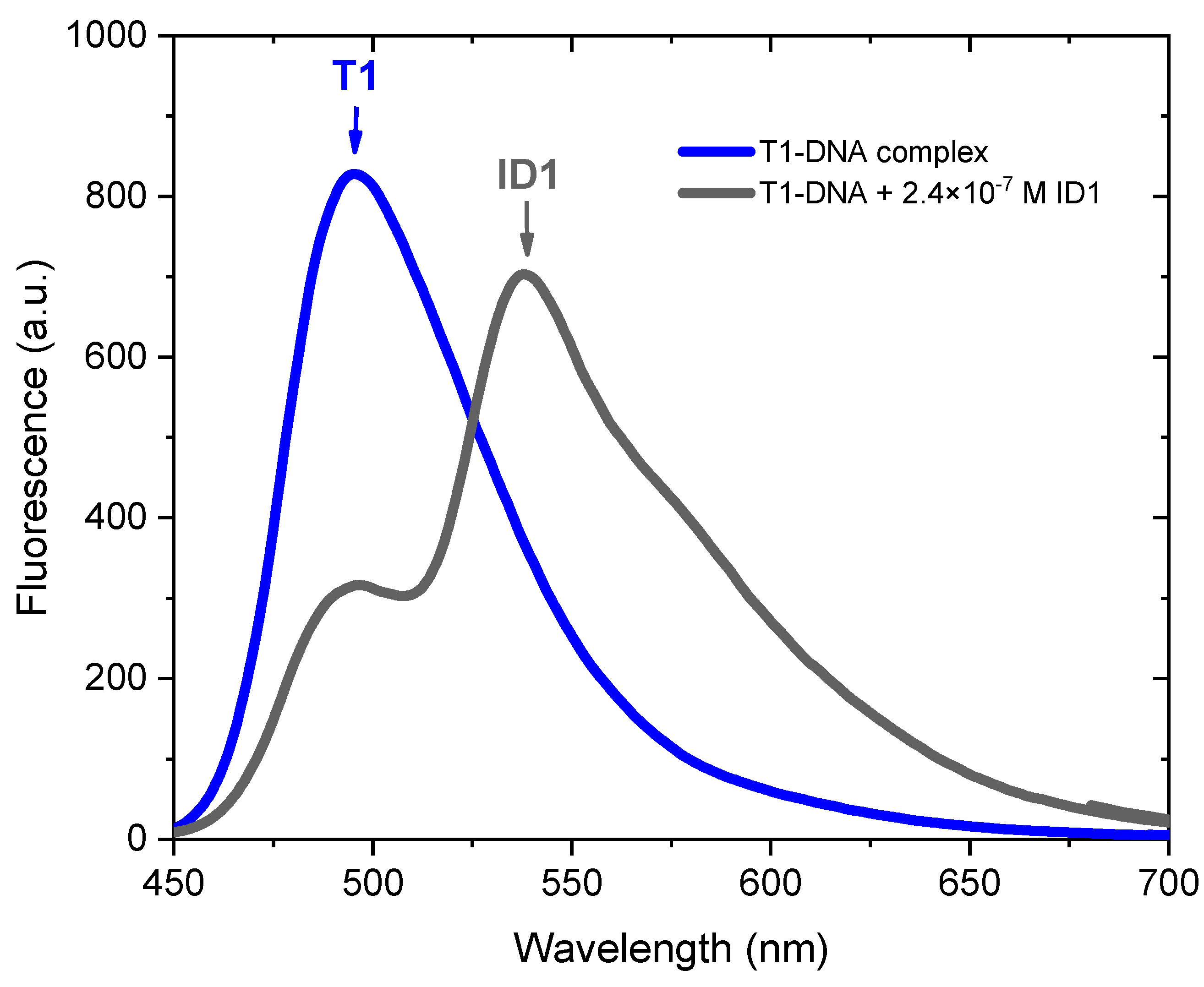
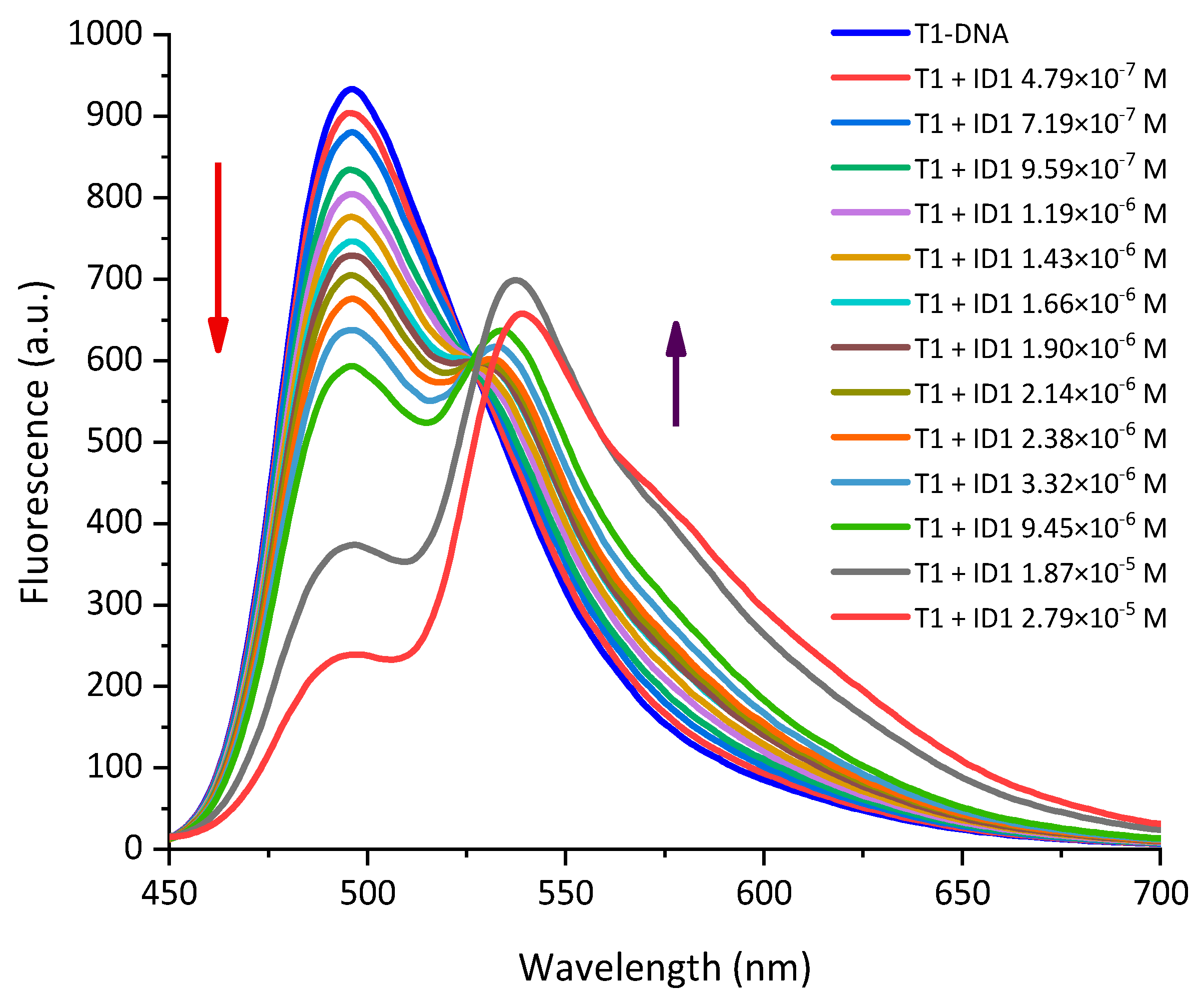
2.2. Photophysical Properties of Dyes and Their Complexes with dsDNA and RNA
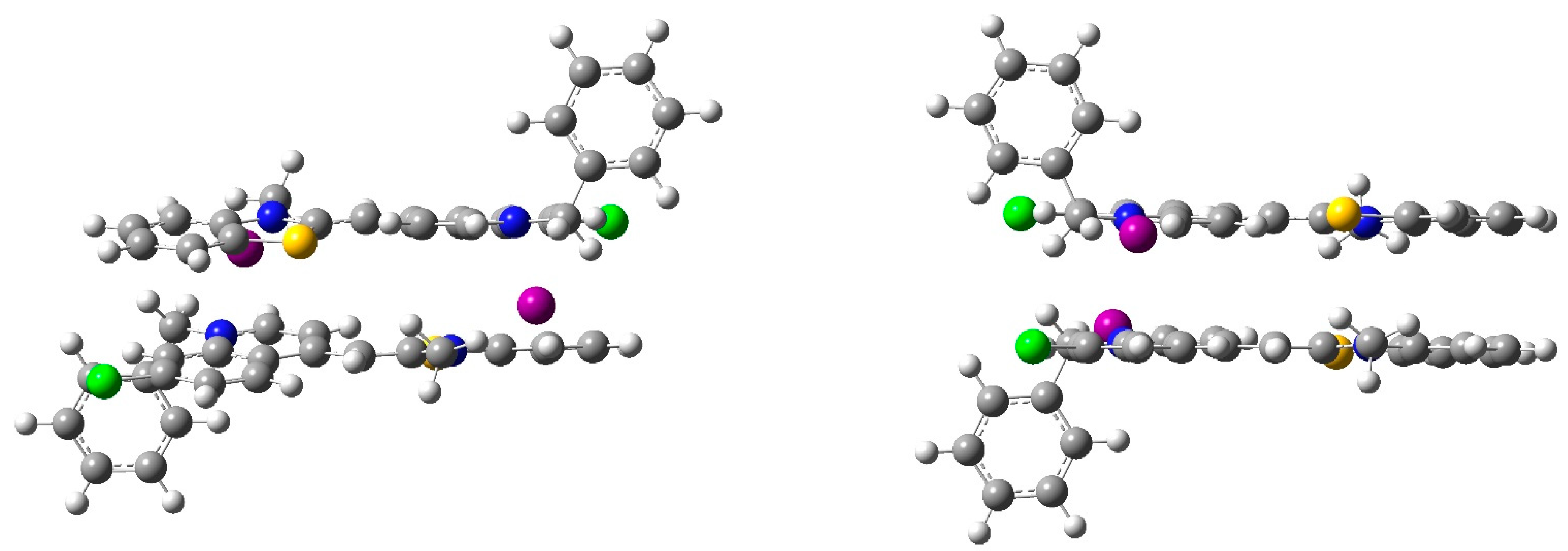
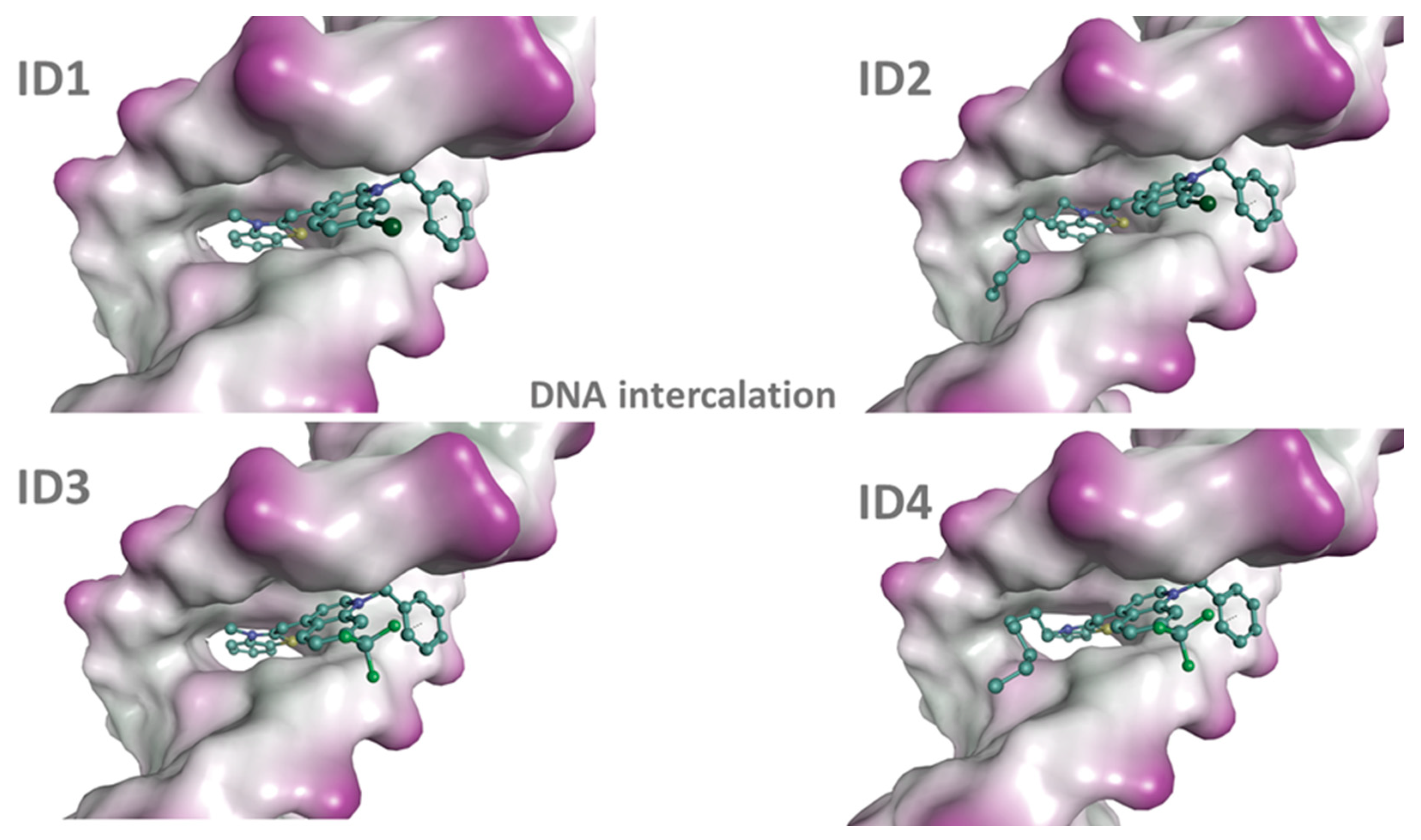
2.3. Molecular Docking
3. Materials and Methods
3.1. Procedure A
3.2. Procedure B
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- A guide to fluorescent probes and labeling technologies. In Molecular Probes, 11th ed.; Thermo-Fisher: Eugene, OR, USA, 2010.
- Anastas, P.T.; Warner, J. Green Chemistry: Theory and Practice; Oxford Univesity Press: Oxford, UK, 1988. [Google Scholar]
- Wardencki, W.; Curylo, J.; Namiesnic, J. Green Chemistry—Current and Future Issues. Pol. J. Environ. Stud. 2005, 14, 389–395. [Google Scholar]
- Ahluwalia, V.K.; Kidwai, M. New Trends in Green Chemistry; Kluwer: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Zonjic, I.; Stojkovic, M.R.; Crnolatac, I.; Paic, A.T.; Psenicnik, S.; Vasilev, A.; Kandinska, M.; Mondeshki, M.; Baluschev, S.; Landfester, K.; et al. Styryl dyes with N-Methylpiperazine and N-Phenylpiperazine Functionality: AT-DNA and G-quadruplex binding ligands and theranostic agents. Bioorg. Chem. 2002, 127, 105999. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, A.A.; Kandinska, M.I.; Stoyanov, S.S.; Yordanova, S.B.; Sucunza, D.; Vaquero, J.J.; Castaño, O.D.; Baluschev, S.; Angelova, S.E. Halogen-containing thiazole orange analogues—New fluorogenic DNA stains. Beilstein J. Org. Chem. 2017, 13, 2902–2914. [Google Scholar] [CrossRef] [PubMed]
- Brooker, L.G.S.; Myers, G.; Williams, W. Color and Constitution. V.1 The Absorption of Unsymmetrical Cyanines. Resonance as a Basis for a Classification of Dyes. J. Am. Chem. Sos. 1942, 64, 199. [Google Scholar] [CrossRef]
- West, W.; Pearce, S. The Dimeric State of Cyanine Dyes. J. Phys. Chem. 1965, 69, 1894–1903. [Google Scholar] [CrossRef]
- Ilieva, S.; Kandinska, M.; Vasilev, A.; Cheshmedzhieva, D. Theoretical Modeling of Absorption and Fluorescent Characteristics of Cyanine Dyes. Photochem 2022, 2, 202–216. [Google Scholar] [CrossRef]
- Evenson, W.E.; Boden, L.M.; Muzikar, K.A.; Oleary, D.J. 1H and 13C NMR assignments for the cyanine dyes SYBR safe and thiazole orange. J. Org. Chem. 2012, 77, 10967–10971. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Kandinska, M.I.; Cheshmedzhieva, D.V.; Kostadinov, A.; Rusinov, K.; Rangelov, M.; Todorova, N.; Ilieva, S.; Ivanov, D.P.; Videva, V.; Lozanov, V.S.; et al. Tricationic asymmetric monomeric monomethine cyanine dyes with chlorine and trifluoromethyl functionality—Fluorogenic nucleic acids probes. J. Mol. Liq. 2021, 342, 117501–117516. [Google Scholar] [CrossRef]
- OriginPro, Version 2023b; OriginLab Corporation: Northampton, MA, USA, 2023.
- Guan, L.; Zhao, J.; Sun, W.; Deng, W.; Wang, L. Meso-Substituted Thiazole Orange for Selective Fluorescence Detection to G-Quadruplex DNA and Molecular Docking Simulation. ACS Omega 2020, 5, 26056–26062. [Google Scholar] [CrossRef] [PubMed]
- Ihmels, H.; Otto, D. Intercalation of organic dye molecules into double-stranded DNA general principles and recent developments. Top. Curr. Chem. 2005, 258, 161–204. [Google Scholar]
- Garbett, N.; Ragazzon, P.; Chaires, J. Circular dichroism to determine binding mode and affinity of ligand–DNA interactions. Nat. Protoc. 2007, 2, 3166. [Google Scholar] [CrossRef] [PubMed]
- Rodger, A.; Taylor, S.; Adlam, G.; Blagbrough, I.S.; Haworth, I.S. Multiple DNA binding modes of anthracene-9-carbonyl-N1-spermine. Bioorg. Med. Chem. 1995, 3, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Rodger, A.; Blagbrough, I.S.; Adlam, G.; Carpenter, M.L. DNA binding of a spermine derivative: Spectroscopic study of anthracene-9-carbonyl-N1-spermine with poly[d(G-C).(d(G-C)] and poly[d(A-T).d(A-T)]. Biopolymers 1994, 34, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Fink, B.E.; Brunette, S.R.; Tse, W.C.; Hedrick, M.P. A Simple, High-Resolution Method for Establishing DNA Binding Affinity and Sequence Selectivity. J. Am. Chem. Soc. 2001, 123, 5878–5891. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Fink, B.E.; Hedrick, M.P. Total Synthesis of Distamycin A and 2640 Analogues: A Solution-Phase Combinatorial Approach to the Discovery of New, Bioactive DNA Binding Agents and Development of a Rapid, High-Throughput Screen for Determining Relative DNA Binding Affinity or DNA Binding Sequence Selectivity. J. Am. Chem. Soc. 2000, 122, 6382–6394. [Google Scholar] [CrossRef]
- Boger, D.L.; Dechantsreiter, M.A.; Ishii, T.; Fink, B.E.; Hedrick, M.P. Assessment of solution-phase positional scanning libraries based on distamycin A for the discovery of new DNA binding agents. Bioorg. Med. Chem. 2000, 8, 2049–2057. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Chen, Z.-F.; Song, X.-Y.; Peng, Y.; Qin, Q.-P.; Liang, H. Synthesis, crystal structure, cytotoxicity and DNA interaction of 5,7-dibromo-8-quinolinolato-lanthanides. Eur. J. Med. Chem. 2013, 59, 168–175. [Google Scholar] [CrossRef]
- Ishkitiev, N.; Miteva, M.; Micheva, M.; Stoyanova, T.; Lozanova, V.V.; Lozanov, V.S.; Mihaylova, Z.; Cheshmedzhieva, D.V.; Kandinska, M.; Rangelov, M.; et al. Aggregation induced nucleic acids recognition by homodimeric asymmetric monomethyne cyanine fluorochromes in mesenchymal stem cells. Int. J. Biol. Macromol. 2023, 250, 126094–126110. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 01; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2021. 2019. Available online: https://www.chemcomp.com/index.htm (accessed on 20 December 2023).
- Naїm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.A.; Wang, M.; Niu, X.; Sun, W.; Jiao, K. Spectrophotometric studies on the interaction of yeast RNA with crystal violet and its analytical application. J. Chil. Chem. Soc. 2008, 53, 1594–1598. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | Buffer | dsDNA λabs | RNA λabs | ||
|---|---|---|---|---|---|
| λabs [nm] ε [L/mol.cm] | λ1 [nm] ε [L/mol.cm] | λ2 [nm] ε [L/mol.cm] | λ1 [nm] ε [L/mol.cm] | λ2 [nm] ε [L/mol.cm] | |
| ID1 | 512 (55 921) 489 (50981) | 486 (31 043) | 518 (47 917) | 486 (35 954) | 515 (45 580) |
| ID2 | 514 (59 359) 496 (44 737) | 492 (44 804) | 526 (53 762) | 492 (45 403) | 523 (41 894) |
| ID3 | 518 (45 088) 496 (35454) | 489 (31 518) | 526 (38 826) | 489 (30 344) | 522 (40 287) |
| ID4 | 520 (43 035) 498 (38 075) | 492 (33 651) | 529 (38 589) | 492 (29 962) | 529 (33 020) |
| Dye | Monomer | Dimer | ||
|---|---|---|---|---|
| λcalc [nm] | λexp [nm] | λcalc [nm] | λexp [nm] | |
| ID1 | 507 | 512 | 475 485 | 489 |
| ID2 | 502 | 514 | 477 487 | 496 |
| ID3 | 513 | 518 | 479 493 | 496 |
| ID4 | 506 | 520 | 482 493 | 498 |
| Dye | Buffer λ(nm) | Dye + dsDNA λ(nm) | Dye + RNA λ(nm) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λex | λem | I0(au) | λex | λem | I(au) | I/I0 | λex | λem | I(au) | I/I0 | |
| ID1 | 486 | 631 | 20.2 | 486 | 542 | 843.1 | 183.9 | 486 | 543 | 853 | 223.2 |
| 542 | 4.6 | ||||||||||
| ID2 | 492 | 641 | 25.4 | 492 | 543.5 | 219.3 | 61 | 492 | 544.5 | 216.7 | 86.8 |
| 544 | 3.6 | ||||||||||
| ID3 | 489 | 632 | 7 | 489 | 550 | 221.1 | 87.1 | 489 | 550 | 152 | 57.9 |
| 550 | 1.3 | ||||||||||
| ID4 | 492 | 626 | 13.5 | 492 | 552 | 22 | 4.7 | 492 | 552 | 28.2 | 7.3 |
| 552 | 4.7 | ||||||||||
| Dye | nm | nm | nm | nm | ||
|---|---|---|---|---|---|---|
| ID1 | 486 | 631 | 542 | 543 | 2.9 × 105 | 1.3 × 105 |
| ID2 | 492 | 641 | 544 | 545 | 5.2 × 105 | 5.3 × 105 |
| ID3 | 489 | 638 | 550 | 550 | 2.7 × 105 | 2.7 × 105 |
| ID4 | 492 | 626 | 552 | 552 | 2.8 × 105 | 2.2 × 105 |
| Dye | dsDNA | dsRNA | ||
|---|---|---|---|---|
| In Groove | Intercalator | In Groove | Intercalator | |
| ID1 | −11.6 | −11.9 | −8.5 | −9.6 |
| ID2 | −13.5 | −13.8 | −8.2 | −10.9 |
| ID3 | −11.7 | −11.9 | −8.4 | −10.1 |
| ID4 | −12.8 | −13.2 | −9.7 | −11.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilieva, S.; Bozova, N.; Rangelov, M.; Todorova, N.; Vasilev, A.; Cheshmedzhieva, D. Asymmetric Monomethine Cyanine Dyes with Hydrophobic Functionalities for Fluorescent Intercalator Displacement Assay. Molecules 2024, 29, 114. https://doi.org/10.3390/molecules29010114
Ilieva S, Bozova N, Rangelov M, Todorova N, Vasilev A, Cheshmedzhieva D. Asymmetric Monomethine Cyanine Dyes with Hydrophobic Functionalities for Fluorescent Intercalator Displacement Assay. Molecules. 2024; 29(1):114. https://doi.org/10.3390/molecules29010114
Chicago/Turabian StyleIlieva, Sonia, Nadezhda Bozova, Miroslav Rangelov, Nadezhda Todorova, Aleksey Vasilev, and Diana Cheshmedzhieva. 2024. "Asymmetric Monomethine Cyanine Dyes with Hydrophobic Functionalities for Fluorescent Intercalator Displacement Assay" Molecules 29, no. 1: 114. https://doi.org/10.3390/molecules29010114
APA StyleIlieva, S., Bozova, N., Rangelov, M., Todorova, N., Vasilev, A., & Cheshmedzhieva, D. (2024). Asymmetric Monomethine Cyanine Dyes with Hydrophobic Functionalities for Fluorescent Intercalator Displacement Assay. Molecules, 29(1), 114. https://doi.org/10.3390/molecules29010114