
Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum–Classical Vibronic Approaches—Application to Coumarin C153 in Methanol
 ,
,  , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
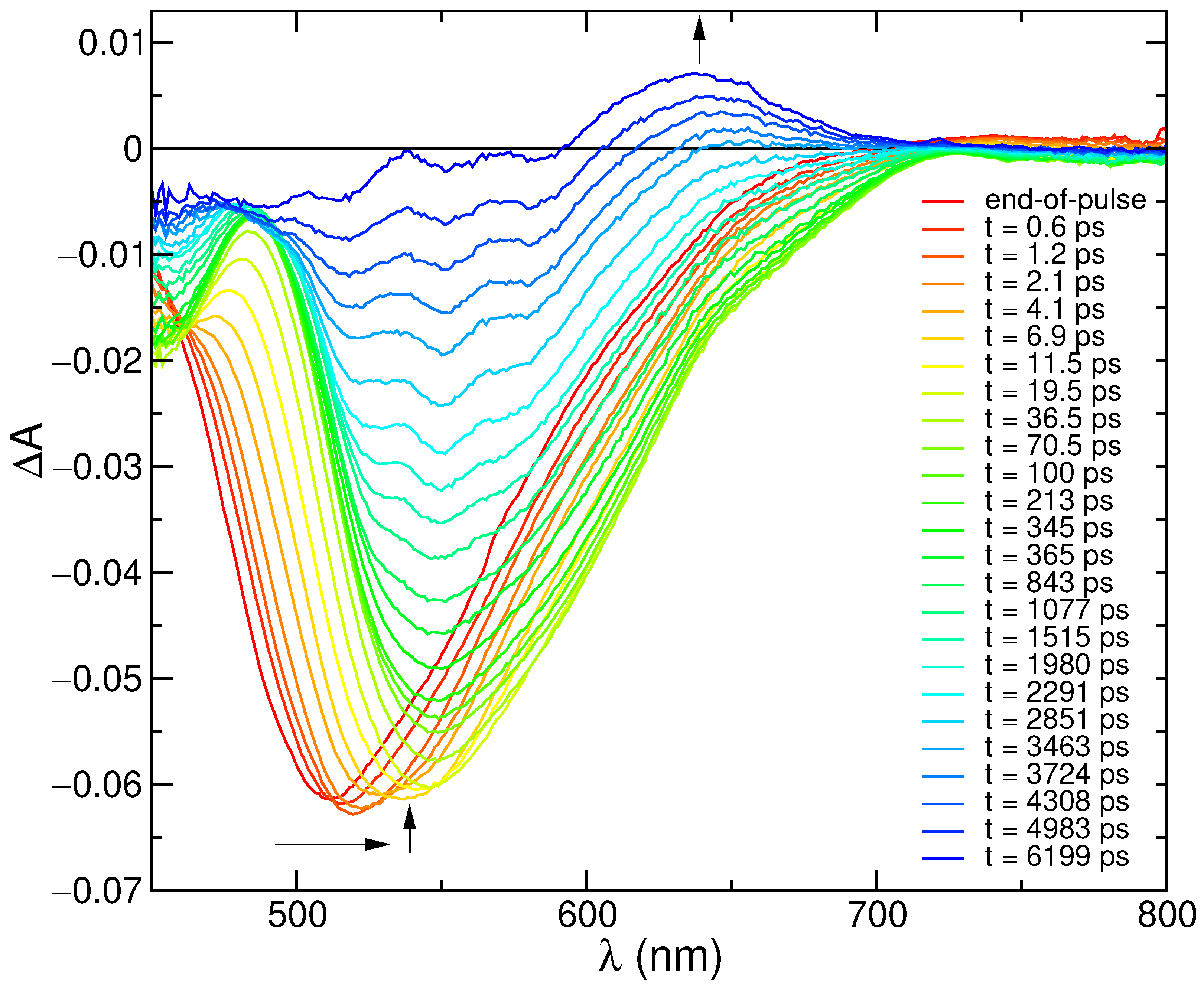
2.1. Experimental Steady-State Absorption and Emission Spectra and Transient Absorption Analysis
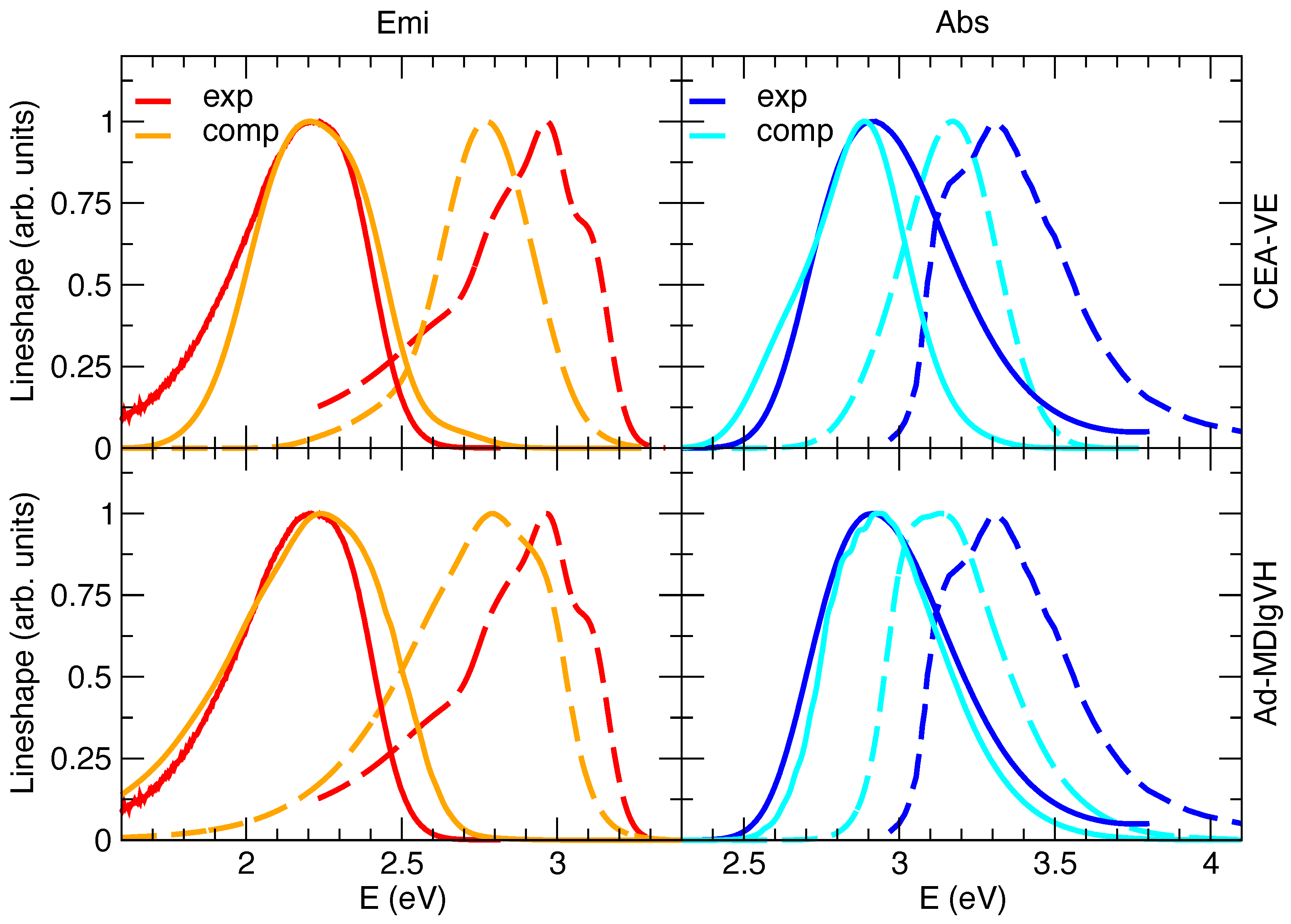
2.2. Simulated Spectra
Steady-State
- I.
- Only the solute is accounted for at QM level, while all methanol molecules within a radius of 18 Å with respect to C153’s geometrical center are treated at MM level through the QMD-FF parameters. Van der Waals terms are only used for water molecules within the first solvation shell while Coulomb terms are considered for all water molecules in the model.
- II.
- The solute and all methanol molecules within a 6 Å radius with respect to C153’s geometrical center are accounted for at QM level, and all methanol molecules found in between 6 Å and 18 Å from C153’s geometrical center are also included in the frame as point charges.
- III.
- The solute and all methanol molecules within a 6 Å radius with respect to C153’s geometrical center are accounted for at QM level, whereas PCM is employed for the rest of the solvent.
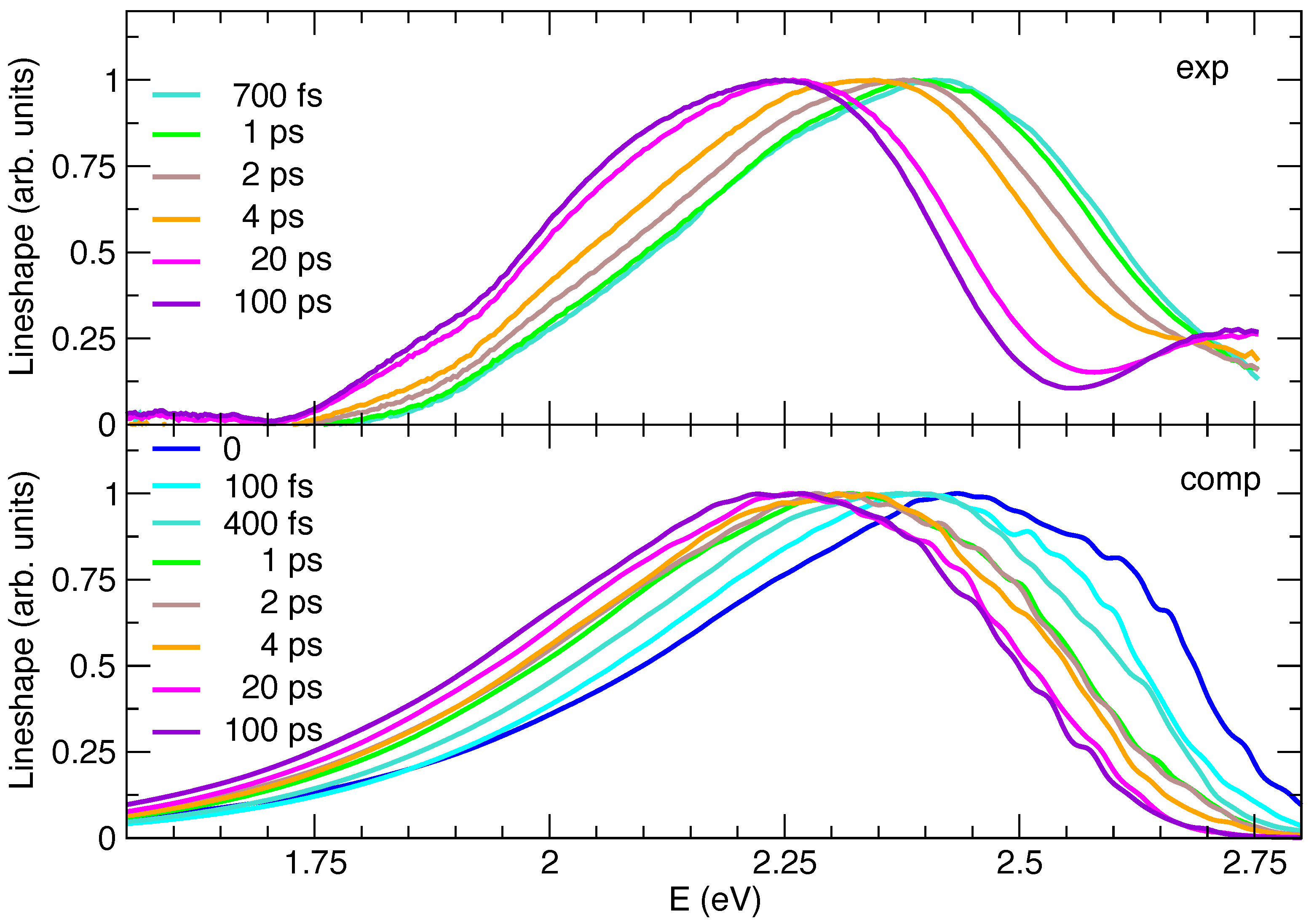
2.3. Simulated Time-Resolved Emission Spectra
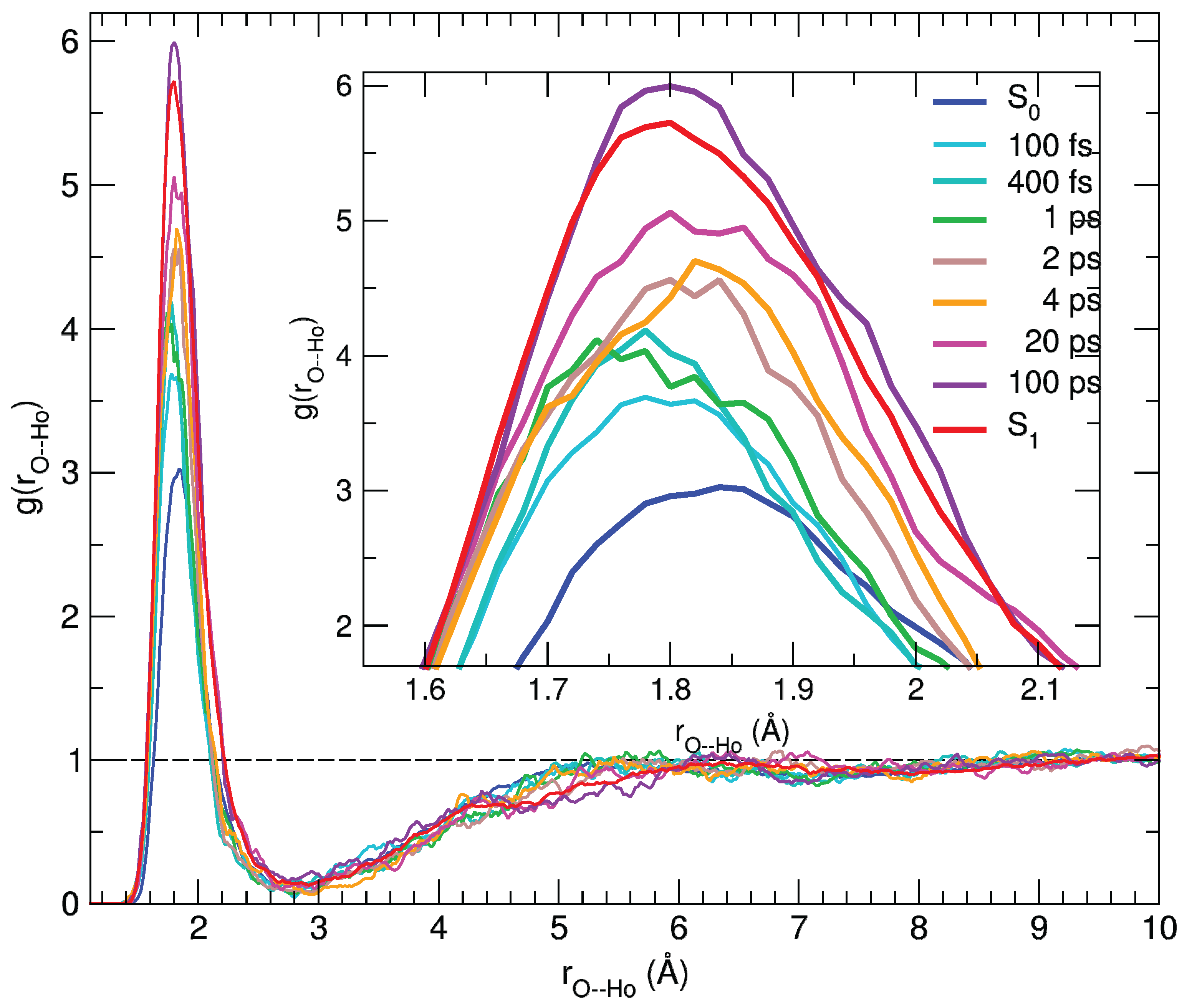
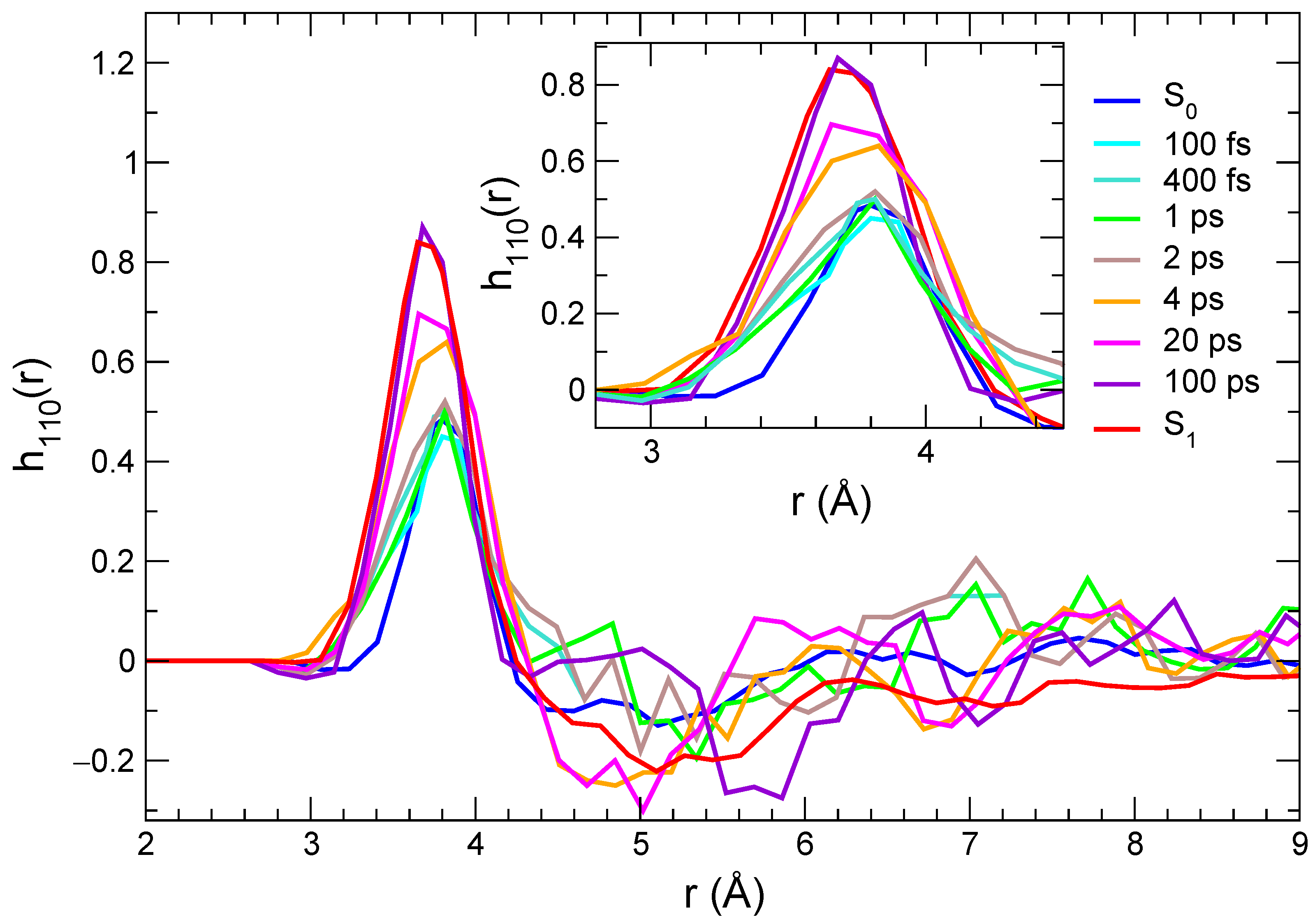
2.4. Solvation Structure during Time-Resolved Emission
3. Materials and Methods
3.1. Computational Protocols
3.1.1. Steady-State Absorption and Emission Spectra
- (i)
- Static vibronic approachThis approach accounts for the effect of intra-molecular vibrations at quantum level and it is based on a single molecular structure, namely the optimized geometry, which is separately considered in its ground or first electronic excited state, for the simulation of absorption or emission spectra, respectively. Since it does not require MD to explicitly simulate the system dynamics, it will be hereafter referred to as “static”. The optimized, state-specific geometries were employed together with their corresponding Hessian matrices to build up harmonic model PESs and compute the vibronically resolved static spectra with the vertical Hessian (VH) model, which accounts for the effects of normal mode mixings (Duschinsky rotation) [92] on the spectra and it is based on a Taylor expansion of both the initial- and final-state PESs at the initial-state geometry up to the quadratic terms. Moreover, the Franck–Condon (FC) approach was also applied, an approximation fully adequate due to the brightness of the electronic transition.
- (ii)
- Classical ensemble average of vertical energies (CEA-VE) approachThis approach is based on the FC classical principle and exploits classical MD to sample the configurational space in the initial state. MD trajectories were first carried out with QMD-FFs, specifically tailored for the target molecule in S and S. Absorption and emission spectra in gas phase and in methanol were thereafter computed over 100 snapshots sampled along equilibrated MD trajectories, obtained in vacuo and on the solvated C153 dye, with either the S or S QMD-FFs. The final spectral shape is retrieved by a classical ensemble average (CEA) of the vertical excitations (VE) computed at each extracted snapshot.
- (iii)
- Ad-MDVH approachThis is a MQC method designed to re-introduce the quantum effect of the relevant molecular vibrations on top of a classical MD sampling of the remaining nuclear DoFs. In practice, for each of the snapshots extracted from either the S or S MD runs, a frame-specific vibronic spectrum is also obtained according to the TD formulation, based on analytical correlation functions [59,87,93,94,95,96,97], implemented in the 3.0 software [98,99]. The latter code exploits QM energies, gradients and Hessians, for the initial and final electronic states, and a partition of the nuclear DoFs into soft and stiff modes [31,99]. At each snapshot , vibronic spectra are computed with the so-called generalized VH (gVH) model, building up reduced-dimensionality harmonic potential energy surface (PES) for the stiff modes (r), obtained by projecting out the soft modes from gradients and Hessians. The vibronic signals specific to each different snapshot are eventually averaged to obtain the final Ad-MDVH spectrum, ), which readswhere in the subscript r indicates that the spectrum is computed including the contributions of the fast stiff coordinates only, and the superscripts Q and that it is computed at quantum vibronic level (Q) specifically for the configuration of the soft modes.
3.1.2. Time-Resolved Emission Spectra
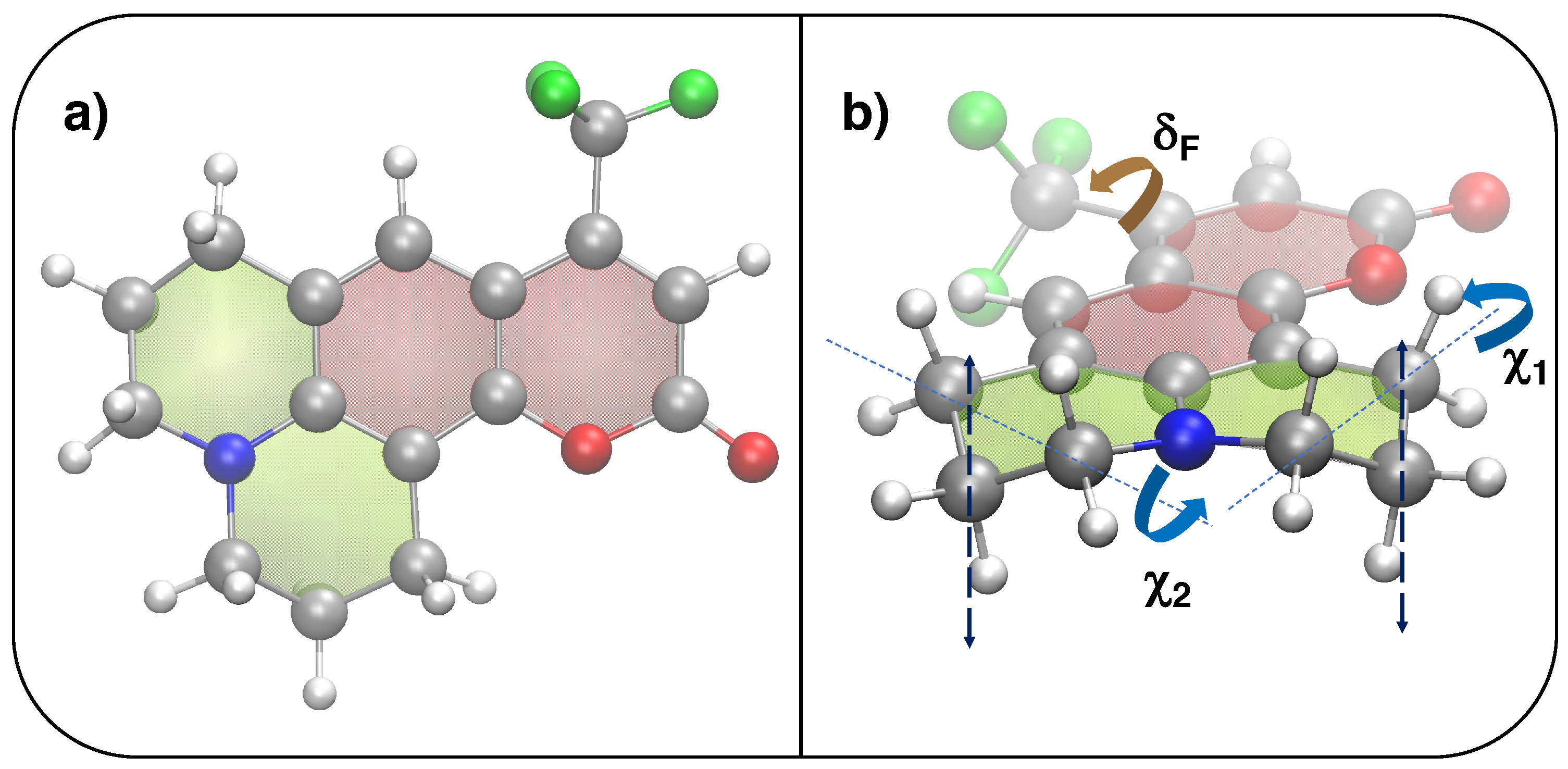
3.2. Computational Details
3.3. Experimental Set Up
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MQC | Mixed quantum–classical |
| MD | Molecular dynamics |
| TR | Time-resolved |
| GSB | Ground-state bleaching |
| SE | Stimulated emission |
| ESA | Excited-state absorption |
| UV-Vis | Ultraviolet–visible |
| TD-DFT | Time-dependent density functional theory |
| QMD-FF | Quantum mechanical derived force field |
| Ad-MDVH | Adiabatic molecular dynamics generalized vertical Hessian |
| DOF | Degree of freedom |
| C153 | Coumarin 153 |
| S | Ground electronic state |
| S | First excited state |
| VH | Vertical Hessian |
| PES | Potential energy surface |
| FC | Franck–Condon |
| CEA-VE | Classical ensemble average of vertical energies |
| AH | Adiabatic Hessian |
| QM | Quantum mechanics |
| MM | Molecular mechanics |
| HWHM | Half width at half maximum |
| PCM | Polarizable continuum model |
References
- He, Y.; Tang, L.; Wu, X.; Hou, X.; Lee, Y.I. Spectroscopy: The best way toward green analytical chemistry? Appl. Spectrosc. Rev. 2007, 42, 119–138. [Google Scholar] [CrossRef]
- Laane, J. Frontiers of Molecular Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Drummen, G.P. Fluorescent probes and fluorescence (microscopy) techniques—illuminating biological and biomedical research. Molecules 2012, 17, 14067–14090. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Drummen, G.P.; Konishi, G.i. Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef]
- Suzuki, S.; Sasaki, S.; Sairi, A.S.; Iwai, R.; Tang, B.Z.; Konishi, G.I. Principles of aggregation-induced emission: Design of deactivation pathways for advanced AIEgens and applications. Angew. Chem. 2020, 132, 9940–9951. [Google Scholar] [CrossRef]
- Yang, M.; Guo, X.; Mou, F.; Guan, J. Lighting up Micro-/Nanorobots with Fluorescence. Chem. Rev. 2023, 123, 3944–3975. [Google Scholar] [CrossRef] [PubMed]
- Oggianu, M.; Figus, C.; Ashoka-Sahadevan, S.; Monni, N.; Marongiu, D.; Saba, M.; Mura, A.; Bongiovanni, G.; Caltagirone, C.; Lippolis, V.; et al. Silicon-based fluorescent platforms for copper (ii) detection in water. RSC Adv. 2021, 11, 15557–15564. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Frei, H.; Park, J.Y. Advancing the frontiers in nanocatalysis, biointerfaces, and renewable energy conversion by innovations of surface techniques. J. Am. Chem. Soc. 2009, 131, 16589–16605. [Google Scholar] [CrossRef]
- Schulze, T.F.; Schmidt, T.W. Photochemical upconversion: Present status and prospects for its application to solar energy conversion. Energy Environ. Sci. 2015, 8, 103–125. [Google Scholar] [CrossRef]
- El-Zohry, A.M.; Diez-Cabanes, V.; Pastore, M.; Ahmed, T.; Zietz, B. Highly Emissive Biological Bilirubin Molecules: Shedding New Light on the Phototherapy Scheme. J. Phys. Chem. B 2021, 125, 9213–9222. [Google Scholar] [CrossRef]
- Pastore, M.; Mosconi, E.; De Angelis, F.; Grätzel, M. A computational investigation of organic dyes for dye-sensitized solar cells: Benchmark, strategies, and open issues. J. Phys. Chem. C 2010, 114, 7205–7212. [Google Scholar] [CrossRef]
- Bertrand, O.; Gohy, J.F. Photo-responsive polymers: Synthesis and applications. Polym. Chem. 2017, 8, 52–73. [Google Scholar] [CrossRef]
- Karuthedath, S.; Gorenflot, J.; Firdaus, Y.; Chaturvedi, N.; De Castro, C.S.; Harrison, G.T.; Khan, J.I.; Markina, A.; Balawi, A.H.; Peña, T.A.D.; et al. Intrinsic efficiency limits in low-bandgap non-fullerene acceptor organic solar cells. Nat. Mater. 2021, 20, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C.; Pastore, M.; Monari, A.; Assfeld, X.; Gros, P.C.; Haacke, S. Ultrafast Spectroscopy of Fe (II) Complexes Designed for Solar-Energy Conversion: Current Status and Open Questions. ChemPhysChem 2022, 23, e202100659. [Google Scholar] [CrossRef] [PubMed]
- El-Zohry, A.M.; Agrawal, S.; De Angelis, F.; Pastore, M.; Zietz, B. Critical Role of Protons for Emission Quenching of Indoline Dyes in Solution and on Semiconductor Surfaces. J. Phys. Chem. C 2020, 124, 21346–21356. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Preat, J.; Wathelet, V.; Fontaine, M.; Perpète, E.A. Thioindigo dyes: Highly accurate visible spectra with TD-DFT. J. Am. Chem. Soc. 2006, 128, 2072–2083. [Google Scholar] [CrossRef]
- Romero, E.; Novoderezhkin, V.I.; van Grondelle, R. Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 2017, 543, 355–365. [Google Scholar] [CrossRef]
- Zaleśny, R.; Alam, M.M.; Day, P.N.; Nguyen, K.A.; Pachter, R.; Lim, C.K.; Prasad, P.N.; Ågren, H. Computational design of two-photon active organic molecules for infrared responsive materials. J. Mater. Chem. C 2020, 8, 9867–9873. [Google Scholar] [CrossRef]
- Berraud-Pache, R.; Neese, F.; Bistoni, G.; Izsak, R. Computational design of near-infrared fluorescent organic dyes using an accurate new wave function approach. J. Phys. Chem. Lett. 2019, 10, 4822–4828. [Google Scholar] [CrossRef]
- Olivier, Y.; Sancho-Garcia, J.C.; Muccioli, L.; D’Avino, G.; Beljonne, D. Computational design of thermally activated delayed fluorescence materials: The challenges ahead. J. Phys. Chem. Lett. 2018, 9, 6149–6163. [Google Scholar] [CrossRef]
- Ashley, D.C.; Jakubikova, E. Ironing out the photochemical and spin-crossover behavior of Fe (II) coordination compounds with computational chemistry. Coord. Chem. Rev. 2017, 337, 97–111. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpete, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT performance for the visible absorption spectra of organic dyes: Conventional versus long-range hybrids. J. Chem. Theor. Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, M.; Aschi, M.; Mazzuca, C.; Palleschi, A.; Amadei, A. Theoretical modeling of UV-Vis absorption and emission spectra in liquid state systems including vibrational and conformational effects: The vertical transition approximation. J. Chem. Phys. 2013, 139, 114102. [Google Scholar] [CrossRef] [PubMed]
- D’Abramo, M.; Aschi, M.; Amadei, A. Theoretical modeling of UV-Vis absorption and emission spectra in liquid state systems including vibrational and conformational effects: Explicit treatment of the vibronic transitions. J. Chem. Phys. 2014, 140, 164104. [Google Scholar] [CrossRef]
- Cerezo, J.; Ferrer, F.J.A.; Prampolini, G.; Santoro, F. Modeling Solvent Broadening on the Vibronic Spectra of a Series of Coumarin Dyes. From Implicit to Explicit Solvent Models. J. Chem. Theory Comput. 2015, 11, 5810–5825. [Google Scholar] [CrossRef]
- Cerezo, J.; Santoro, F.; Prampolini, G. Comparing classical approaches with empirical or quantum-mechanically derived force fields for the simulation electronic lineshapes: Application to coumarin dyes. Theor. Chem. Accounts 2016, 135, 143. [Google Scholar] [CrossRef]
- Zuehlsdorff, T.J.; Isborn, C.M. Combining the ensemble and Franck-Condon approaches for calculating spectral shapes of molecules in solution. J. Chem. Phys. 2018, 148, 024110. [Google Scholar] [CrossRef]
- Zuehlsdorff, T.J.; Isborn, C.M. Modeling absorption spectra of molecules in solution. Int. J. Quantum Chem. 2019, 119, e25719. [Google Scholar] [CrossRef]
- Loco, D.; Cupellini, L. Modeling the absorption lineshape of embedded systems from molecular dynamics: A tutorial review. Int. J. Quant. Chem. 2019, 119, e25726. [Google Scholar] [CrossRef]
- Zuehlsdorff, T.J.; Montoya-Castillo, A.; Napoli, J.A.; Markland, T.E.; Isborn, C.M. Optical spectra in the condensed phase: Capturing anharmonic and vibronic features using dynamic and static approaches. J. Chem. Phys. 2019, 151, 074111. [Google Scholar] [CrossRef]
- Cerezo, J.; Aranda, D.; Avila Ferrer, F.J.; Prampolini, G.; Santoro, F. Adiabatic-Molecular Dynamics Generalized Vertical Hessian Approach: A Mixed Quantum Classical Method to Compute Electronic Spectra of Flexible Molecules in the Condensed Phase. J. Chem. Theory Comput. 2020, 16, 1215–1231. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Zemmouche, M.; Carmona-García, J.; Francés-Monerris, A.; Mulet, P.; Navizet, I.; Roca-Sanjuán, D. Multiconfigurational Quantum Chemistry Determinations of Absorption Cross Sections (σ) in the Gas Phase and Molar Extinction Coefficients (ε) in Aqueous Solution and Air–Water Interface. J. Chem. Theory Comput. 2021, 17, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Segalina, A.; Aranda, D.; Green, J.A.; Cristino, V.; Caramori, S.; Prampolini, G.; Pastore, M.; Santoro, F. How the Interplay among Conformational Disorder, Solvation, Local, and Charge-Transfer Excitations Affects the Absorption Spectrum and Photoinduced Dynamics of Perylene Diimide Dimers: A Molecular Dynamics/Quantum Vibronic Approach. J. Chem. Theory Comput. 2022, 18, 3718–3736. [Google Scholar] [CrossRef]
- Gómez, S.; Giovannini, T.; Cappelli, C. Multiple Facets of Modeling Electronic Absorption Spectra of Systems in Solution. ACS Phys. Chem. Au 2022, in press. [CrossRef] [PubMed]
- Cerezo, J.; García-Iriepa, C.; Santoro, F.; Navizet, I.; Prampolini, G. Unraveling the contributions to the spectral shape of flexible dyes in solution: Insights on the absorption spectrum of an oxyluciferin analogue. Phys. Chem. Chem. Phys. 2023, 25, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Avila Ferrer, F.J.; Cerezo, J.; Stendardo, E.; Improta, R.; Santoro, F. Insights for an Accurate Comparison of Computational Data to Experimental Absorption and Emission Spectra: Beyond the Vertical Transition Approximation. J. Chem. Theory Comput. 2013, 9, 2072–2082. [Google Scholar] [CrossRef]
- Maiuri, M.; Garavelli, M.; Cerullo, G. Ultrafast spectroscopy: State of the art and open challenges. J. Am. Chem. Soc. 2019, 142, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.A.; Cramer, C.J.; Govind, N. Excited State Absorption from Real-Time Time-Dependent Density Functional Theory. J. Chem. Theor. Comput. 2015, 11, 4294–4303. [Google Scholar] [CrossRef]
- Conti, I.; Cerullo, G.; Nenov, A.; Garavelli, M. Ultrafast Spectroscopy of Photoactive Molecular Systems from First Principles: Where We Stand Today and Where We Are Going. J. Am. Chem. Soc. 2020, 142, 16117–16139. [Google Scholar] [CrossRef]
- Mukamel, S. Principles of Nonlinear Optical Spectroscopy; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Domcke, W.; Stock, G. Theory of ultrafast nonadiabatic excited-state processes and their spectroscopic detection in real time. Adv. Chem. Phys. 1997, 100, 1–169. [Google Scholar]
- Gelin, M.F.; Chen, L.; Domcke, W. Equation-of-Motion Methods for the Calculation of Femtosecond Time-Resolved 4-Wave-Mixing and N-Wave-Mixing Signals. Chem. Rev. 2022, 122, 17339–17396. [Google Scholar] [CrossRef]
- Xu, C.; Lin, K.; Hu, D.; Gu, F.L.; Gelin, M.F.; Lan, Z. Ultrafast Internal Conversion Dynamics through the on-the-Fly Simulation of Transient Absorption Pump–Probe Spectra with Different Electronic Structure Methods. J. Phys. Chem. Lett. 2022, 13, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.Y.; Zuehlsdorff, T.J.; Hong, H.; Aguirre, V.P.; Isborn, C.M.; Shi, L. The Influence of Electronic Polarization on Nonlinear Optical Spectroscopy. J. Phys. Chem. B 2021, 125, 12214–12227. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C.; Cina, J.A. What can short-pulse pump-probe spectroscopy tell us about Franck-Condon dynamics? J. Chem. Phys. 1999, 110, 9793–9806. [Google Scholar] [CrossRef]
- Avagliano, D.; Bonfanti, M.; Nenov, A.; Garavelli, M. Automatized protocol and interface to simulate QM/MM time-resolved transient absorption at TD-DFT level with COBRAMM. J. Comput. Chem. 2022, 43, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Segatta, F.; Russo, M.; Nascimento, D.R.; Presti, D.; Rigodanza, F.; Nenov, A.; Bonvicini, A.; Arcioni, A.; Mukamel, S.; Maiuri, M.; et al. In Silico Ultrafast Nonlinear Spectroscopy Meets Experiments: The Case of Perylene Bisimide Dye. J. Chem. Theory Comput. 2021, 17, 7134–7145. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Peng, J.; Chen, L.; Gelin, M.F.; Lan, Z. Spectral Fingerprint of Excited-State Energy Transfer in Dendrimers through Polarization-Sensitive Transient-Absorption Pump–Probe Signals: On-the-Fly Nonadiabatic Dynamics Simulations. J. Phys. Chem. Lett. 2021, 12, 9710–9719. [Google Scholar] [CrossRef]
- Cusati, T.; Granucci, G.; Persico, M. Photodynamics and Time-Resolved Fluorescence of Azobenzene in Solution: A Mixed Quantum-Classical Simulation. J. Am. Chem. Soc. 2011, 133, 5109–5123. [Google Scholar] [CrossRef]
- Borrego-Varillas, R.; Nenov, A.; Kabaciński, P.; Conti, I.; Ganzer, L.; Oriana, A.; Jaiswal, V.K.; Delfino, I.; Weingart, O.; Manzoni, C.; et al. Tracking excited state decay mechanisms of pyrimidine nucleosides in real time. Nat. Commun. 2021, 12, 7285. [Google Scholar] [CrossRef]
- Prampolini, G.; Ingrosso, F.; Segalina, A.; Caramori, S.; Foggi, P.; Pastore, M. Dynamical and Environmental Effects on the Optical Properties of an Heteroleptic Ru(II)–Polypyridine Complex: A Multilevel Approach Combining Accurate Ground and Excited State QM-Derived Force Fields, MD and TD-DFT. J. Chem. Theory Comput. 2019, 15, 529–545. [Google Scholar] [CrossRef]
- Prampolini, G.; Ingrosso, F.; Cerezo, J.; Iagatti, A.; Foggi, P.; Pastore, M. Short- and Long-Range Solvation Effects on the Transient UV–Vis Absorption Spectra of a Ru(II)–Polypyridine Complex Disentangled by Nonequilibrium Molecular Dynamics. J. Phys. Chem. Lett. 2019, 10, 2885–2891. [Google Scholar] [CrossRef]
- Chergui, M. Ultrafast photophysics of transition metal complexes. Acc. Chem. Res. 2015, 48, 801–808. [Google Scholar] [CrossRef] [PubMed]
- McCusker, J.K. Femtosecond Absorption Spectroscopy of Transition Metal Charge-Transfer Complexes. Acc. Chem. Res. 2003, 36, 876–887. [Google Scholar] [CrossRef]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and photophysics of coordination compounds: Ruthenium. In Photochemistry and Photophysics of Coordination Compounds I; Springer: Berlin/Heidelberg, Germany, 2007; pp. 117–214. [Google Scholar]
- Kalyanasundaram, K.; Grätzel, M. Applications of functionalized transition metal complexes in photonic and optoelectronic devices. Coord. Chem. Rev. 1998, 177, 347–414. [Google Scholar] [CrossRef]
- Stennett, E.M.S.; Ciuba, M.A.; Levitus, M. Photophysical processes in single molecule organic fluorescent probes. Chem. Soc. Rev. 2014, 43, 1057–1075. [Google Scholar] [CrossRef]
- Mai, S.; González, L. Unconventional two-step spin relaxation dynamics of [Re(CO)3(im)(phen)]+ in aqueous solution. Chem. Sci. 2019, 10, 10405–10411. [Google Scholar] [CrossRef] [PubMed]
- Avila Ferrer, F.J.; Cerezo, J.; Soto, J.; Improta, R.; Santoro, F. First-principle computation of absorption and fluorescence spectra in solution accounting for vibronic structure, temperature effects and solvent inhomogenous broadening. Comput. Theoret. Chem. 2014, 1040–1041, 328–337. [Google Scholar] [CrossRef]
- Zuehlsdorff, T.J.; Shedge, S.V.; Lu, S.Y.; Hong, H.; Aguirre, V.P.; Shi, L.; Isborn, C.M. Vibronic and environmental effects in simulations of optical spectroscopy. Ann. Rev. Phys. Chem. 2021, 72, 165–188. [Google Scholar] [CrossRef]
- Mennucci, B. Modeling absorption and fluorescence solvatochromism with QM/Classical approaches. Int. J. Quant. Chem. 2015, 115, 1202–1208. [Google Scholar] [CrossRef]
- Pápai, M.; Abedi, M.; Levi, G.; Biasin, E.; Nielsen, M.M.; Møller, K.B. Theoretical evidence of solvent-mediated excited-state dynamics in a functionalized iron sensitizer. J. Phys. Chem. C 2019, 123, 2056–2065. [Google Scholar] [CrossRef]
- Kunnus, K.; Vacher, M.; Harlang, T.C.; Kjær, K.S.; Haldrup, K.; Biasin, E.; van Driel, T.B.; Pápai, M.; Chabera, P.; Liu, Y.; et al. Vibrational wavepacket dynamics in Fe carbene photosensitizer determined with femtosecond X-ray emission and scattering. Nat. Commun. 2020, 11, 634. [Google Scholar] [CrossRef]
- Herbert, J.M. Dielectric continuum methods for quantum chemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1519. [Google Scholar] [CrossRef]
- Biasin, E.; Fox, Z.W.; Andersen, A.; Ledbetter, K.; Kjær, K.S.; Alonso-Mori, R.; Carlstad, J.M.; Chollet, M.; Gaynor, J.D.; Glownia, J.M.; et al. Direct observation of coherent femtosecond solvent reorganization coupled to intramolecular electron transfer. Nat. Chem. 2021, 13, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Aherne, D.; Tran, V.; Schwartz, B.J. Nonlinear, Nonpolar Solvation Dynamics in Water: The Roles of Electrostriction and Solvent Translation in the Breakdown of Linear Response. J. Phys. Chem. B 2000, 104, 5382–5394. [Google Scholar] [CrossRef]
- Cacelli, I.; Prampolini, G. Parametrization and Validation of Intramolecular Force Fields Derived from DFT Calculations. J. Chem. Theory Comput. 2007, 3, 1803–1817. [Google Scholar] [CrossRef]
- Cerezo, J.; Prampolini, G.; Cacelli, I. Developing accurate intramolecular force fields for conjugated systems through explicit coupling terms. Theor. Chem. Accounts 2018, 137, 80. [Google Scholar] [CrossRef]
- Segalina, A.; Cerezo, J.; Prampolini, G.; Santoro, F.; Pastore, M. Accounting for Vibronic Features through a Mixed Quantum-Classical Scheme: Structure, Dynamics, and Absorption Spectra of a Perylene Diimide Dye in Solution. J. Chem. Theor. Comput. 2020, 16, 7061–7077. [Google Scholar] [CrossRef]
- Reynolds, G.; Drexhage, K. New coumarin dyes with rigidized structure for flashlamp-pumped dye lasers. Opt. Commun. 1975, 13, 222–225. [Google Scholar] [CrossRef]
- Schimitschek, E.; Trias, J.; Hammond, P.; Henry, R.; Atkins, R. New laser dyes with blue-green emission. Opt. Commun. 1976, 16, 313–316. [Google Scholar] [CrossRef]
- Horng, M.; Gardecki, J.; Papazyan, A.; Maroncelli, M. Subpicosecond measurements of polar solvation dynamics: Coumarin 153 revisited. J. Phys. Chem. 1995, 99, 17311–17337. [Google Scholar] [CrossRef]
- Maroncelli, M.; Fleming, G.R. Picosecond solvation dynamics of coumarin 153: The importance of molecular aspects of solvation. J. Chem. Phys. 1987, 86, 6221–6239. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V.; Santoro, F. Ab Initio Calculations of Absorption Spectra of Large Molecules in Solution: Coumarin C153. Angew. Chem. Int. Ed. 2007, 46, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Maroncelli, M. On the (uninteresting) dependence of the absorption and emission transition moments of coumarin 153 on solvent. Chem. Phys. Lett. 1998, 282, 197–203. [Google Scholar] [CrossRef]
- Chapman, C.; Fee, R.; Maroncelli, M. Measurements of the solute dependence of solvation dynamics in 1-propanol: The role of specific hydrogen-bonding interactions. J. Phys. Chem. 1995, 99, 4811–4819. [Google Scholar] [CrossRef]
- Kovalenko, S.; Ruthmann, J.; Ernsting, N. Ultrafast strokes shift and excited-state transient absorption of coumarin 153 in solution. Chem. Phys. Lett. 1997, 271, 40–50. [Google Scholar] [CrossRef]
- Dobek, K.; Karolczak, J. The influence of temperature on C153 steady-state absorption and fluorescence kinetics in hydrogen bonding solvents. J. Fluoresc. 2012, 22, 1647–1657. [Google Scholar] [CrossRef]
- Sayed, M.; Maity, D.K.; Pal, H. A comparative photophysical study on the structurally related coumarin 102 and coumarin 153 dyes. J. Photochem. Photobiol. A Chem. 2023, 434, 114265. [Google Scholar] [CrossRef]
- Agmon, N. Dynamic Stokes shift in coumarin: Is it only relaxation? J. Phys. Chem. 1990, 94, 2959–2963. [Google Scholar] [CrossRef]
- Boens, N.; Qin, W.; Basarić, N.; Hofkens, J.; Ameloot, M.; Pouget, J.; Lefèvre, J.P.; Valeur, B.; Gratton, E.; vandeVen, M.; et al. Fluorescence Lifetime Standards for Time and Frequency Domain Fluorescence Spectroscopy. Anal. Chem. 2007, 79, 2137–2149. [Google Scholar] [CrossRef]
- Priyadarsini, K.I.; Mittal, J.P.; Naik, D.B.; Moorthy, P.N. Triplet–triplet and singlet–singlet energy-transfer studies between t-stilbene and 7-aminocoumarin laser dyes. J. Chem. Soc. Faraday Trans. 1991, 87, 269–272. [Google Scholar] [CrossRef]
- Karunakaran, V.; Senyushkina, T.; Saroja, G.; Liebscher, J.; Ernsting, N.P. 2-Amino-7-nitro-fluorenes in Neat and Mixed SolventsOptical Band Shapes and Solvatochromism. J. Phys. Chem. 2007, 111, 10944–10952. [Google Scholar] [CrossRef]
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence: Principles and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Karunakaran, V.; Pérez Lustres, J.L.; Zhao, L.; Ernsting, N.P.; Seitz, O. Large Dynamic Stokes Shift of DNA Intercalation Dye Thiazole Orange has Contribution from a High-Frequency Mode. J. Am. Chem. Soc. 2006, 128, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Loco, D.; Gelfand, N.; Jurinovich, S.; Protti, S.; Mezzetti, A.; Mennucci, B. Polarizable QM/Classical Approaches for the Modeling of Solvation Effects on UV-Vis and Fluorescence Spectra: An Integrated Strategy. J. Phys. Chem. A 2018, 122, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.; Berger, R. Coherent state-based generating function approach for Franck-Condon transitions and beyond. J. Phys. Conf. Ser. 2012, 380, 012019. [Google Scholar] [CrossRef]
- von Cosel, J.; Cerezo, J.; Kern-Michler, D.; Neumann, C.; van Wilderen, L.J.G.W.; Bredenbeck, J.; Santoro, F.; Burghardt, I. Vibrationally resolved electronic spectra including vibrational pre-excitation: Theory and application to VIPER spectroscopy. J. Chem. Phys. 2017, 147, 164116. [Google Scholar] [CrossRef] [PubMed]
- Stell, G.; Patey, G.; Høye, J. Dielectric constants of fluid models: Statistical mechanical theory and its quantitative implementation. Adv. Chem. Phys. 1981, 48, 183–328. [Google Scholar]
- Fonseca, T.; Ladanyi, B.M. Breakdown of linear response for solvation dynamics in methanol. J. Phys. Chem. 1991, 95, 2116–2119. [Google Scholar] [CrossRef]
- Fonseca, T.; Ladanyi, B.M. Solvation dynamics in methanol: Solute and perturbation dependence. J. Mol. Liq. 1994, 60, 1–24. [Google Scholar] [CrossRef]
- Duschinsky, F. On the Interpretation of Electronic Spectra of Polyatomic Molecules. I. The Franck-Condon Principle. Acta Physicochim. URSS 1937, 7, 551–566. [Google Scholar]
- Ianconescu, R.; Pollak, E. Photoinduced Cooling of Polyatomic Molecules in an Electronically Excited State in the Presence of Dushinskii Rotations. J. Phys. Chem. A 2004, 108, 7778–7784. [Google Scholar] [CrossRef]
- Baiardi, A.; Bloino, J.; Barone, V. General Time Dependent Approach to Vibronic Spectroscopy Including Franck-Condon, Herzberg-Teller, and Duschinsky Effects. J. Chem. Theory Comput. 2013, 9, 4097–4115. [Google Scholar] [CrossRef]
- Borrelli, R.; Capobianco, A.; Peluso, A. Generating Function Approach to the Calculation of Spectral Band Shapes of Free-Base Chlorin Including Duschinsky and Herzberg-Teller Effects. J. Phys. Chem. A 2012, 116, 9934–9940. [Google Scholar] [CrossRef] [PubMed]
- Tatchen, J.; Pollak, E. Ab initio spectroscopy and photoinduced cooling of the trans-stilbene molecule. J. Chem. Phys. 2008, 128, 164303. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Niu, Y.; Deng, C.; Shuai, Z. Vibration correlation function formalism of radiative and non-radiative rates for complex molecules. Chem. Phys. 2010, 370, 215–222. [Google Scholar] [CrossRef]
- Cerezo, J.; Santoro, F. FCclasses3: Vibrationally-resolved spectra simulated at the edge of the harmonic approximation. J. Comput. Chem. 2023, 44, 626–643. [Google Scholar] [CrossRef]
- Santoro, F.; Cerezo, J. FCclasses3, a Code for Vibronic Calculations. 2022. Available online: http://www.iccom.cnr.it/en/fcclasses (accessed on 1 February 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Avila Ferrer, F.J.; Santoro, F. Comparison of vertical and adiabatic harmonic approaches for the calculation of the vibrational structure of electronic spectra. Phys. Chem. Chem. Phys. 2012, 14, 13549–13563. [Google Scholar] [CrossRef]
- Santoro, F. FCclasses, a Fortran 77 Code for Vibronic Calculations. Available online: http://www.pi.iccom.cnr.it/fcclasses (accessed on 1 November 2019).
- Cacelli, I.; Cerezo, J.; De Mitri, N.; Prampolini, G. Joyce2.10, a Fortran 77 Code for Intra-Molecular Force Field Parameterization. Available online: http://www.pi.iccom.cnr.it/joyce (accessed on 1 July 2020).
- Barone, V.; Cacelli, I.; De Mitri, N.; Licari, D.; Monti, S.; Prampolini, G. Joyce and Ulysses: Integrated and User-Friendly Tools for the Parameterization of Intramolecular Force Fields from Quantum Mechanical Data. Phys. Chem. Chem. Phys. 2013, 15, 3736–3751. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genge, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Prot. 2013, 8, 1535–1550. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 7863, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential Energy Functions for Atomic-Level Simulations of Water and Organic and Biomolecular Systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- M’uhlpfordt, A.; Schanz, R.; Ernsting, N.P.; Farztdinov, V.; Grimme, S. Coumarin 153 in the gas phase: Optical spectra and quantum chemical calculations. Phys. Chem. Chem. Phys. 1999, 1, 3209–3218. [Google Scholar] [CrossRef]
- Lax, M. The Franck-Condon Principle and Its Application to Crystals. J. Chem. Phys. 1952, 20, 1752–1760. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorption a (298 K) | |||
| (nm) | (10 M cm) | ||
| 221, 266, 424 | 2.14, 0.77, 1.76 | ||
| Emission (298 K) | |||
| c (nm) | d (ns) | e (%) | |
| 537 | 4.0 | 42.2 | |
| Emission (77 K) | |||
| a,c (nm) | b,c (nm) | a,d (ns) | b,d (s) |
| 497 | 603, 633 | 5.8 | 0.9 (52%), 7.4 (48%) |
| Ad-MD|gVH | Experiments | ||||
|---|---|---|---|---|---|
| Time (ps) | E (eV) | E (eV) | Time (ps) | E (eV) | E (eV) |
| 0 | 2.43 | 0.00 | |||
| 0.1 | 2.39 | −0.04 | |||
| 0.4 | 2.37 | −0.06 | 0.7 | 2.41 | 0.00 |
| 1 | 2.32 | −0.11 | 1 | 2.39 | −0.02 |
| 2 | 2.28 | −0.15 | 2 | 2.38 | −0.03 |
| 4 | 2.31 | −0.12 | 4 | 2.34 | −0.07 |
| 20 | 2.26 | −0.17 | 20 | 2.26 | −0.15 |
| 100 | 2.22 | −0.21 | 100 | 2.24 | −0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerezo, J.; Gao, S.; Armaroli, N.; Ingrosso, F.; Prampolini, G.; Santoro, F.; Ventura, B.; Pastore, M. Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum–Classical Vibronic Approaches—Application to Coumarin C153 in Methanol. Molecules 2023, 28, 3910. https://doi.org/10.3390/molecules28093910
Cerezo J, Gao S, Armaroli N, Ingrosso F, Prampolini G, Santoro F, Ventura B, Pastore M. Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum–Classical Vibronic Approaches—Application to Coumarin C153 in Methanol. Molecules. 2023; 28(9):3910. https://doi.org/10.3390/molecules28093910
Chicago/Turabian StyleCerezo, Javier, Sheng Gao, Nicola Armaroli, Francesca Ingrosso, Giacomo Prampolini, Fabrizio Santoro, Barbara Ventura, and Mariachiara Pastore. 2023. "Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum–Classical Vibronic Approaches—Application to Coumarin C153 in Methanol" Molecules 28, no. 9: 3910. https://doi.org/10.3390/molecules28093910
APA StyleCerezo, J., Gao, S., Armaroli, N., Ingrosso, F., Prampolini, G., Santoro, F., Ventura, B., & Pastore, M. (2023). Non-Phenomenological Description of the Time-Resolved Emission in Solution with Quantum–Classical Vibronic Approaches—Application to Coumarin C153 in Methanol. Molecules, 28(9), 3910. https://doi.org/10.3390/molecules28093910