

Structural, Electric and Dynamic Properties of (Pyrrolidinium)3[Bi2I9] and (Pyrrolidinium)3[Sb2I9]: New Lead-Free, Organic–Inorganic Hybrids with Narrow Band Gaps

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
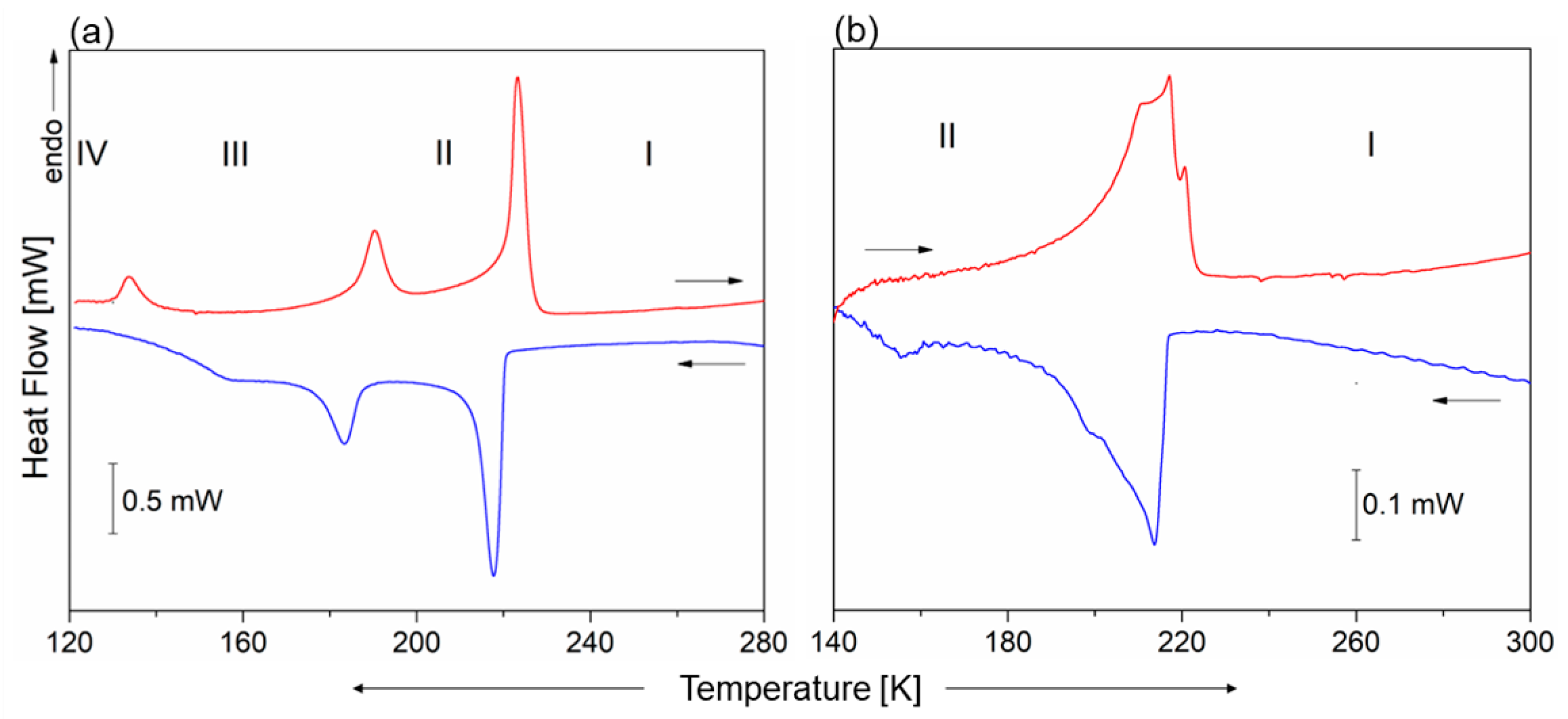
2.1. Thermal Properties of 1 and 2
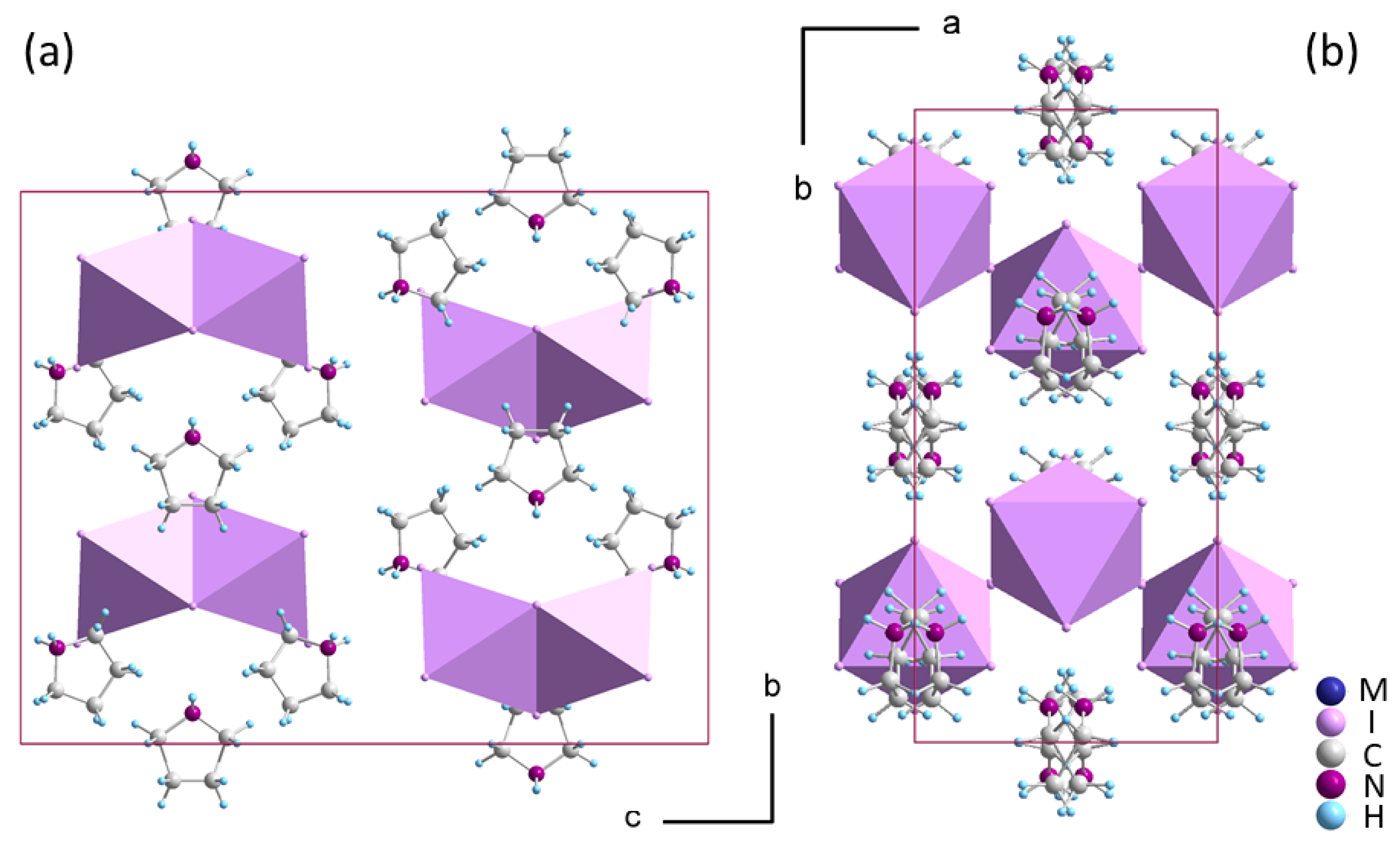
2.2. Crystal Structure of Phase I
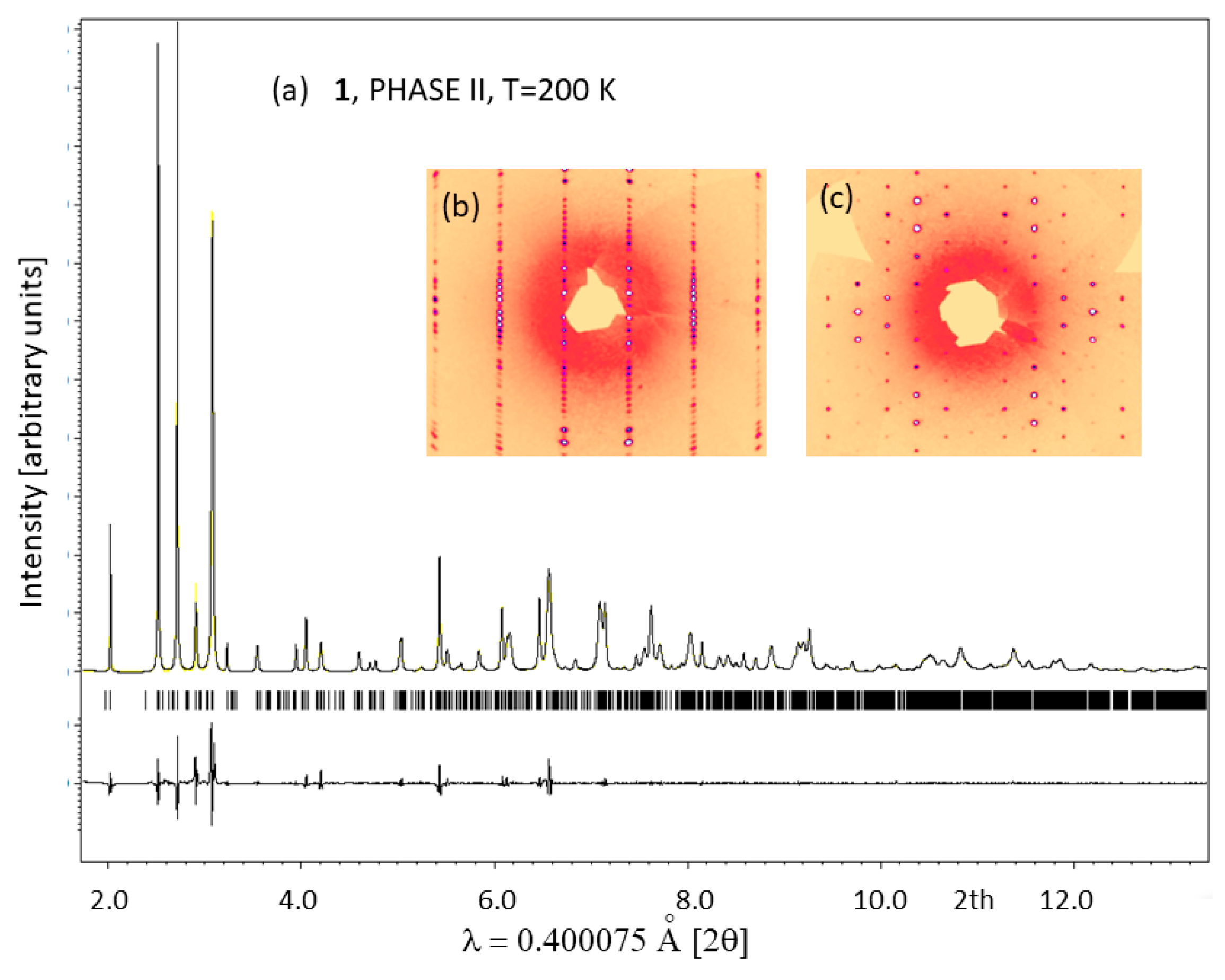
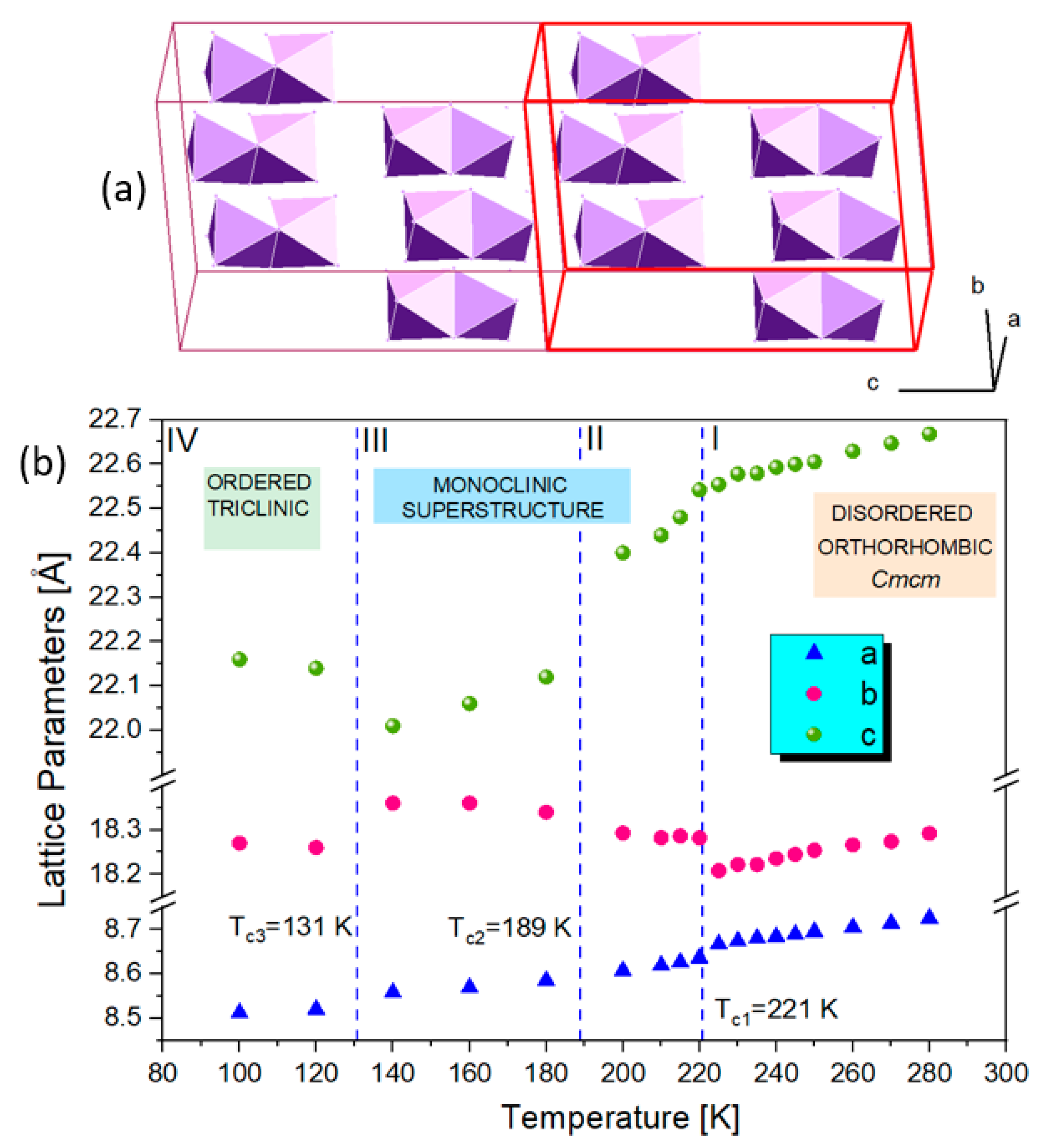
2.3. Low-Temperature Phases in 1
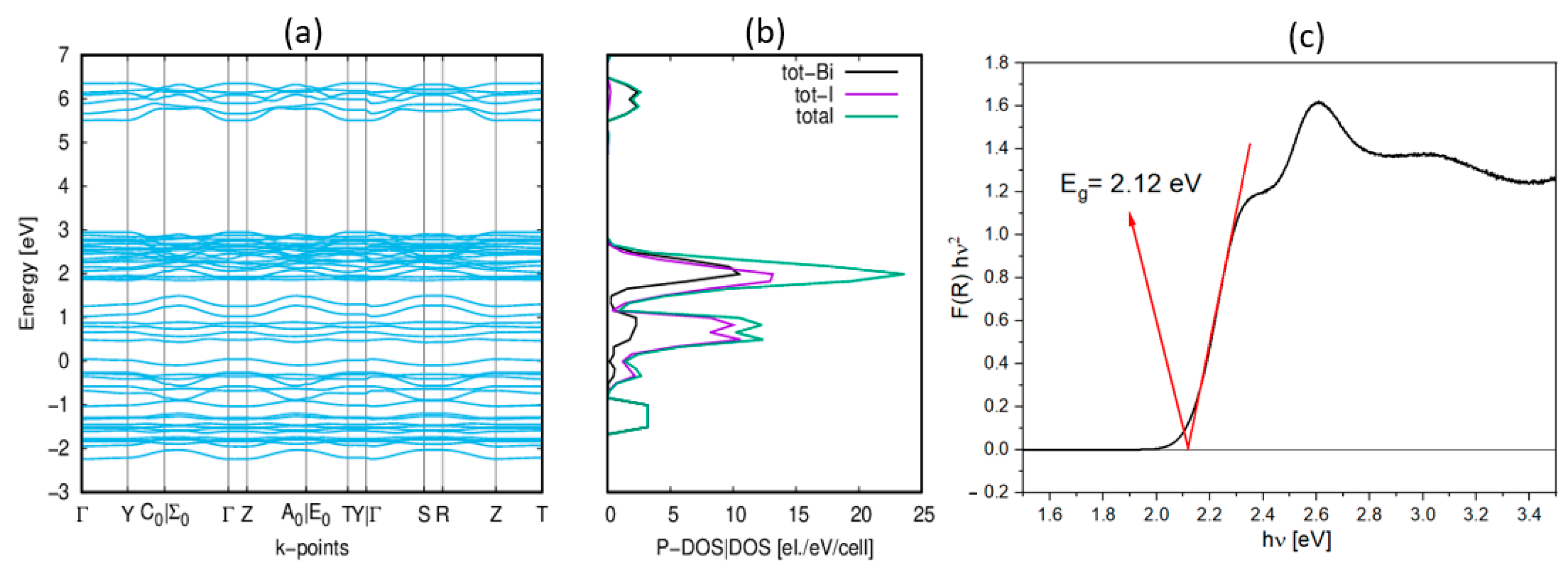
2.4. Electronic Properties
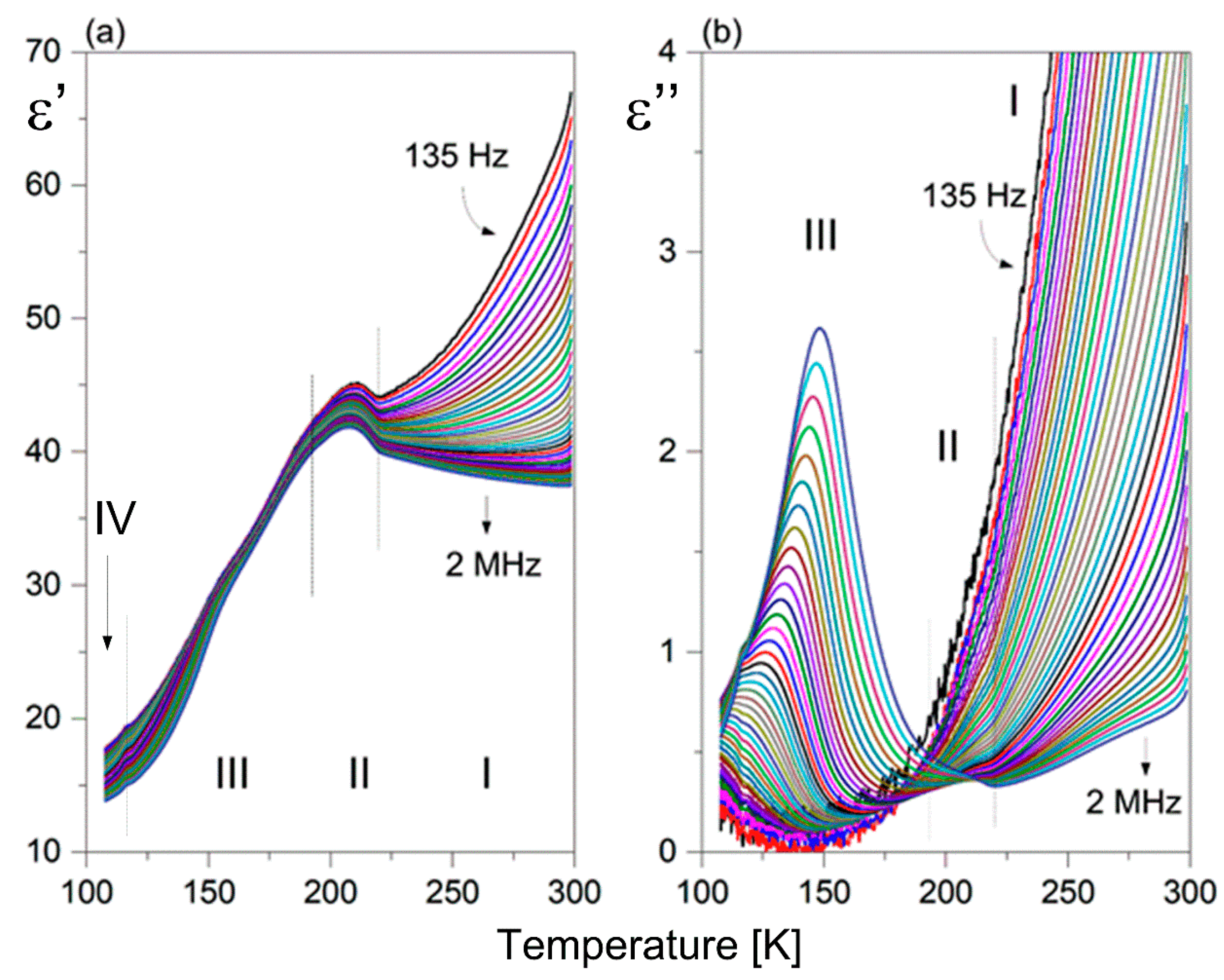
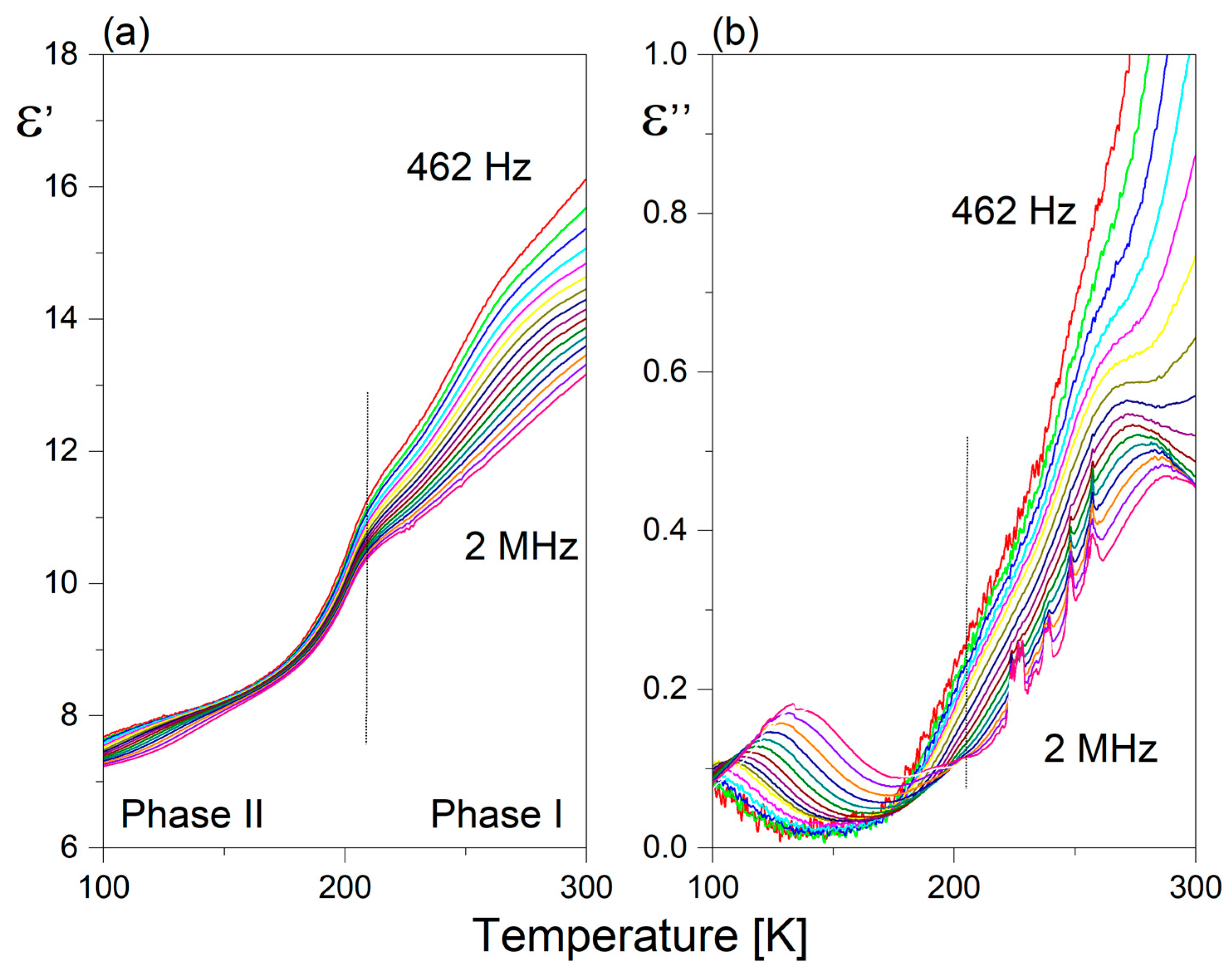
2.5. Dielectric Response
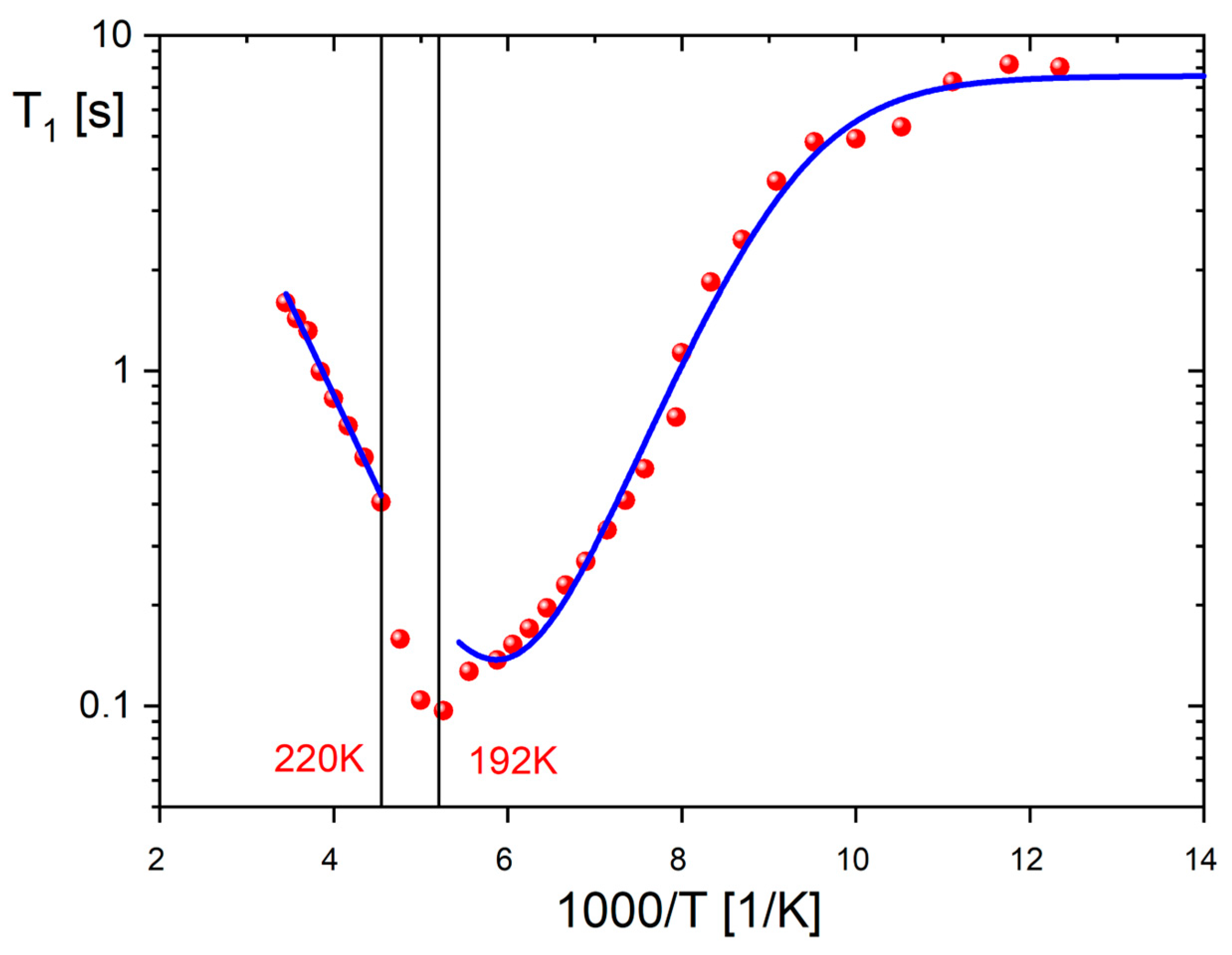
2.6. Cations Dynamics in 1 from Proton Magnetic Resonance (1H NMR)
- (i).
- Small-angle rotations of the pentagonal ring;
- (ii).
- Changes of the pyrrolidinium ring conformation (twisted/envelope conformation);
- (iii).
- Rotation about the pseudo-C5 axis perpendicular to the pyrrolidine ring;
- (iv).
- Isotropic rotation and possibly cationic self-diffusion/rotational diffusion at the highest temperatures.
3. Experimental Section
3.1. Synthesis
3.2. Single-Crystal X-ray Diffraction
3.3. Synchrotron Powder Diffraction
3.4. Optical Measurements
3.5. Thermal Analysis
3.6. Electrical Measurements
3.7. NMR
3.8. Calculation Details
4. Conclusions

Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Snaith, H.J. Present Status and Future Prospects of Perovskite Photovoltaics. Nat. Mater. 2018, 17, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Stranks, S.D.; Eperon, G.E.; Grancini, G.; Menelaou, C.; Alcocer, M.J.P.; Leijtens, T.; Herz, L.M.; Petrozza, A.; Snaith, H.J. Electron-Hole Diffusion Lengths Exceeding 1um in an Organometal Trihalide Perovskite Absorber. Science 2013, 342, 341–344. [Google Scholar] [CrossRef] [PubMed]
- D’Innocenzo, V.; Grancini, G.; Alcocer, M.J.P.; Srimath Kandada, A.R.; Stranks, S.D.; Lee, M.M.; Lanzani, G.; Snaith, H.J.; Petrozza, A. Excitons v. Free Charges in Organo-Lead Trihalide Perovskites. Nat. Commun. 2014, 5, 3586. [Google Scholar] [CrossRef]
- Shi, Z.; Guo, J.; Chen, Y.; Li, Q.; Pan, Y.; Zhang, H.; Xia, Y.; Huang, W. Lead-Free Organic–Inorganic Hybrid Perovskites for Photovoltaic Applications: Recent Advances and Perspectives. Adv. Mater. 2017, 29, 1605005. [Google Scholar] [CrossRef]
- Xiao, Z.; Song, Z.; Yan, Y. From Lead Halide Perovskites to Lead-Free Metal Halide Perovskites and Perovskite Derivatives. Adv. Mater. 2019, 31, 1803792. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Pal, A.J. Substitution in (CH3NH3)3Sb2I9:Toward Low-Band-Gap Defect-Ordered Hybrid Perovskite Solar Cells. ACS Tin(IV) Appl. Mater. Interfaces 2018, 10, 35194. [Google Scholar] [CrossRef]
- Jain, S.M.; Edvinsson, T.; Durrant, J.R. Green Fabrication of Stable Lead-Free Bismuth Based Perovskite Solar Cells Using a Non-Toxic Solvent. Commun. Chem. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Sanders, S.; Stümmler, D.; Pfeiffer, P.; Ackermann, N.; Simkus, G.; Heuken, M.; Baumann, K.; Vescan, A.; Kalisch, H. Chemical Vapor Deposition of Organic-Inorganic Bismuth-Based Perovskite Films for Solar Cell Application. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Hoye, R.L.Z.; Brandt, R.E.; Osherov, A.; Stevanovic, V.; Stranks, S.D.; Wilson, M.W.B.; Kim, H.; Akey, A.J.; Perkins, J.D.; Kurchin, R.C.; et al. Methylammonium Bismuth Iodide as a Lead-Free, Stable Hybrid Organic-Inorganic Solar Absorber. Chem.-A Eur. J. 2016, 22, 2605–2610. [Google Scholar] [CrossRef]
- Park, B.-W.; Philippe, B.; Zhang, X.; Rensmo, H.; Boschloo, G.; Johansson, E.M.J. Bismuth Based Hybrid Perovskites A3Bi2I9 (A: Methylammonium or Cesium) for Solar Cell Application. Adv. Mater. 2015, 27, 6806–6813. [Google Scholar] [CrossRef]
- Ganose, A.M.; Savory, C.N.; Scanlon, D.O. Beyond Methylammonium Lead Iodide: Prospects for the Emergent Field of Ns2 Containing Solar Absorbers. Chem. Commun. 2017, 53, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Xia, X.; Wang, Z.; Huang, Z.; Lei, B.; Gao, Y. High-Quality (CH3NH3)3Bi2I9 Film-Based Solar Cells: Pushing Efficiency up to 1.64%. J. Phys. Chem. Lett. 2017, 8, 4300–4307. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chu, K.B.; Zhang, L.; Ding, X.; Sun, J.; Liu, J.Z.; Song, J.L.; Zheng, C.; Deng, J. Lead-Free Molecular Ferroelectric [N,N-Dimethylimidazole]3Bi2I9 with Narrow Bandgap. Mater. Des. 2020, 193, 108868. [Google Scholar] [CrossRef]
- Szklarz, P.; Gagor, A.; Jakubas, R.; Zieliński, P.; Piecha-Bisiorek, A.; Cichos, J.; Karbowiak, M.; Bator, G.; Ciżman, A. Lead-Free Hybrid Ferroelectric Material Based on Formamidine: [NH2CHNH2]3Bi2I9. J. Mater. Chem. C Mater. 2019, 7, 3003–3014. [Google Scholar] [CrossRef]
- Jakubas, R.; Rok, M.; Mencel, K.; Bator, G.; Piecha-Bisiorek, A. Correlation between Crystal Structures and Polar (Ferroelectric) Properties of Hybrids of Haloantimonates(III) and Halobismuthates(III). Inorg. Chem. Front. 2020, 7, 2128. [Google Scholar] [CrossRef]
- Liang, L.; Gao, P. Lead-Free Hybrid Perovskite Absorbers for Viable Application: Can We Eat the Cake and Have It Too? Adv. Sci. 2018, 5, 1700331. [Google Scholar] [CrossRef]
- Adonin, S.A.; Sokolov, M.N.; Fedin, V.P. Polynuclear Halide Complexes of Bi(III): From Structural Diversity to the New Properties. Coord. Chem. Rev. 2016, 312, 1–21. [Google Scholar] [CrossRef]
- Adonin, S.A.; Usoltsev, A.N.; Novikov, A.S.; Kolesov, B.A.; Fedin, V.P.; Sokolov, M.N. One-and Two-Dimensional Iodine-Rich Iodobismuthate(III) Complexes: Structure, Optical Properties, and Features of Halogen Bonding in the Solid State. Inorg. Chem. 2020, 59, 3290–3296. [Google Scholar] [CrossRef]
- Wȩcławik, M.; Gągor, A.; Jakubas, R.; Piecha-Bisiorek, A.; Medycki, W.; Baran, J.; Zieliński, P.; Gałązka, M. Structure-Property Relationships in Hybrid (C3H5N2)3[Sb2I9] and (C3H5N2)3[Bi2I9] Isomorphs. Inorg. Chem. Front. 2016, 3, 1306–1316. [Google Scholar] [CrossRef]
- Piecha-Bisiorek, A.; Jakubas, R.; Medycki, W.; Florek-Wojciechowska, M.; Wojciechowski, M.; Kruk, D. Dynamics of Ferroelectric Bis(Imidazolium) Pentachloroantimonate(III) by Means of Nuclear Magnetic Resonance 1 H Relaxometry and Dielectric Spectroscopy. J. Chem. Phys. Chem. A 2014, 118, 3564–3571. [Google Scholar] [CrossRef]
- Szklarz, P.; Pietraszko, A.; Jakubas, R.; Bator, G.; Zieliński, P.; Gałazka, M. Structure, Phase Transitions and Molecular Dynamics of [C(NH 2)3]3[M2I9], M = Sb, Bi. J. Phys. Condens. Matter 2008, 20, 1–12. [Google Scholar] [CrossRef]
- Zaleski, J.; Jakubas, R.; Sobczyk, L.; Mroz, J. Properties and Structural Phase Transitions of (CH3NH3)3Sb2I9. Ferroelectrics 1990, 103, 83–90. [Google Scholar] [CrossRef]
- Aizu, K. Possible Species of “Ferroelastic” Crystals and of Simultaneusly Ferroelectric and Ferroelastic Crystals. J. Phys. Soc. Jpn. 1969, 27, 387–396. [Google Scholar] [CrossRef]
- Izumi, K.M.F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar]
- Szklarz, P.; Jakubas, R.; Medycki, W.; Gągor, A.; Cichos, J.; Karbowiak, M.; Bator, G. (C3N2H5)3Sb2I9and (C3N2H5)3Bi2I9: Ferroelastic Lead-Free Hybrid Perovskite-like Materials as Potential Semiconducting Absorbers. Dalton Trans. 2022, 51, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, P.; Munk, F. Ein Beitrag Zur Optik Der Farbanstriche. Z. Techn. Phys. 1931, 12, 593–601. [Google Scholar]
- Baranov, A.I.; Kabanov, O.A.; Merinov, B.V.; Shuvalov, L.A.; Dolbinina, V.V. Glass-like Dielectric Relaxation in Cs5H3(SO4)4H2O Crystal. Ferroelectrics 1992, 127, 257–262. [Google Scholar] [CrossRef]
- Shin, H.K. Glass-like Dielectric Relaxation in C5NH6ClO4. Sol. State Comm. 2007, 143, 313–316. [Google Scholar] [CrossRef]
- Ono, H.; Ishimaru, S.; Ikeda, R.; Ishida, H. 1H, 2H, 19F, 31P, and 35Cl NMR Studies on Molecular Motions in Ionic Plastic Phases of Pyrrolidinium Perchlorate and Hexafluorophosphate. Bull. Chem. Soc. Jpn. 1999, 72, 2049–2054. [Google Scholar] [CrossRef]
- Bednarska-Bolek, B.; Jakubas, R.; Medycki, W.; Nowak, D.; Zaleski, J. Structure, Phase Transitions and Molecular Motions in Ferroelastic (C4H8NH2)SbCl6·(C4H8NH2)Cl. J. Phys. Condens. Matter 2002, 14, 3129–3142. [Google Scholar] [CrossRef]
- Medycki, W.; Świergiel, J.; Hołderna-Natkaniec, K.; Jurga, K.; Jakubas, R. 1H NMR Studies on Molecular Motions of 4-Aminopyridinium and Pyrrolidinium Cations in New Ferroics. Solid. State Nucl. Magn. Reson. 2004, 25, 129–132. [Google Scholar] [CrossRef]
- Jakubas, R.; Bednarska-Bolek, B.; Zaleski, J.; Medycki, W.; Hołderna-Natkaniec, K.; Zieliński, P.; Gała̧zka, M. Structure, Phase Transitions and Molecular Dynamics in Ferroelastic Crystal Pyrrolidinium Hexachloroantimonate(V), [C4H8NH2][SbCl6]. Solid State Sci. 2005, 7, 381–390. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Gągor, A.; Piecha-Bisiorek, A.; Jakubas, R.; Ciżman, A.; Zaręba, J.K.; Nyk, M.; Zieliński, P.Z.; Medycki, W.; Bil, A.; et al. Ferroelectricity and Ferroelasticity in Organic Inorganic Hybrid (Pyrrolidinium) 3 [Sb2Cl9]. Chem. Mater. 2018, 30, 45. [Google Scholar] [CrossRef]
- Ksiądzyna, M.; Kinzhybalo, V.; Bieńko, A.; Medycki, W.; Jakubas, R.; Rajnák, C.; Boča, R.; Ozarowski, A.; Ozerov, M.; Piecha-Bisiorek, A. Symmetry-Breaking Phase Transitions, Dielectric and Magnetic Properties of Pyrrolidinium-Tetrahalidocobaltates. Inorg. Chem. Front. 2022, 9, 2353–2364. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL Electronic Structure Theory > Density Functional Theory. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; University of Torino: Turin, Italy, 2017. [Google Scholar]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the Exchange Screening Parameter on the Performance of Screened Hybrid Functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J. Special Points for Brillouin-Zone Integrations. Phys. Rev. 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Heifets, E.; Kotomin, E.A.; Bagaturyants, A.A.; Maier, J. Ab Initio Study of BiFeO 3 : Thermodynamic Stability Conditions. J. Phys. Chem. Lett 2015, 6, 2847–2851. [Google Scholar] [CrossRef]
- Heifets, E.; Kotomin, E.A.; Bagaturyants, A.A.; Maier, J. Thermodynamic Stability of Stoichiometric LaFeO3 and BiFeO3 : A Hybrid DFT Study. Phys. Chem. Chem. Phys 2017, 3738, 3738. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar]
- Hay, P.J.; Wadt, W.R. No Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hinuma, Y.; Pizzi, G.; Kumagai, Y.; Oba, F.; Tanaka, I. Band Structure Diagram Paths Based on Crystallography. Comput. Mater. Sci. 2017, 128, 140–184. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot Homepage—An Interactive Plotting Program. Available online: http://gnuplot.info (accessed on 20 March 2023).
- Van Vleck, J.H. The dipolar broadening of magnetic resonance lines in crystals. Phys. Rev. 1948, 74, 1168. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (PYR)3[Bi2I9] [24] | (PYR)3[Sb2I9] [24] | (IM)3[Bi2I9] [19] | (IM)3[Sb2I9] [19] | 1 (This Work) | 2 (This Work) | |
|---|---|---|---|---|---|---|
| Average bond length (Å) | 3.105 | 3.061 | 3.101 | 3.065 | 3.095 | 3.045 |
| Polyhedral volume (Å3) | 39.51 | 37.891 | 39.36 | 38.05 | 39.17 | 37.30 |
| Distortion index (bond length) * | 0.042 | 0.049 | 0.049 | 0.057 | 0.047 | 0.060 |
| Bond angle variance (deg.2) | 23.98 | 21.82 | 22.85 | 19.38 | 25.08 | 20.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rowińska, M.; Piecha-Bisiorek, A.; Medycki, W.; Durlak, P.; Jakubas, R.; Gagor, A. Structural, Electric and Dynamic Properties of (Pyrrolidinium)3[Bi2I9] and (Pyrrolidinium)3[Sb2I9]: New Lead-Free, Organic–Inorganic Hybrids with Narrow Band Gaps. Molecules 2023, 28, 3894. https://doi.org/10.3390/molecules28093894
Rowińska M, Piecha-Bisiorek A, Medycki W, Durlak P, Jakubas R, Gagor A. Structural, Electric and Dynamic Properties of (Pyrrolidinium)3[Bi2I9] and (Pyrrolidinium)3[Sb2I9]: New Lead-Free, Organic–Inorganic Hybrids with Narrow Band Gaps. Molecules. 2023; 28(9):3894. https://doi.org/10.3390/molecules28093894
Chicago/Turabian StyleRowińska, Magdalena, Anna Piecha-Bisiorek, Wojciech Medycki, Piotr Durlak, Ryszard Jakubas, and Anna Gagor. 2023. "Structural, Electric and Dynamic Properties of (Pyrrolidinium)3[Bi2I9] and (Pyrrolidinium)3[Sb2I9]: New Lead-Free, Organic–Inorganic Hybrids with Narrow Band Gaps" Molecules 28, no. 9: 3894. https://doi.org/10.3390/molecules28093894
APA StyleRowińska, M., Piecha-Bisiorek, A., Medycki, W., Durlak, P., Jakubas, R., & Gagor, A. (2023). Structural, Electric and Dynamic Properties of (Pyrrolidinium)3[Bi2I9] and (Pyrrolidinium)3[Sb2I9]: New Lead-Free, Organic–Inorganic Hybrids with Narrow Band Gaps. Molecules, 28(9), 3894. https://doi.org/10.3390/molecules28093894