

Biological Evaluation of Triorganotin Derivatives as Potential Anticancer Agents

 ,
,  , ,
, ,  ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
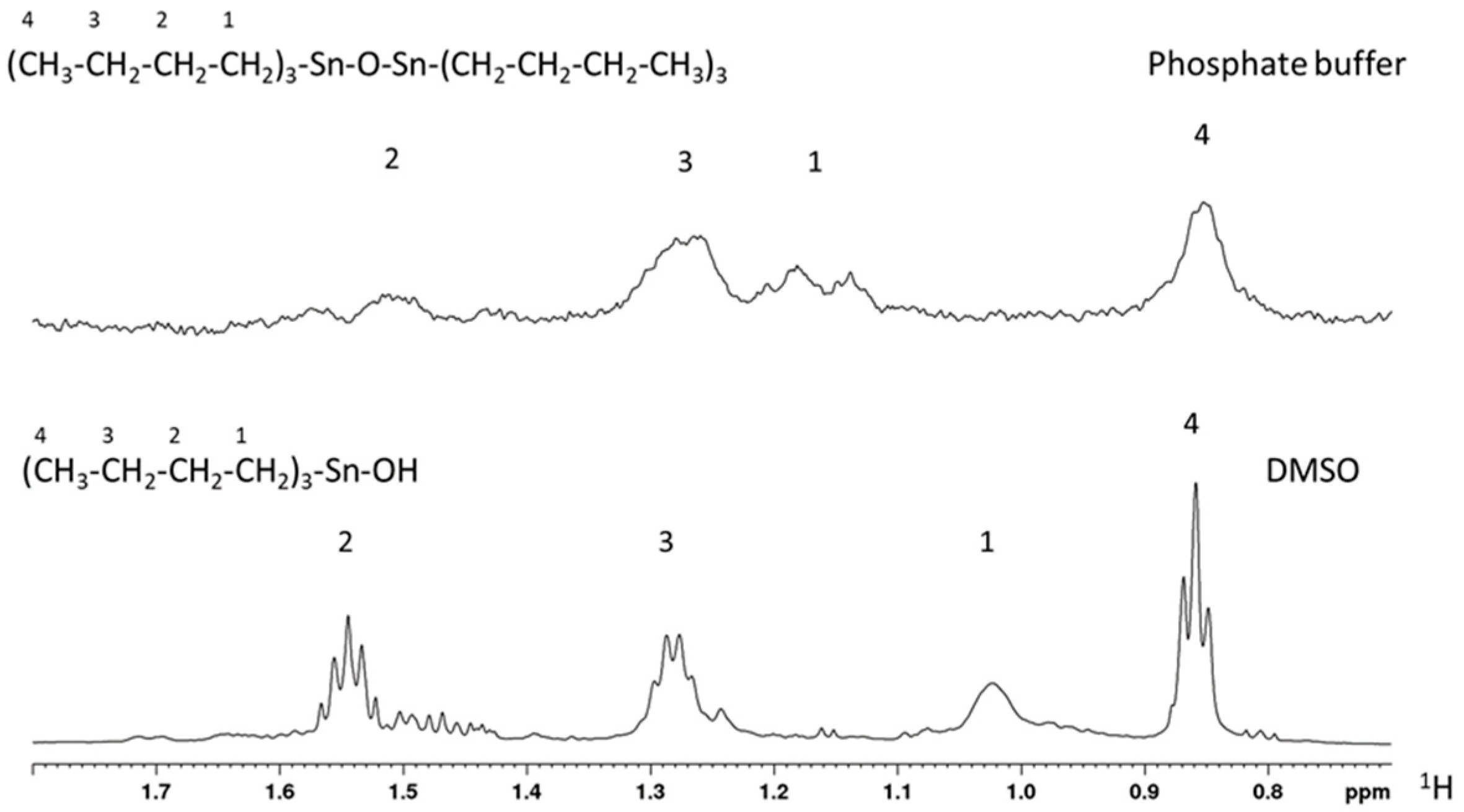
2.1. NMR Characterization of Structure, Solubility, and Aggregation
2.2. Effect of Organotin Compounds on the Metabolic Activity of CAL27, MCF-10A, U937 Cells, and PBMCs
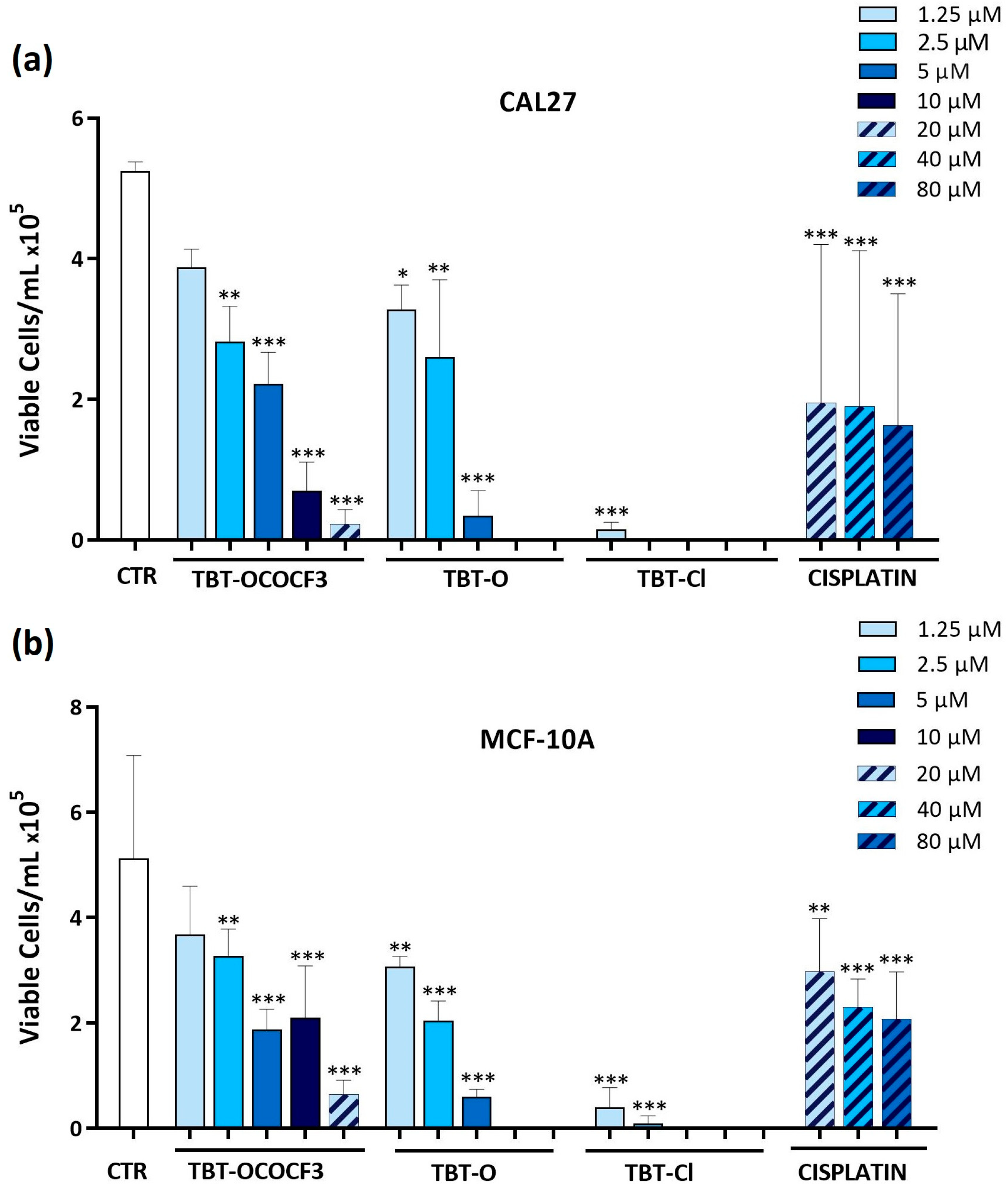
2.3. Effects of Triorganotin Compounds on Cell Viabilities of CAL-27 and MCF-10A Cells
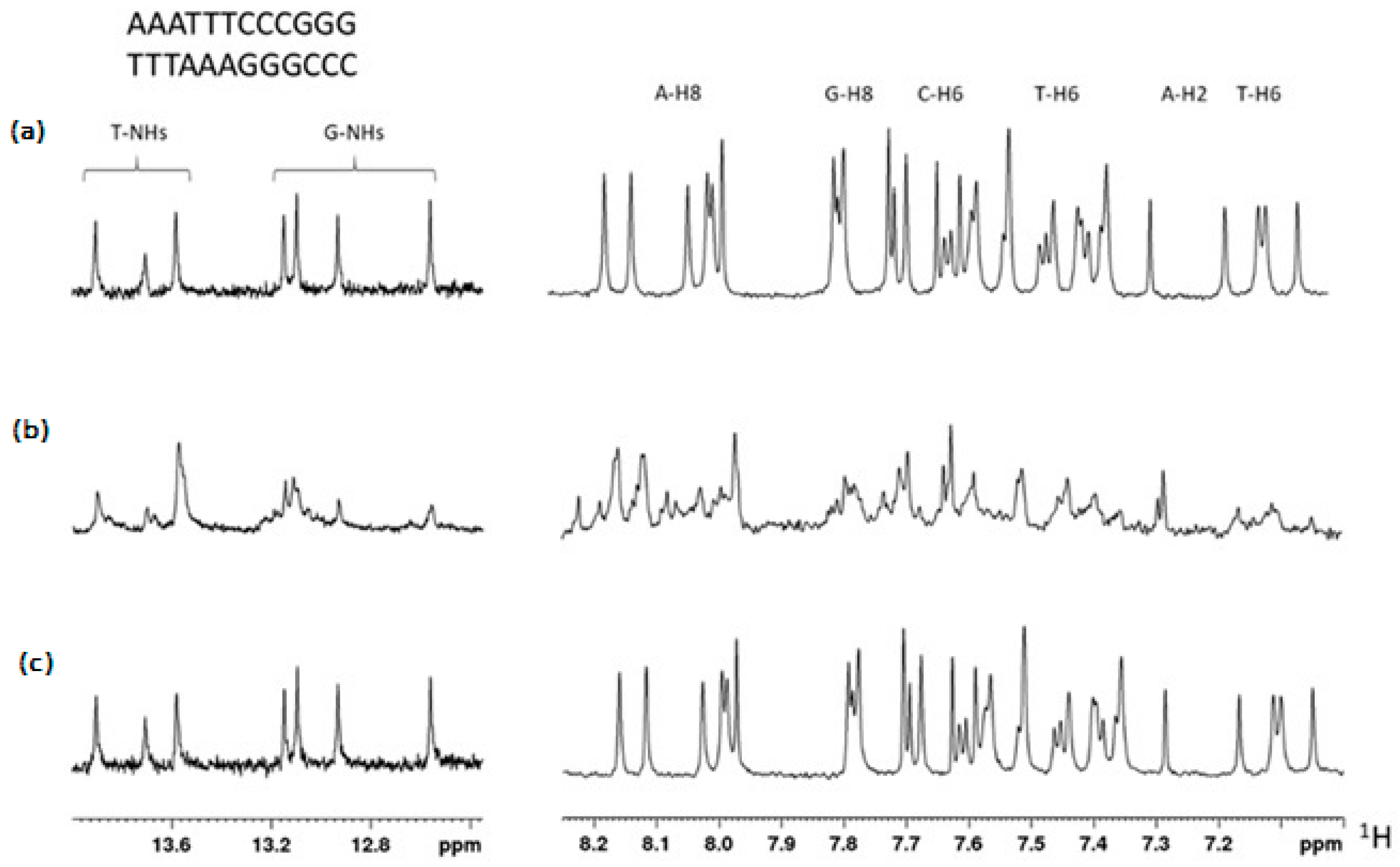
2.4. Interaction of TBT-OCOCF3 with 12 bp-DNA
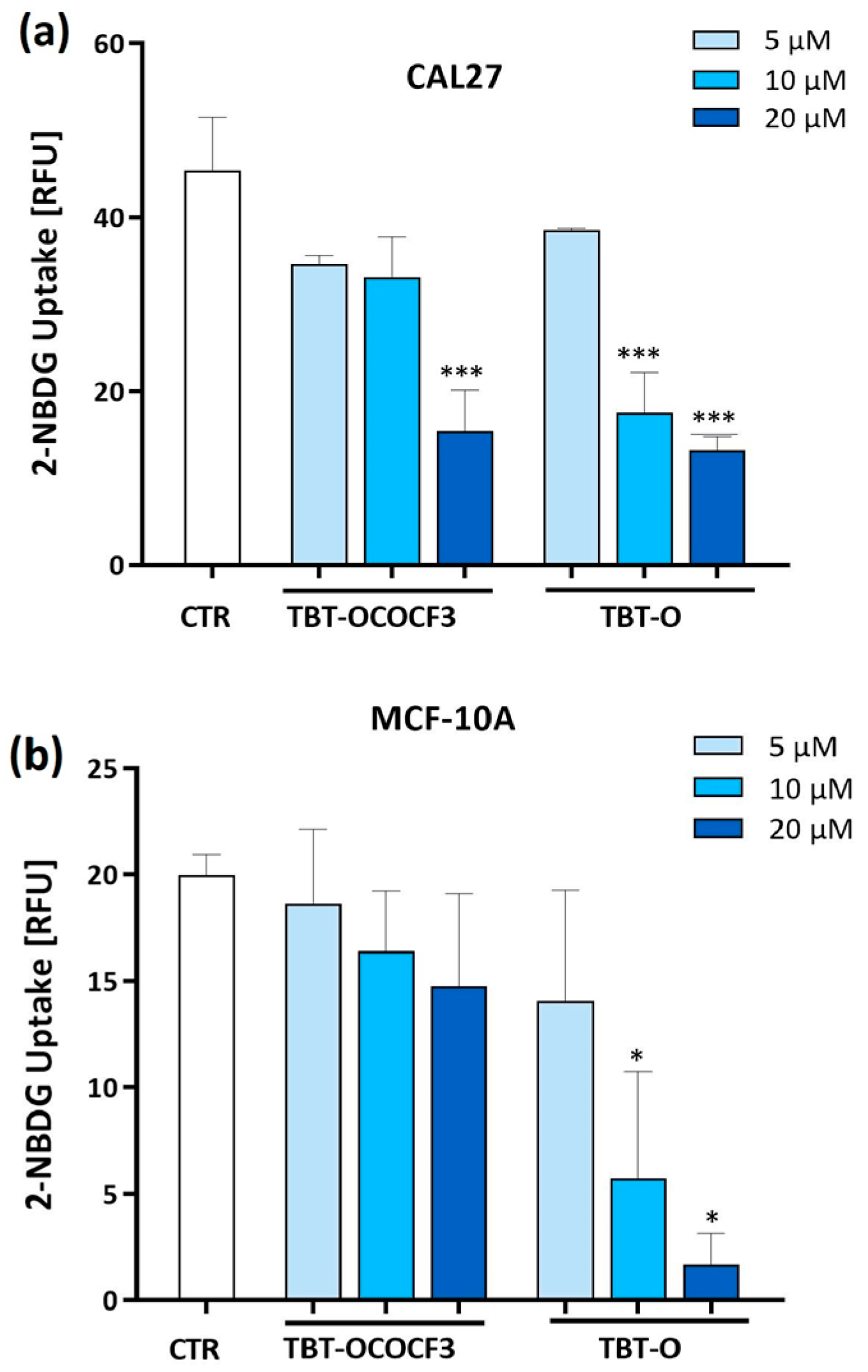
2.5. Inhibition of Glucose Uptake by Triorganotins
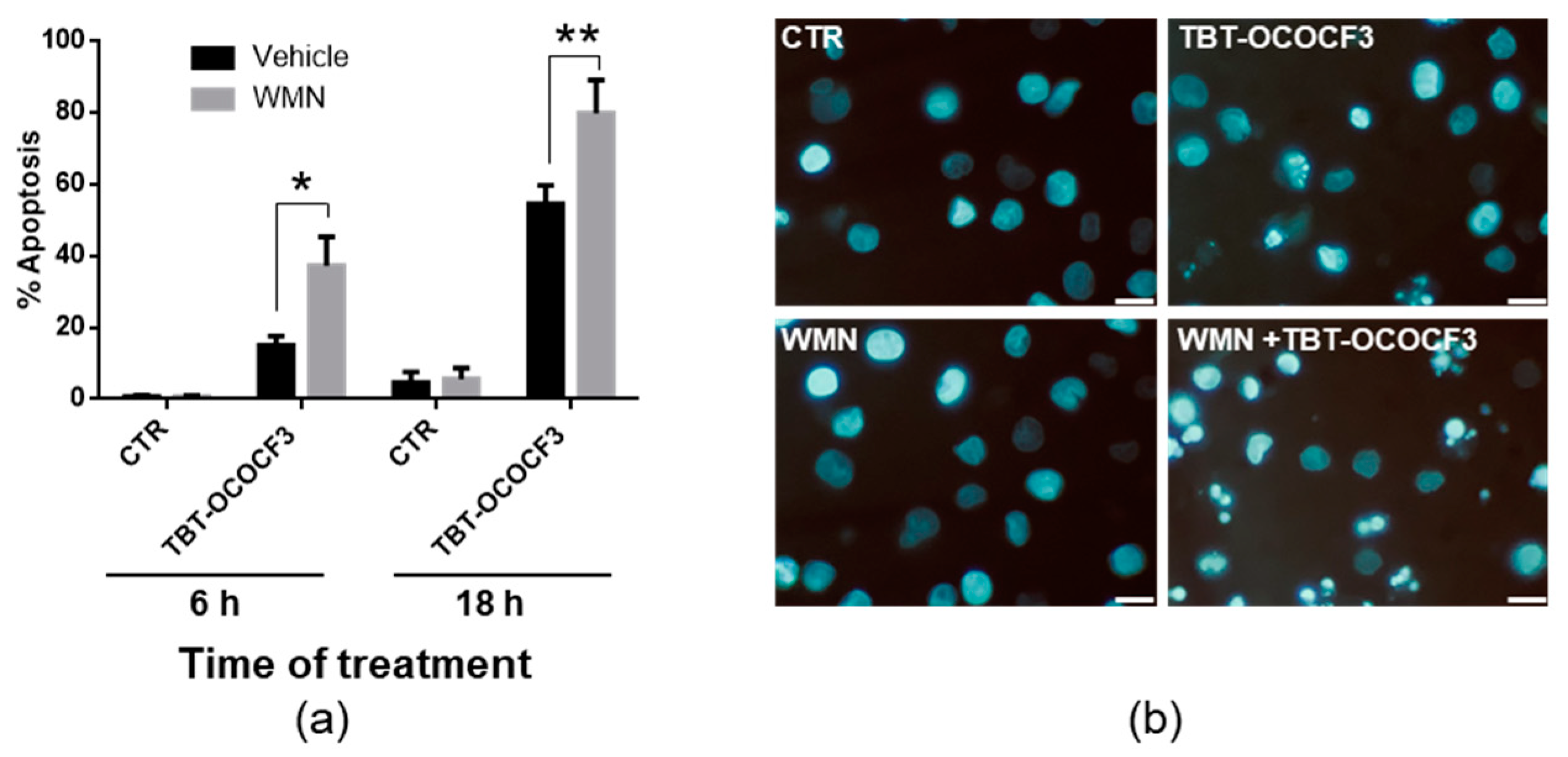
2.6. Effects of TBT-OCOCF3 on Cell Death in CAL-27 and MCF-10A Cells
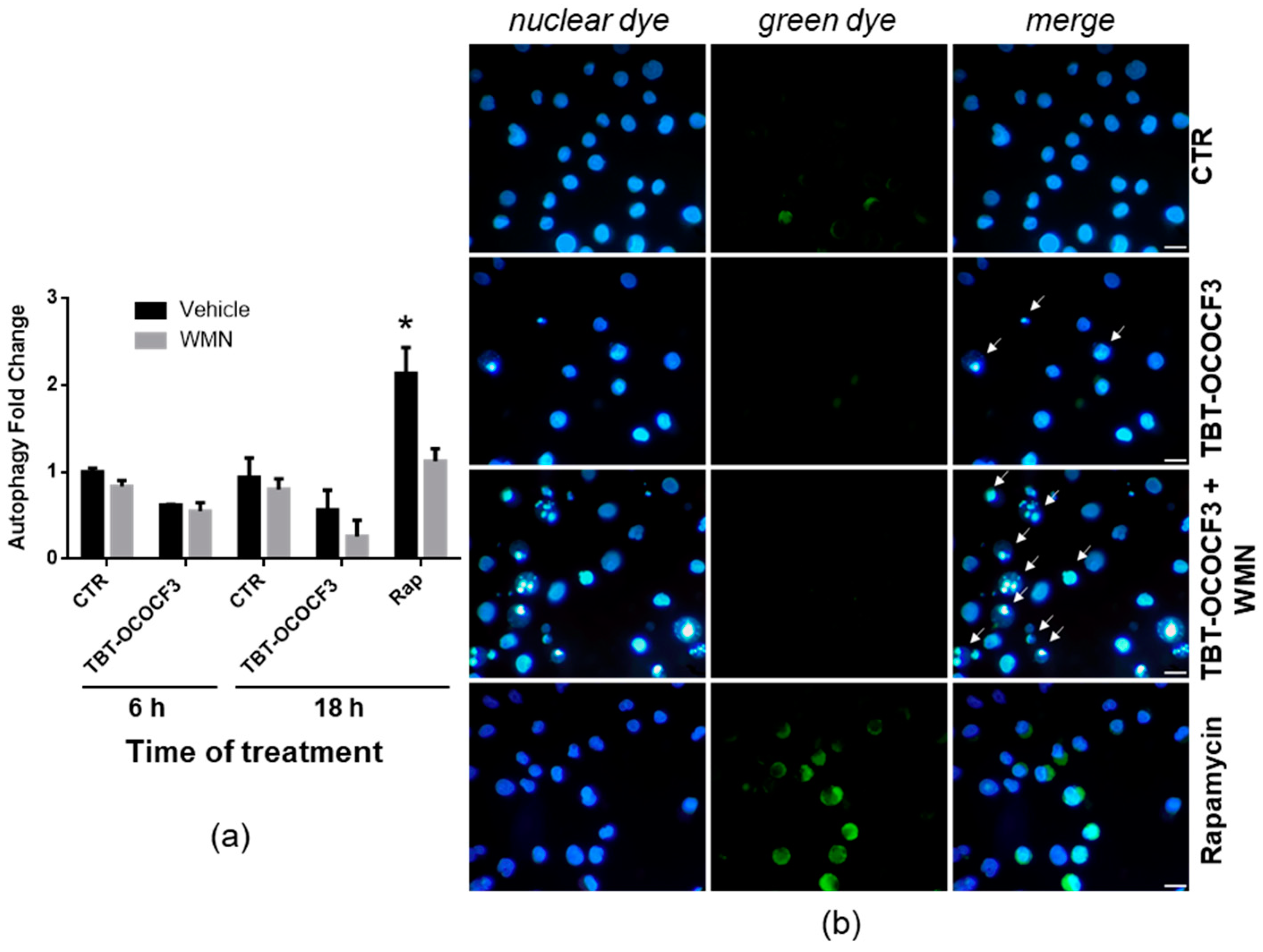
2.7. Effect of TBT-OCOCF3 on Cell Death and Autophagy in U937 Cells
3. Discussion
4. Materials and Methods
4.1. Synthesis of Tributyltin Trifluoroacetate, Chemicals, and Reagents
4.2. Cells
4.3. Metabolic Activity and Viability Assay
4.4. NMR Spectroscopy
4.5. Glucose Uptake
4.6. Cell Death and Autophagy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Syed Annuar, S.N.; Kamaludin, N.F.; Awang, N.; Chan, K.M. Cellular basis of organotin(iv) derivatives as anticancer metallodrugs: A review. Front. Chem. 2021, 9, 657599. [Google Scholar] [CrossRef]
- Hoch, M.; Alonso-Azcarate, J.; Lischick, M. Adsorption behavior of toxic tributyltin to clay-rich sediments under various environmental conditions. Environ. Toxicol. Chem. 2002, 21, 1390–1397. [Google Scholar] [CrossRef]
- Delgado Filho, V.S.; Lopes, P.F.; Podratz, P.L.; Graceli, J.B. Triorganotin as a compound with potential reproductive toxicity in mammals. Braz. J. Med. Biol. Res. 2011, 44, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Hobler, C.; Andrade, A.J.; Grande, S.W.; Gericke, C.; Talsness, C.E.; Appel, K.E.; Chahoud, I.; Grote, K. Sex-dependent aromatase activity in rat offspring after pre- and postnatal exposure to triphenyltin chloride. Toxicology 2010, 276, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Nishikawa, J.; Hiromori, Y.; Yokoyama, H.; Koyanagi, M.; Takasuga, S.; Ishizaki, J.; Watanabe, M.; Isa, S.; Utoguchi, N.; et al. Trialkyltin compounds bind retinoid x receptor to alter human placental endocrine functions. Mol. Endocrinol. 2005, 19, 2502–2516. [Google Scholar] [CrossRef]
- Banti, C.N.; Hadjikakou, S.K.; Sismanoglu, T.; Hadjiliadis, N. Anti-proliferative and antitumor activity of organotin(iv) compounds. An overview of the last decade and future perspectives. J. Inorg. Biochem. 2019, 194, 114–152. [Google Scholar] [CrossRef]
- Alama, A.; Tasso, B.; Novelli, F.; Sparatore, F. Organometallic compounds in oncology: Implications of novel organotins as antitumor agents. Drug Discov. Today 2009, 14, 500–508. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Kotake, Y.; Takai, N.; Ohta, S. Involvement of autophagy via mammalian target of rapamycin (mtor) inhibition in tributyltin-induced neuronal cell death. J. Toxicol. Sci. 2010, 35, 245–251. [Google Scholar] [CrossRef]
- Katika, M.R.; Hendriksen, P.J.M.; van Loveren, H.; Peijnenburg, A. Exposure of jurkat cells to bis (tri-n-butyltin) oxide (tbto) induces transcriptomics changes indicative for er- and oxidative stress, t cell activation and apoptosis. Toxicol. Appl. Pharm. 2011, 254, 311–322. [Google Scholar] [CrossRef]
- Fickova, M.; Macho, L.; Brtko, J. A comparison of the effects of tributyltin chloride and triphenyltin chloride on cell proliferation, proapoptotic p53, bax, and antiapoptotic bcl-2 protein levels in human breast cancer mcf-7 cell line. Toxicol. In Vitro 2015, 29, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Basu Baul, T.S.; Dutta, D.; Duthie, A.; Guchhait, N.; Rocha, B.G.M.; Guedes da Silva, M.F.C.; Mokhamatam, R.B.; Raviprakash, N.; Manna, S.K. New dibutyltin(iv) ladders: Syntheses, structures and, optimization and evaluation of cytotoxic potential employing a375 (melanoma) and hct116 (colon carcinoma) cell lines in vitro. J. Inorg. Biochem. 2017, 166, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Anasamy, T.; Thy, C.K.; Lo, K.M.; Chee, C.F.; Yeap, S.K.; Kamalidehghan, B.; Chung, L.Y. Tribenzyltin carboxylates as anticancer drug candidates: Effect on the cytotoxicity, motility and invasiveness of breast cancer cell lines. Eur. J. Med. Chem. 2017, 125, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Waseem, D.; Butt, A.F.; Haq, I.-u.; Bhatti, M.H.; Khan, G.M. Carboxylate derivatives of tributyltin (iv) complexes as anticancer and antileishmanial agents. DARU J. Pharm. Sci. 2017, 25, 8. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kotake, Y.; Sekino, Y.; Kanda, Y. Amp-activated protein kinase-mediated glucose transport as a novel target of tributyltin in human embryonic carcinoma cells. Metallomics 2013, 5, 484–491. [Google Scholar] [CrossRef]
- Bohacova, V.; Seres, M.; Pavlikova, L.; Kontar, S.; Cagala, M.; Bobal, P.; Otevrel, J.; Brtko, J.; Sulova, Z.; Breier, A. Triorganotin derivatives induce cell death effects on l1210 leukemia cells at submicromolar concentrations independently of p-glycoprotein expression. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Latsis, G.K.; Banti, C.N.; Kourkoumelis, N.; Papatriantafyllopoulou, C.; Panagiotou, N.; Tasiopoulos, A.; Douvalis, A.; Kalampounias, A.G.; Bakas, T.; Hadjikakou, S.K. Poly organotin acetates against DNA with possible implementation on human breast cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Pantelic, N.D.; Zmejkovski, B.B.; Bozic, B.; Dojcinovic, B.; Banjac, N.R.; Wessjohann, L.A.; Kaluderovic, G.N. Synthesis, characterization and in vitro biological evaluation of novel organotin(iv) compounds with derivatives of 2-(5-arylidene-2,4-dioxothiazolidin-3-yl)propanoic acid. J. Inorg. Biochem. 2020, 211, 111207. [Google Scholar] [CrossRef]
- Pantelic, N.D.; Bozic, B.; Zmejkovski, B.B.; Banjac, N.R.; Dojcinovic, B.; Wessjohann, L.A.; Kaluderovic, G.N. In vitro evaluation of antiproliferative properties of novel organotin(iv) carboxylate compounds with propanoic acid derivatives on a panel of human cancer cell lines. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Attanzio, A.; Ippolito, M.; Girasolo, M.A.; Saiano, F.; Rotondo, A.; Rubino, S.; Mondello, L.; Capobianco, M.L.; Sabatino, P.; Tesoriere, L.; et al. Anti-cancer activity of di- and tri-organotin(iv) compounds with d-(+)-galacturonic acid on human tumor cells. J. Inorg. Biochem. 2018, 188, 102–112. [Google Scholar] [CrossRef]
- Pellerito, C.; Emanuele, S.; Ferrante, F.; Celesia, A.; Giuliano, M.; Fiore, T. Tributyltin(iv) ferulate, a novel synthetic ferulic acid derivative, induces autophagic cell death in colon cancer cells: From chemical synthesis to biochemical effects. J. Inorg. Biochem. 2020, 205, 110999. [Google Scholar] [CrossRef]
- Celesia, A.; Morana, O.; Fiore, T.; Pellerito, C.; D’Anneo, A.; Lauricella, M.; Carlisi, D.; De Blasio, A.; Calvaruso, G.; Giuliano, M.; et al. Ros-dependent er stress and autophagy mediate the anti-tumor effects of tributyltin (iv) ferulate in colon cancer cells. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Waseem, D.; Khan, G.M.; Haq, I.U.; Rashid, U.; Syed, D.N. The triphenyltin carboxylate derivative triphenylstannyl 2-(benzylcarbamoyl)benzoate impedes prostate cancer progression via modulation of akt/foxo3a signaling. Toxicol. Appl. Pharmacol. 2020, 401, 115091. [Google Scholar] [CrossRef] [PubMed]
- Hubner, D.; Kaluderovic, M.R.; Gomez-Ruiz, S.; Kaluderovic, G.N. Anionic chlorido(triphenyl)tin(iv) bearing n-phthaloylglycinato or 1,2,4-benzenetricarboxylato 1,2-anhydride ligands: Potential cytotoxic and apoptosis-inducing agents against several types of cancer. Chem. Biol. Drug Des. 2017, 89, 628–633. [Google Scholar] [CrossRef]
- Zhou, M.; Feng, M.; Fu, L.L.; Ji, L.D.; Zhao, J.S.; Xu, J. Toxicogenomic analysis identifies the apoptotic pathway as the main cause of hepatotoxicity induced by tributyltin. Food Chem. Toxicol. 2016, 97, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Gera, R.; Siddiqui, W.A.; Khandelwal, S. Tributyltin induces oxidative damage, inflammation and apoptosis via disturbance in blood-brain barrier and metal homeostasis in cerebral cortex of rat brain: An in vivo and in vitro study. Toxicology 2013, 310, 39–52. [Google Scholar] [CrossRef]
- Liu, W.X.; Saito, T.; Li, L.; Rinaldi, P.L.; Hirst, R.; Halasa, A.F.; Visintainer, J. Characterization of sn-containing polymer chain ends of polybutadiene using h-1/c-13/sn-119 triple-resonance 3d-nmr. Macromolecules 2000, 33, 2364–2369. [Google Scholar] [CrossRef]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient nmr techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P.B. Resistance to cis- and carboplatin initiated by epigenetic changes in ovarian cancer patients. Cancer Drug Resist. 2019, 2, 271–296. [Google Scholar] [CrossRef]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Kotake, Y.; Komasaka, K.; Hakozaki, H.; Taguchi, R.; Kume, T.; Akaike, A.; Ohta, S. Glutamate excitotoxicity is involved in cell death caused by tributyltin in cultured rat cortical neurons. Toxicol. Sci. 2006, 89, 235–242. [Google Scholar] [CrossRef]
- Balogova, M.; Sharma, S.; Cherek, P.; Olafsson, S.N.; Jonsdottir, S.; Ogmundsdottir, H.M.; Damodaran, K.K. Cytotoxic effects of halogenated tin phosphinoyldithioformate complexes against several cancer cell lines. Dalton Trans. 2022, 51, 13119–13128. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ancey, P.B.; Contat, C.; Meylan, E. Glucose transporters in cancer—from tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Marino, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 2011, 14, 2251–2269. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014, 26, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Tsuchiya, A.; Shimizu, T.; Tanaka, A.; Nishizaki, T. Huhs1015 suppresses colonic cancer growth by inducing necrosis and apoptosis in association with mitochondrial damage. Anticancer Res. 2016, 36, 39–48. [Google Scholar]
- Fabrizi, C.; Pompili, E.; Somma, F.; De Vito, S.; Ciraci, V.; Artico, M.; Lenzi, P.; Fornai, F.; Fumagalli, L. Lithium limits trimethyltin-induced cytotoxicity and proinflammatory response in microglia without affecting the concurrent autophagy impairment. J. Appl. Toxicol. 2017, 37, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Rehman, S.K.; Zhang, W.; Wen, A.D.; Yao, L.B.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. BBA-Rev Cancer, 2010; 1806, 220–229. [Google Scholar]
- Eberhart, K.; Oral, O.; Gozuacik, D. Induction of autophagic cell death by anticancer agents. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Molecular Mechanisms; Academic Press: Cambridge, MA, USA, 2014; Volume 1, pp. 179–202. [Google Scholar]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mtor and beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.C.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3 ‘-kinases are involved in signaling pathways that control macroautophagy in ht-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Shintani, T.; Hayashido, Y.; Hamada, A.; Higaki, M.; Yoshioka, Y.; Sakamoto, A.; Yanamoto, S.; Okamoto, T. Vegf-a promotes the motility of human melanoma cells through the vegfr1-pi3k/akt signaling pathway. In Vitro Cell Dev. Biol. Anim. 2022, 58, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.T.; Ismail, A.; Tolomeo, C. Targeting the phosphatidylinositol 3-kinase (pi3k)/akt/mammalian target of rapamycin (mtor) pathway: An emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat. Rev. 2014, 40, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Shaka, A.J. Water suppression that works—excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Mastino, A.; Sciortino, M.T.; Medici, M.A.; Perri, D.; Ammendolia, M.G.; Grelli, S.; Amici, C.; Pernice, A.; Guglielmino, S. Herpes simplex virus 2 causes apoptotic infection in monocytoid cells. Cell Death Differ. 1997, 4, 629–638. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1H (ppm) | 1H (ppm) | 13C (ppm) | ||
|---|---|---|---|---|---|
| Position 1 | Buffer | DMSO | |||
 | TBT-OH | 1 | 1.210 | 1.084 | 19.90 |
| 2 | 1.558 | 1.592 | 29.25 | ||
| 3 | 1.323 | 1.329 | 28.09 | ||
| 4 | 0.901 | 0.916 | 15.05 | ||
 | TBT-Cl | 1 | 1.222 | 1.081 | 19.94 |
| 2 | 1.637 | 1.590 | 29.45 | ||
| 3 | 1.349 | 1.321 | 28.19 | ||
| 4 | 0.905 | 0.903 | 15.33 | ||
 | TBT-OCOCF3 | 1 | 1.164 | 1.071 | 19.91 |
| 2 | 1.617 | 1.579 | 29.45 | ||
| 3 | 1.343 | 1.314 | 28.34 | ||
| 4 | 0.899 | 0.890 | 15.33 | ||
| Compound | Solubility (μM) | DMSO (Å) | Phosphate Buffer (Å) |
|---|---|---|---|
| TBT-OH/TBTO | 42 | 5.4 | 7.8 |
| TBT-Cl | 61 | 5.6 | 13.0 |
| TBT-OCOCF3 | 69 | 5.4 | 4.6 |
| Cells | ||||
|---|---|---|---|---|
| Compound | CAL-27 | MCF-10A | PBMCs | U937 |
| TBT-O | 13.18 ± 3.70 1 | 4.93 ± 2.33 | 2.96 ± 1.35 | 1.82 ± 0.21 |
| TBT-Cl | 0.91 ± 0.53 | 1.22 ± 0.41 | ≤1 | 0.42 ± 0.30 |
| TBT-OCOF3 | 2.45 ± 0.14 | 3.14 ± 2.53 | 2.68 ± 1.17 | 1.32 ± 0.32 |
| Cisplatin | 138.60 ± 59.52 | 21.50 ± 0.04 | 52.48 ± 5.88 | 38.13 ± 3.23 |
| Cells | Treatment | Anx-/7AAD- | Anx+/7AAD- | Anx+/7AAD+ | Anx-/7AAD+ |
|---|---|---|---|---|---|
| CAL-27 | CTR | 88.01 ± 2.35 1 | 10.53 ± 1.35 | 0.06 ± 0.01 | 0.58 ± 0.02 |
| TBT-OCOCF3 | 86.80 ± 0.01 | 4.01 ± 0.21 | 1.26 ± 0.42 | 7.85 ± 1.01 2 | |
| WMN | 89.8 ± 4.63 | 6.42 ± 2.45 | 0.78 ± 0.04 | 2.99 ± 0.02 | |
| WMN+ TBT-OCOF3 | 58.8 ± 8.01 3 | 6.46 ± 0.40 | 5.56 ± 0.21 4 | 29.14 ± 3.02 3 | |
| MCF-10 | CTR | 75.96 ± 5.55 | 3.68 ± 1.25 | 9.36 ± 3.24 | 12.00 ± 2.56 |
| TBT-OCOCF3 | 14.16 ± 2.56 5 | 24.68 ± 5.45 5 | 47.59 ± 3.65 5 | 13.75 ± 1.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanizzi, V.; Minutolo, A.; Valletta, E.; Carlini, M.; Cordero, F.M.; Ranzenigo, A.; Prete, S.P.; Cicero, D.O.; Pitti, E.; Petrella, G.; et al. Biological Evaluation of Triorganotin Derivatives as Potential Anticancer Agents. Molecules 2023, 28, 3856. https://doi.org/10.3390/molecules28093856
Stefanizzi V, Minutolo A, Valletta E, Carlini M, Cordero FM, Ranzenigo A, Prete SP, Cicero DO, Pitti E, Petrella G, et al. Biological Evaluation of Triorganotin Derivatives as Potential Anticancer Agents. Molecules. 2023; 28(9):3856. https://doi.org/10.3390/molecules28093856
Chicago/Turabian StyleStefanizzi, Valeria, Antonella Minutolo, Elena Valletta, Martina Carlini, Franca M. Cordero, Anna Ranzenigo, Salvatore Pasquale Prete, Daniel Oscar Cicero, Erica Pitti, Greta Petrella, and et al. 2023. "Biological Evaluation of Triorganotin Derivatives as Potential Anticancer Agents" Molecules 28, no. 9: 3856. https://doi.org/10.3390/molecules28093856
APA StyleStefanizzi, V., Minutolo, A., Valletta, E., Carlini, M., Cordero, F. M., Ranzenigo, A., Prete, S. P., Cicero, D. O., Pitti, E., Petrella, G., Matteucci, C., Marino-Merlo, F., Mastino, A., & Macchi, B. (2023). Biological Evaluation of Triorganotin Derivatives as Potential Anticancer Agents. Molecules, 28(9), 3856. https://doi.org/10.3390/molecules28093856