3.2. Chemical Syntheses
(3R,4R,5S,6S)-6-methyltetrahydro-2H-pyran-2,3,4,5-tetrayl tetraacetate (8). The synthetic process followed the literature [
27].
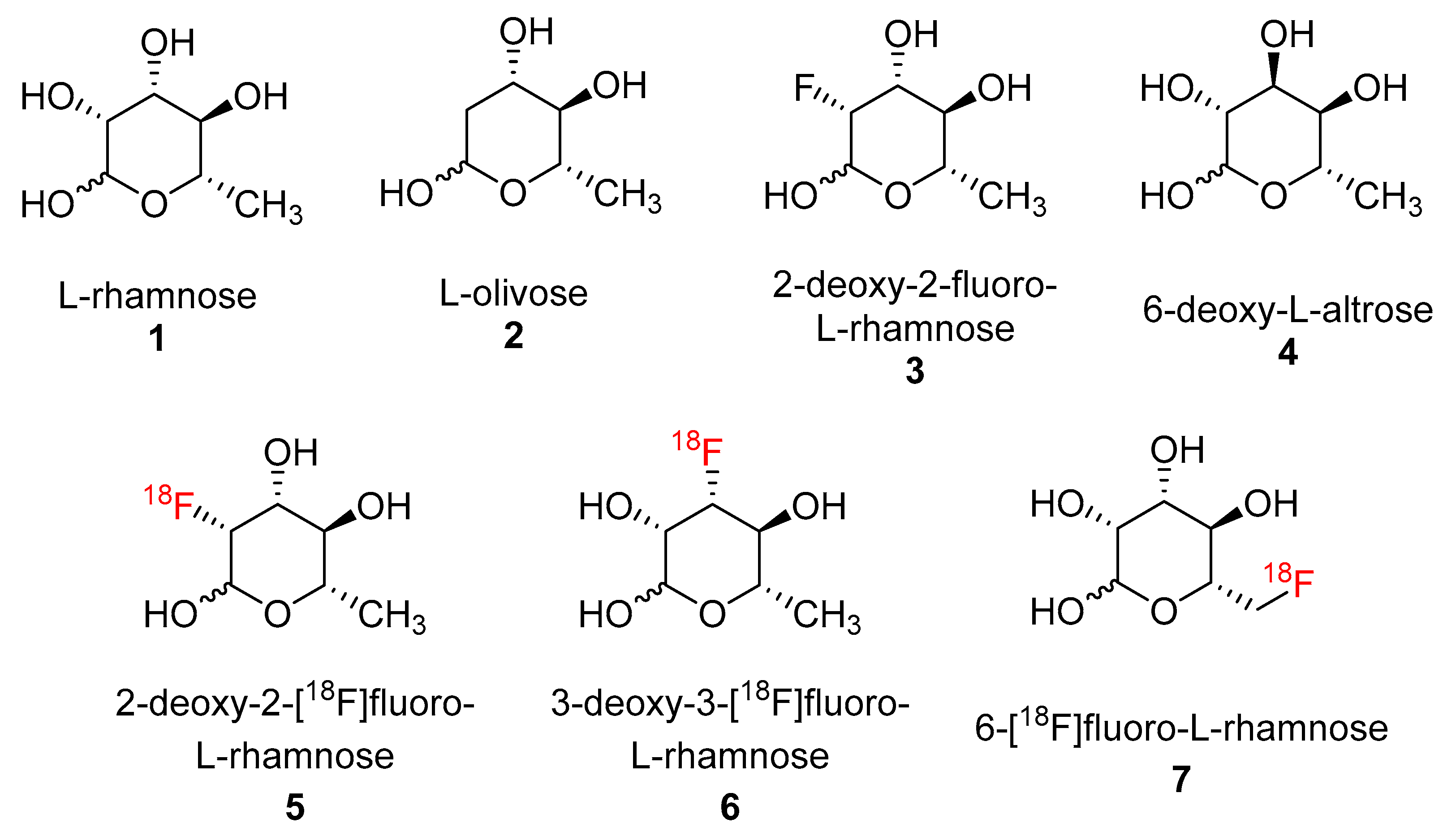
L-Rhamnose (
1, 10 g, 54.9 mmol) was carefully added in portions (3 portions in 15 min) to a stirring solution of iodine (0.125 g, 0.49 mmol) in acetic anhydride (60 mL) in a cool-water bath (10–15 °C). The resulting mixture was allowed to warm up to room temperature and stirred for 2 h. The mixture was then poured onto a mixture of crushed ice and saturated aqueous Na
2S
2O
3 (250 mL, 1:1 mixture) with vigorous stirring. To the resulting light-yellow mixture in an ice-water bath, NaHCO
3 was added portion wise until no more CO
2 was released. The crude product was extracted with CH
2Cl
2 (150 mL × 3). The organic layer was combined, washed with saturated NaHCO
3 solution and water (400 mL each), and dried over anhydrous Na
2SO
4. Crude product
8 was obtained by removing the volatiles under reduced pressure (19.33 g, 95.4% yield, α:β anomer ratio = 3:1).
1H NMR (400 MHz, Chloroform-
d) δ 6.02 (d,
J = 1.9 Hz, 1H), 5.83 (s, 0.34H), 5.48 (s, 0.34H), 5.36–5.28 (m, 1H), 5.26 (dd,
J = 3.5, 2.0 Hz, 1H), 5.18–5.05 (m, 1.7H), 4.00–3.90 (m, 1H), 3.72–3.62 (m, 0.36H), 2.23 (s, 4H), 2.22, (s, 1H), 2.20 (s, 3H), 2.15 (s, 3H), 2.11 (s, 1H), 2.07 (s, 4H), 2.01 (s, 4H), 1.30 (d,
J = 6.2 Hz, 1H), 1.25 (d,
J = 6.2 Hz, 3H). MS (ESI) calculated mass for the parent C
14H
20O
9 332.11 [M], found 355.00 [M + Na].
(3R,4R,5S,6S)-2-bromo-6-methyltetrahydro-2H-pyran-3,4,5-triyl triacetate (9). To a solution of compound 8 (19.33 g, 58.2 mmol) in glacial acetic acid (20 mL) and acetic anhydride (1.6 mL), HBr in acetic acid (30%, 20 mL) was added dropwise in an ice-water bath and vigorous stirring. The resulting mixture was stirred at rt overnight and slowly quenched with a pre-cooled saturated NaHCO3 solution (500 mL). The brominated intermediate was extracted with CHCl3 (200 mL × 2). The organic layer was combined and dried over anhydrous Na2SO4. The bromide intermediate 9 was obtained by removing the volatiles under reduced pressure as a yellow oil (17.8 g).
(5S,6S,7R,7aR)-2-ethoxy-2,5-dimethyltetrahydro-5H-[1,3]dioxolo [4,5-b]pyran-6,7-diyl diacetate (10). The oily bromide intermediate 9 was dissolved in a mixture of anhydrous acetonitrile (8 mL), and 2,4,6-collidine (11 mL) and ethanol (200 proof, 13 mL) was added. The resulting mixture was stirred at rt overnight, diluted with CH2Cl2 (300 mL), and washed with water (300 mL × 2) and brine (200 mL). The organic layer was dried over anhydrous Na2SO4. Crude product was obtained by removing the volatiles under reduced pressure. Product 10 was purified using flash column chromatography with hexane/ethyl acetate 4/1 to 2/1 gradient (7.8 g, 42.2% yield for 2 steps). 1H NMR (400 MHz, Chloroform-d) δ 5.41 (d, J = 2.4 Hz, 1H), 5.16–5.02 (m, 2H), 4.59 (dd, J = 3.8, 2.4 Hz, 1H), 3.65–3.47 (m, 3H), 2.12 (s, 3H), 2.07 (s, 3H), 1.75 (s, 3H), 1.30–1.14 (m, 6H). MS (ESI) calculated mass for the parent C14H22O8 318.13 [M], found 341.00 [M + Na].
(3R,4S,5S,6S)-3-hydroxy-6-methyltetrahydro-2H-pyran-2,4,5-triyl triacetate (11). Hydrochloric acid (1 N, 10 mL) was added to a solution of the orthoester 10 (7 g, 22.0 mmol) and acetone (15 mL). The mixture was stirred at rt for 10 min and volatiles were removed under reduced pressure. The resulting crude product was dissolved in CH2Cl2 (150 mL) and washed with water (150 mL × 2). The organic layer was dried over anhydrous Na2SO4. Crude product was obtained by removing the volatiles under reduced pressure. Product 11 was purified using flash column chromatography with hexane/ethyl acetate 4/1 to 1/1 gradient (3.15 g, 49.3% yield). 1H NMR (400 MHz, Chloroform-d) δ 5.76 (s, 1H), 5.15 (t, J = 9.8 Hz, 1H), 4.99 (dd, J = 9.9, 3.0 Hz, 1H), 4.22–4.15 (m, 1H), 3.65 (dq, J = 9.3, 6.2 Hz, 1H), 2.5–2.25 (br s, 1H), 2.17 (s, 3H), 2.11 (s, 3H), 2.06 (s, 3H), 1.27 (d, J = 6.2 Hz, 3H). MS (ESI) calculated mass for the parent C12H18O8 290.10 [M], found 313.00 [M + Na].
(3S,4S,5S,6S)-3-hydroxy-6-methyltetrahydro-2H-pyran-2,4,5-triyl triacetate (13). The triacetate 11 (1.0 g, 3.19 mmol) was dissolved in anhydrous CH2Cl2 (20 mL) and anhydrous pyridine (3.5 mL) and cooled with an ice-salt bath. Trifluoromethanesulfonic anhydride (4.5 g, 15.97 mmol) in CH2Cl2 (10 mL) was added dropwise. The mixture was stirred at rt for 20 min, and then sequentially washed with HCl (0.3 M, 30 mL), saturated NaHCO3 (30 mL), and brine (30 mL). The organic layer was dried over anhydrous Na2SO4. The crude triflate was obtained by removing the volatiles under reduced pressure.
The crude triflate (12, 1.35 g) was stirred with acetonitrile (30 mL) and tetrabutylammonium nitrate (4.59 g, 16.0 mmol) at rt for 1 h. Crude product was obtained by removing the volatiles under reduced pressure. Product 13 was purified using flash column chromatography with hexane/ethyl acetate 3/1 to 1/1 gradient (0.65 g, 65% yield for 2 steps). 1H NMR (400 MHz, Chloroform-d) δ 5.58 (d, J = 8.3 Hz, 1H), 5.07 (t, J = 9.5 Hz, 1H), 4.77 (t, J = 9.6 Hz, 1H), 3.76–3.60 (m, 2H), 3.05 (s, 1H), 2.15 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 1.20 (d, J = 6.2 Hz, 3H). MS (ESI) calculated mass for the parent C12H18O8 290.10 [M], found 313.00 [M + Na].
(3S,4R,5S,6S)-6-methyl-3-(((trifluoromethyl)sulfonyl)oxy)tetrahydro-2H-pyran-2,4,5-triyl triacetate (14). The triacetate 13 (0.52 g, 1.79 mmol) was dissolved in anhydrous CH2Cl2 (20 mL) and anhydrous pyridine (1.2 mL) and cooled with an ice-salt bath. Trifluoromethanesulfonic anhydride (1.52 g, 5.37 mmol) in CH2Cl2 (10 mL) was added dropwise. The mixture was stirred at rt for 20 min, and then sequentially washed with HCl (0.3 M, 30 mL), saturated NaHCO3 (30 mL), and brine (30 mL). The organic layer was dried over anhydrous Na2SO4. Crude product was obtained by removing the volatiles under reduced pressure. Flash column chromatography was used to purify product 14 with hexane/ethyl acetate 3/1 to 1/1 gradient (0.75 g, quant. yield). 1H NMR (400 MHz, Chloroform-d) δ 5.80 (d, J = 8.3 Hz, 1H), 5.38 (t, J = 9.6 Hz, 1H), 4.89–4.76 (m, 2H), 3.78 (dq, J = 9.7, 6.1 Hz, 1H), 2.16 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 1.25 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.53, 169.51, 168.44, 118.21 (q, J = 319.0 Hz), 90.21, 80.92, 77.35, 77.23, 77.03, 76.71, 73.02, 71.27, 71.24, 20.49, 20.37, 20.26, 17.02. MS (ESI) calculated mass for the parent C13H17F3O10S 422.05 [M], found 362.90 [M-OAc].
(3R,4R,5R,6S)-3-fluoro-6-methyltetrahydro-2H-pyran-2,4,5-triol (2-deoxy-2-fluoro-L-rhamnose,
(3). To a solution of 2-hydroxyl analogue
14 (50 mg, 0.118 mmol) in anhydrous acetonitrile (2 mL), TBAF in THF (1.0 M, 0.177 mL, 0.177 mmol) was added. The solution was stirred at 65 °C overnight. The volatiles were removed under reduced pressure. Flash column chromatography was used to purify product
7 with hexane/ethyl acetate 5/1 to 2/1 gradient (3.5 mg, 10% yield, α:β anomer ratio = 1:1).
1H NMR (400 MHz, Chloroform-
d) δ 6.02 (s, 1H), 5.78 (d,
J = 60 Hz, 1H), 5.35–5.30 (m, 1 H), 3.30–5.25 (m, 1H), 5.18–5.10 (m, 2H), 5.08–4.95 (m, 1H), 4.88 (dd,
J = 120, 4.0 Hz, 1H), 4.00–3.80 (m, 1H), 3.73–3.65 (m, 1H), 2.20 (s, 3H), 2.09 (s, 3H), 2.08 (s, 3H), 2.12 (s, 3H), 2.08 (s, 3H), 2.02 (s, 3H), 1.30 (d,
J = 6.0 Hz, 3H), 1.25 (d,
J = 6.0 Hz, 3H). MS (ESI) calculated mass for the parent C
12H
17FO
7 292.10 [M], found 273.10 [M-F]. Triacetate (3.5 mg, 0.012 mmol) was dissolved in TFA (1.0 mL) and stirred at 50 °C for 1 h. The volatiles were removed under a reduced pressure to yield 2-deoxy-2-fluoro-L-rhamnose (
25) as a yellow oil (1.2 mg, 6%). The
19F NMR chromatogram was compared with the literature which found an identical result [
22].
(3R,4R,5S,6S)-4-(benzyloxy)-2-methoxy-6-methyltetrahydro-2H-pyran-3,5-diol (16). Methyl-rhamnopyranoside 15 (2.85 g, 16.0 mmol), benzyl bromide (2.91 mL, 24 mmol), dimethyltin dichloride (351 mg, 1.6 mmol), and Ag2O (4.07 g, 17.6 mmol) were stirred in anhydrous acetonitrile (90 mL) at room temperature for 15 h. After being filtered through a celite pad, the filtrate was evaporated and the residue was purified using silica gel flash chromatography to afford 16 as a colorless oil (3.41 g, 79%, α:β = 1). β-isomer: 1H NMR (400 MHz, CDCl3) δ 7.30–7.32 (m, 5H), 4.71 (d, 1H, J = 1.6 Hz), 4.70 (d, 1H, J = 11.3 Hz), 4.57 (d, 1H, J = 11.3 Hz), 4.02 (dd, 1H, J = 1.6 and 3.1 Hz), 3.67–3.61 (m, 2H), 3.56 (m, 1H), 3.36 (s, 3H), 1.32 (d, J = 6.3 Hz, 3H). α-isomer: 1H NMR (400 MHz, CDCl3) δ 7.39–7.32 (m, 5H), 4.75 (d, 1H, J = 11.3 Hz), 4.74 (s, 1H), 4.52 (d, 1H, J = 11.7 Hz), 3.72–3.68 (m, 2H), 3.60 (m, 1H), 3.42 (t, 1H, J = 9.0 Hz), 3.35 (s, 3H), 1.34 (d, J = 6.3 Hz, 3H). MS (ESI) calculated mass for the parent C14H20O5 268 [M], found 268 [M].
(3R,4R,5S,6S)-4-(benzyloxy)-5-hydroxy-6-methyltetrahydro-2H-pyran-2,3-diyldiacetate (18). Compound 16 (3.24 g, 12.1 mmol) was dissolved in anhydrous pyridine (12 mL) and Ac2O (7 mL). The solution was stirred at room temperature for 15 h. Solvents were evaporated and the residue was dissolved in EtOAc (300 mL), washed with sat. NaHCO3, 1 N HCl, H2O, and brine, and dried over Na2SO4. After the evaporation of solvents, the crude product 17 was used for next step. H2SO4 (0.6 mL) was added dropwise to a solution of 17 (4.25 g, 12.1 mmol) in Ac2O (20 mL) and the solution was stirred at room temperature for 5 h. The reaction mixture was poured into a stirred mixture of ethyl acetate (150 mL) and sat. NaHCO3 (80 mL). The organic phase was separated and washed with sat. NaHCO3 and brine and dried over Na2SO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford the product 18 as a colorless oil (3.37 g, 73%). 1H NMR (400 MHz, CDCl3): δ 7.37–7.26 (m, 5H), 6.12 (d, 0.27H, J = 2.0 Hz), 6.03 (d, 0.73H, J = 2.0 Hz), 5.34 (dd, 0.73H, J = 2.0 and 3.5 Hz), 5.23 (m, 0.27H), 5.16 (m, 0.27H), 5.07 (t, 0.73H, J = 9.0 Hz), 4.72–4.43 (m, 2H), 3.94–3.79 (m, 2H), 2.16 (s, 2.19H), 2.12 (s, 0.81H), 2.11 (s, 2.19H), 2.10 (s, 0.81H), 2.05 (s, 0.81H), 2.04 (s, 2.19H), 1.23 (d, J = 6.3 Hz, 0.81H), 1.21(d, J = 6.3 Hz, 2.19H). MS (ESI) calculated mass for the parent C19H24O8 380 [M], found 403 [M + Na].
(3R,4R,5R,6S)-4-hydroxy-6-methyltetrahydro-2H-pyran-2,3,5-triyl triacetate (19). 10% Pd/C (1.5 g) was added to 18 (3.15 g, 8.28 mmol) in EtOAc (200 mL). The mixture was stirred at room temperature under a H2 atmosphere for 2 h and filtered through a celite pad. The filtrate was evaporated and the residue was purified using silica gel flash chromatography to afford 19 as a white solid (2.14 g, 89%). 1H NMR (400 MHz, CDCl3): δ 6.10 (d, 0.26H, J = 2.0 Hz), 6.06 (d, 0.74H, J = 1.6 Hz), 5.25 (dd, 0.26H, J = 3.1 and 9.8 Hz), 5.17 (m, 0.26H), 5.09 (dd, 0.74H, J = 1.8 and 13.7 Hz), 4.90 (t, 0.74H, J = 9.8 Hz), 4.10–4.00 (m, 1H), 3.97–3.84 (m, 1H), 2.16 (s, 2.22H), 2.12 (s, 0.78H), 2.11 (s, 2.22H), 2.10 (s, 0.78H), 2.05 (s, 0.78H), 2.04 (s, 2.22H), 1.23 (d, J = 6.3 Hz, 0.78H), 1.21(d, J = 6.3 Hz, 2.22H). MS (ESI) calculated mass for the parent C12H18O8 290 [M], found 313 [M + Na].
(3R,4R,5S,6S)-6-methyl-4-(((trifluoromethyl)sulfonyl)oxy)tetrahydro-2H-pyran-2,3,5-triyl triacetate (20). Trifluoromethanesulfonic anhydride (0.33 mL, 1.94 mmol) was added to a mixture of compound 19 (508 mg, 1.75 mmol) and pyridine (0.22 mL) in dichloromethane (18 mL) at −18 °C. After stirring for 0.5 h, the mixture was warmed up to 0 °C and stirred for an additional 0.5 h. Water (50 mL) was added and the organic layer was separated. The aqueous layer was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with 10% H2SO4, sat. NaHCO3, and brine and dried over MgSO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford product 20 as a colorless oil (494 mg, 67%). 1H NMR (400 MHz, CDCl3): δ 6.06 (d, 1H, J = 2.0 Hz), 5.38 (dd, 1H, J = 2.0 and 3.5 Hz), 5.28 (t, 1H, J = 9.8 Hz), 5.18 (dd, 1H, J = 3.7 and 10.0 Hz), 3.92 (m, 1H), 2.21 (s, 3H), 2.18 (s, 3H), 2.15 (s, 3H), 1.27 (d, J = 6.3 Hz, 3H). 19F NMR (376 MHz, CDCl3): δ −75.0. MS (ESI) calculated mass for the parent C13H17F3O10S 422 [M], found 445 [M + Na].
(3R,4S,5R,6S)-4-hydroxy-6-methyltetrahydro-2H-pyran-2,3,5-triyl triacetate (21). Compound 20 (422 mg, 1.0 mmol) was dissolved in dry CH3CN (2 mL) and solid tetrabutylammonium nitrite (1.44 g, 5 mmol) was added. After stirring for 1 h at rt, the reaction mixture was evaporated. The residue was dissolved in CH2Cl2, washed with brine, and dried over MgSO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford product 21 as a white solid (87 mg, 30%). 1H NMR (400 MHz, CDCl3): δ 5.95 (d, 0.84H, J = 2.3 Hz), 5.91 (s, 0.16H), 5.10 (m, 0.16H), 5.02 (dd, 0.16H, J = 1.6 and 3.5 Hz), 5.00 (dd, 0.84H, J = 2.3 and 4.3 Hz), 4.89 (dd, 0.84H, J = 3.3 and 8.8 Hz), 4.27 (m, 0.84H), 4.12–4.06 (m, 1H), 3.74 (m, 0.16H), 2.15 (s, 0.48H), 2.13 (s, 0.84 × 6H), 2.12 (s, 0.84 × 3H), 2.11 (s, 0.48H), 2.10 (s, 0.48H), 1.33 (d, J = 6.7 Hz, 0.48H), 1.25(d, J = 6.7 Hz, 0.84 × 3H). MS (ESI) calculated mass for the parent C12H18O8 290 [M], found 313 [M + Na].
(3S,4R,5S,6S)-4-fluoro-6-methyltetrahydro-2H-pyran-2,3,5-triyl triacetate (22). DAST (0.12 mL, 0.90 mmol) was slowly added to a solution of 21 (26 mg, 0.090 mmol) in anhydrous CH2Cl2 (1 mL) at −40 °C. The reaction was stirred at room temperature for 24 h. After being cooled down to −20 °C, MeOH (0.2 mL) was added and the solvent was removed under reduced pressure. The residue was diluted with CH2Cl2 (30 mL), washed with water, and dried over MgSO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford product 22 as a colorless oil (14 mg, 54% yield). 1H NMR (400 MHz, CDCl3): δ 6.05 (d, 1H, J = 2.0 Hz), 5.19 (m, 1H), 4.87 (m, 1H), 4.38 (m, 1H), 4.26 (m, 1H), 2.13 (s, 3H), 2.12 (s, 3H), 2.11 (s, 3H), 1.36 (dd, 3H, J = 1.2 and 7.0 Hz). 19F NMR (376 MHz, CDCl3): δ −205.1. MS (ESI) calculated mass for the parent C12H17FO7 292 [M], found 292 [M].
(3S,4R,5S,6S)-4-fluoro-6-methyltetrahydro-2H-pyran-2,3,5-triol (23). NaOMe (10 mg, 0.19 mmol) was added to a suspension of 22 (14 mg, 0.048 mmol) in dry MeOH (1.7 mL). The mixture was stirred at room temperature for 15 h. Then, the reaction mixture was neutralized with Dowex (H+) resin, filtrated, concentrated, and purified using silica gel flash column chromatography to afford 23 as a white solid (3.2 mg, 56% yield). 1H NMR (400 MHz, CD3OD): δ 4.93 (d, 0.41H, J = 1.2 Hz), 4.90 (d, 0.36H, J = 2.0 Hz), 4.22 (m, 0.36H), 4.28 (m, 0.36H), 4.10–4.03 (m, 0.82H), 3.97 (m, 0.41H), 3.80 (m, 0.36H), 3.51 (m, 0.41H), 3.38 (m, 0.36H), 1.30 (dd, 1.26H, J = 1.0 and 6.9 Hz); 1.25 (dd, 1.08H, J = 1.9 and 6.7 Hz); 19F NMR (376 MHz, CDCl3): δ −201.6, −206.1; HRMS (ESI) calculated mass for the parent C6H11FO4 166.0641 [M], found 165.0565 [M − H].
(3R,4S,5S,6S)-6-methyl-4-(((trifluoromethyl)sulfonyl)oxy)tetrahydro-2H-pyran-2,3,5-triyl triacetate (24). Compound 24 (25 mg, 41% yield) was prepared using the same preparation procedure as compound 12. 1H NMR (400 MHz, CDCl3): δ 5.94 (d, 1H, J = 2.0 Hz), 5.35 (m, 1H), 5.09 (dd, 1H), 4.89 (dd, 1H), 4.42 (dd, 1H), 2.16 (s, 3H), 2.15 (s, 3H), 2.12 (s, 3H), 1.46 (d, 3H); 19F NMR (376 MHz, CDCl3): δ −74.9. HRMS (ESI) calculated mass for the parent C13H17F3O10S 422.0495 [M], found 445.0377 [M + Na].
(3S,5S,6R)-6-((trityloxy)methyl)tetrahydro-2H-pyran-2,3,4,5-tetrayl tetraacetate (26). Triphenylmethyl chloride (3.4 g, 12.2 mmol) was added to L-Mannose 25 (2.00 g, 11.1 mmol) in anhydrous pyridine (10 mL). The mixture was stirred at room temperature for 15 h. A total of 6 mL of Ac2O was added afterwards and the solution was stirred for another 15 h. The mixture was poured into ice-cold water and extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with brine and dried over Na2SO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford product 26 as a white solid (5.84 g, 89%). 1H NMR (400 MHz, CDCl3): δ 7.46–7.22 (m, 15 H), 6.10 (s, 0.7H), 5.85 (s, 0.3H), 5.52 (m, 1H), 5.43–5.52 (m, 2H), 3.91 (m, 0.7H), 3.64 (m, 0.3H), 3.34 (m, 1H), 3.18 (0.3H), 3.07 (m, 0.7H), 2.24 (s, 2.1H), 2.23 (s, 0.9H), 2.17 (s, 2.1H), 2.14 (s, 0.9H), 2.00 (s, 2.1H), 1.98 (s, 0.9H), 1.76 (s, 0.9H), 1.75 (s, 2.1H). MS (ESI) calculated mass for the parent C33H34O10 590 [M], found 613 [M + Na].
(3S,5S,6R)-6-(hydroxymethyl)tetrahydro-2H-pyran-2,3,4,5-tetrayl tetraacetate (27). 33% HBr in HOAc (1.6 mL) was added to the solution of compound 26 (4.60 g, 7.80 mmol) in glacial acetic acid (16 mL) at 10 °C. The mixture was stirred for 10 min. The formed triphenylmethyl bromide was immediately removed via filtration. The filtrate was diluted with cold water and extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with water and brine and dried over Na2SO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford product 27 as a white solid (2.23 g, 82%). 1H NMR (400 MHz, CDCl3): δ 6.09 (d, 0.67H, J = 1.6 Hz), 5.87 (d, 0.33H, J = 1.2 Hz), 5.49 (dd, 0.33H, J = 1.2 and 11.5 Hz), 5.40 (dd, 0.67H, J = 3.3 and 10.0 Hz), 5.33 (m, 0.67H), 5.27 (m, 1H), 5.17 (dd, 0.33H, J = 3.3 and 10.5 Hz), 3.85 (m, 0.67H), 3.73 (m, 1H), 3.66–3.58 (m, 1.33H), 2.21 (s, 0.99H), 2.17 (s, 2.01H), 2.16 (s, 2.01H), 2.10 (s, 0.99H), 2.08 (s, 2.01H), 2.04 (s, 0.99H), 2.02 (s, 2.01H), 2.01 (s, 0.99H). MS (ESI) calculated mass for the parent C14H20O10 348 [M], found 371 [M + Na].
(3S,4R,5R,6R)-6-(fluoromethyl)tetrahydro-2H-pyran-2,3,4,5-tetrayl tetraacetate (28). Compound 28 (30 mg, 28%) was prepared using the same preparation procedure as compound 22. 1H NMR (400 MHz, CDCl3): δ 6.11 (d, 0.57H, J = 2.0 Hz), 5.88 (d, 0.43H, J = 2.0 Hz), 5.49 (m, 0.43H), 5.38–5.35 (m, 2 × 0.57H), 5.31 (m, 0.43H), 5.26 (m, 0.57H), 5.15 (m, 0.43H), 4.56 (m, 1H), 4.44 (m, 1H), 4.02 (m, 0.57H), 3.80 (m, 0.43); 2.21 (s, 3 × 0.43 H), 2.17 (s, 3H), 2.16 (s, 3 × 0.57H) 2.11 (s, 3 × 0.43H), 2.07 (s, 3H), 2.01 (s, 3 × 0.57H); 19F NMR (376 MHz, CDCl3): δ −231.9, −232.4. MS (ESI) calculated mass for the parent C14H19FO9 350 [M], found 350 [M].
(3S,4R,5R,6R)-6-(fluoromethyl)tetrahydro-2H-pyran-2,3,4,5-tetraol (29). Compound 29 (10 mg, 87%) was prepared using the same preparation procedure as compound 23. 1H NMR (400 MHz, D2O/CD3OD): δ 5.18 (d, 0.6H, J = 2.0 Hz), 4.92 (d, 0.4H, J = 1.2 Hz), 4.78–4.57 (m, 2H), 3.93 (m, 1H), 3.87 (m, 1H), 3.77 (m, 1H), 3.68 (m, 1H). HRMS (ESI) calculated mass for the parent C6H11FO5 182.0591 [M], found 181.0521 [M − H].
3S,5S,6R)-6-((((trifluoromethyl)sulfonyl)oxy)methyl)tetrahydro-2H-pyran-2,3,4,5-tetrayl tetraacetate (30). Trifluoromethanesulfonic anhydride (0.37 mL, 2.2 mmol) was added to a mixture of compound 27 (696 mg, 2.0 mmol) and pyridine (0.25 mL) in dichloromethane (20 mL) at −10 °C. After stirring for 2 h, water (50 mL) was added. The organic layer was separated and the aqueous layer was extracted with dichloromethane (3 × 50 mL). The organic layers were combined, washed with 10% H2SO4, sat. NaHCO3, and brine, and dried over MgSO4. After the evaporation of solvents, the residue was purified using silica gel flash chromatography to afford product 30 as a white solid (826 mg, 86%). 1H NMR (400 MHz, CDCl3): δ 6.12 (d, 0.62H, J = 2.0 Hz), 5.89 (d, 0.38H, J = 1.2 Hz), 5.49 (dd, 0.38H, J = 1.2 and 3.1 Hz), 5.39 (dd, 0.62H, J = 3.1 and 10.2 Hz), 5.33 (m, 0.62H), 5.30 (m, 0.38H), 5.26 (dd, 0.62H, J = 1.2 and 11.5 Hz), 5.16 (dd, 0.38H, J = 3.1 and 9.8 Hz), 4.58–4.54 (m, 2H), 4.14 (m, 0.62H), 3.92 (m, 0.38), 2.22 (s, 1.14H), 2.19 (s, 1.86H × 2), 2.12 (s, 1.14H), 2.10 (s, 1.86H), 2.05 (s, 1.14H), 2.03 (s, 1.86H), 2.02 (s, 1.14H). 19F NMR (376 MHz, CDCl3): δ −74.3, −74.4. HRMS (ESI) calculated mass for the parent C15H19F3O12S 480.0549 [M], found 503.0433 [M + Na].













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}