

Chiral Indolizinium Salts Derived from 2-Pyridinecarbaldehyde—First Diastereoselective Syntheses of (−)-1-epi-lentiginosine
 , , and
, , and
Abstract
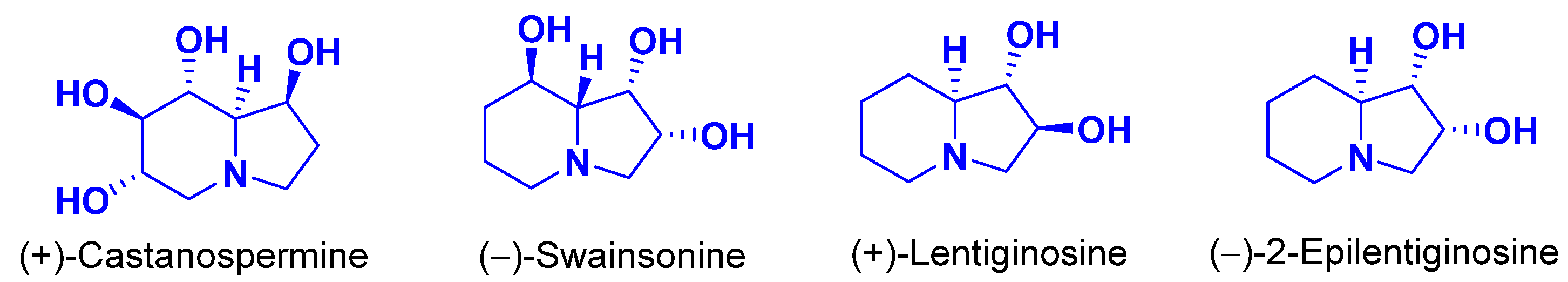
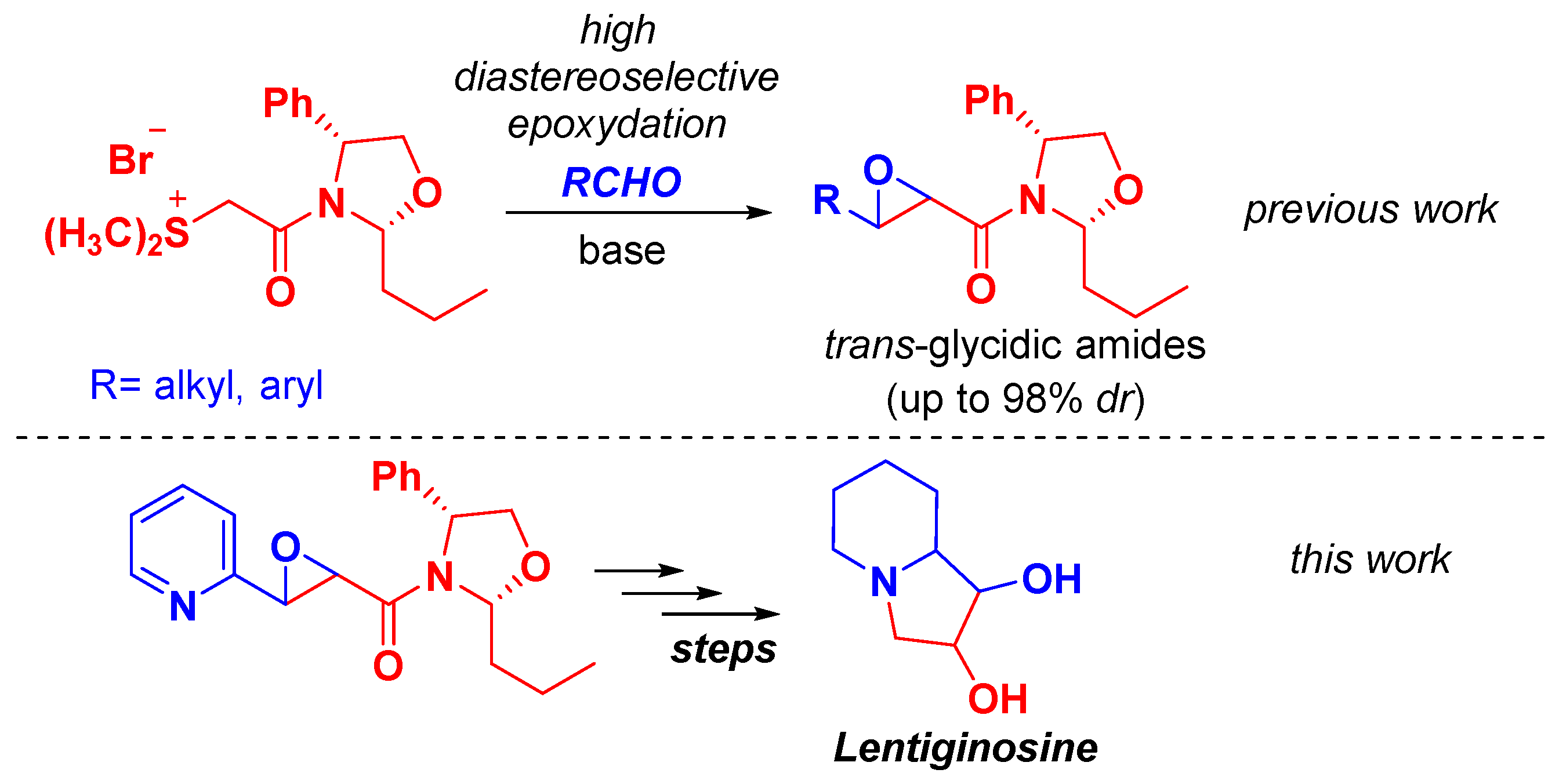
1. Introduction
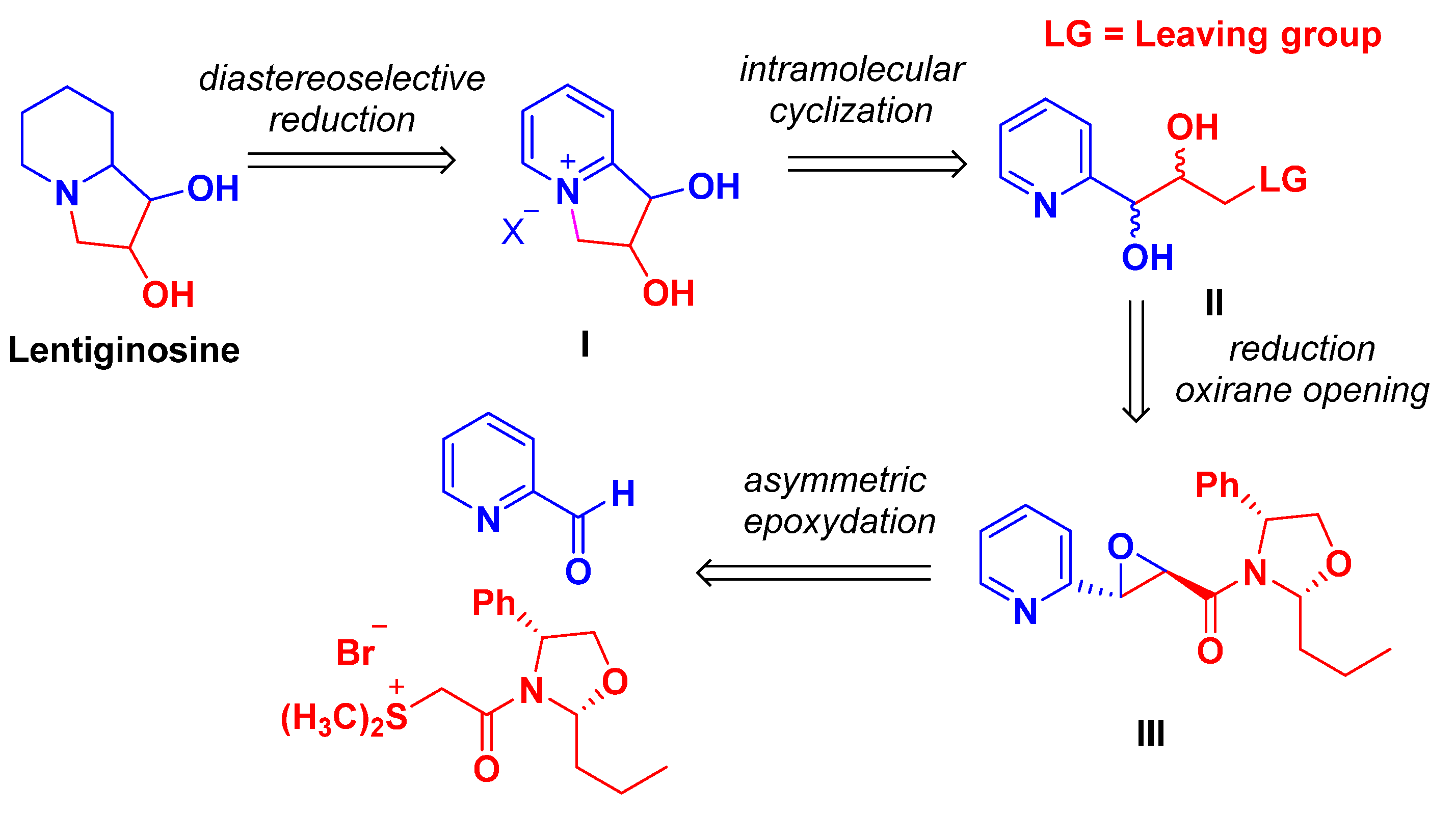
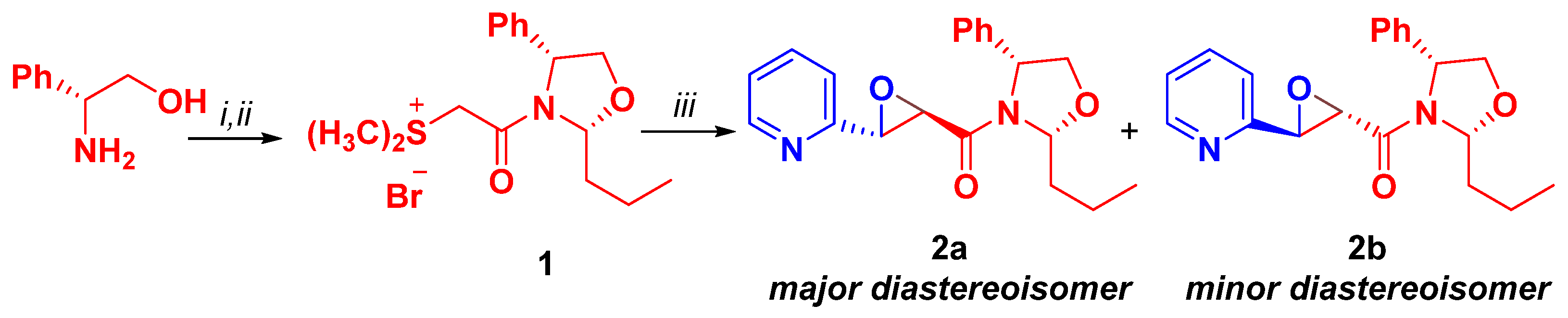
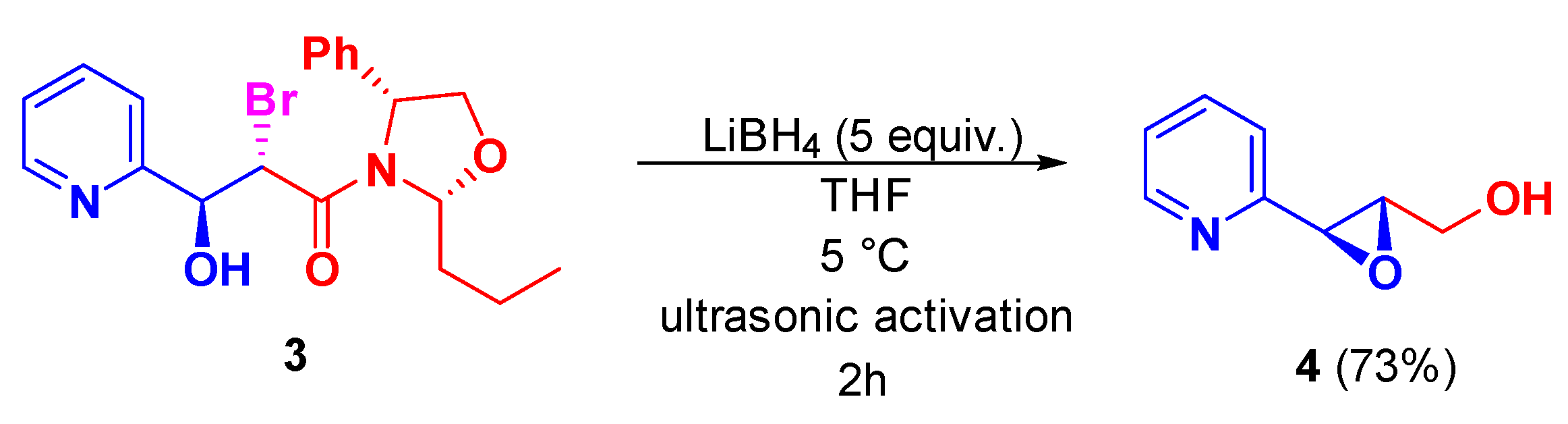
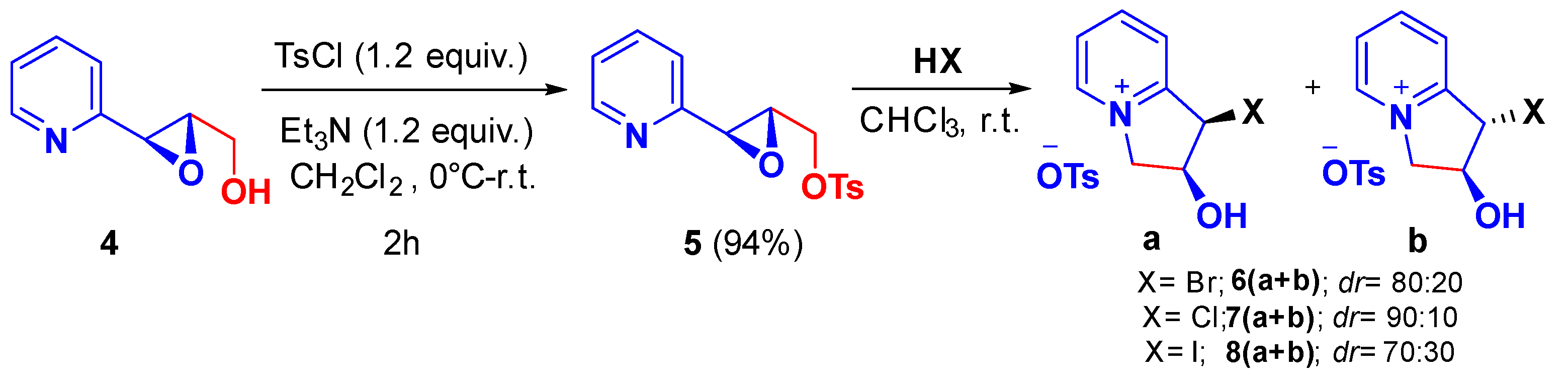
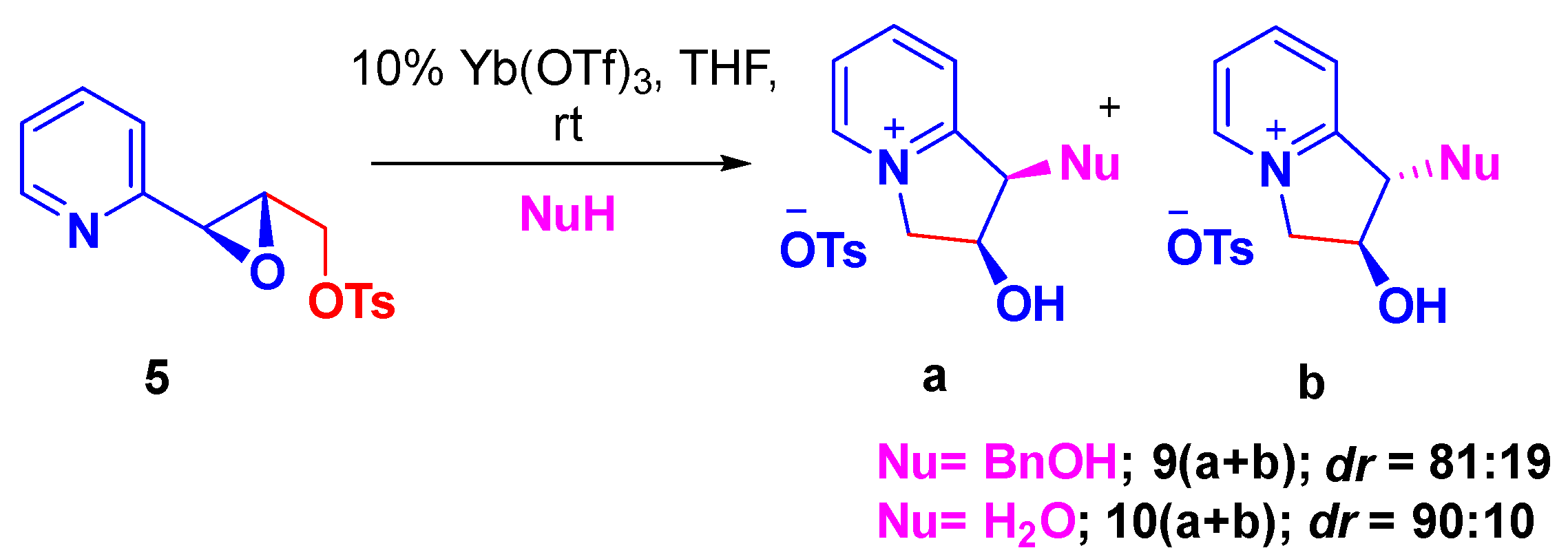
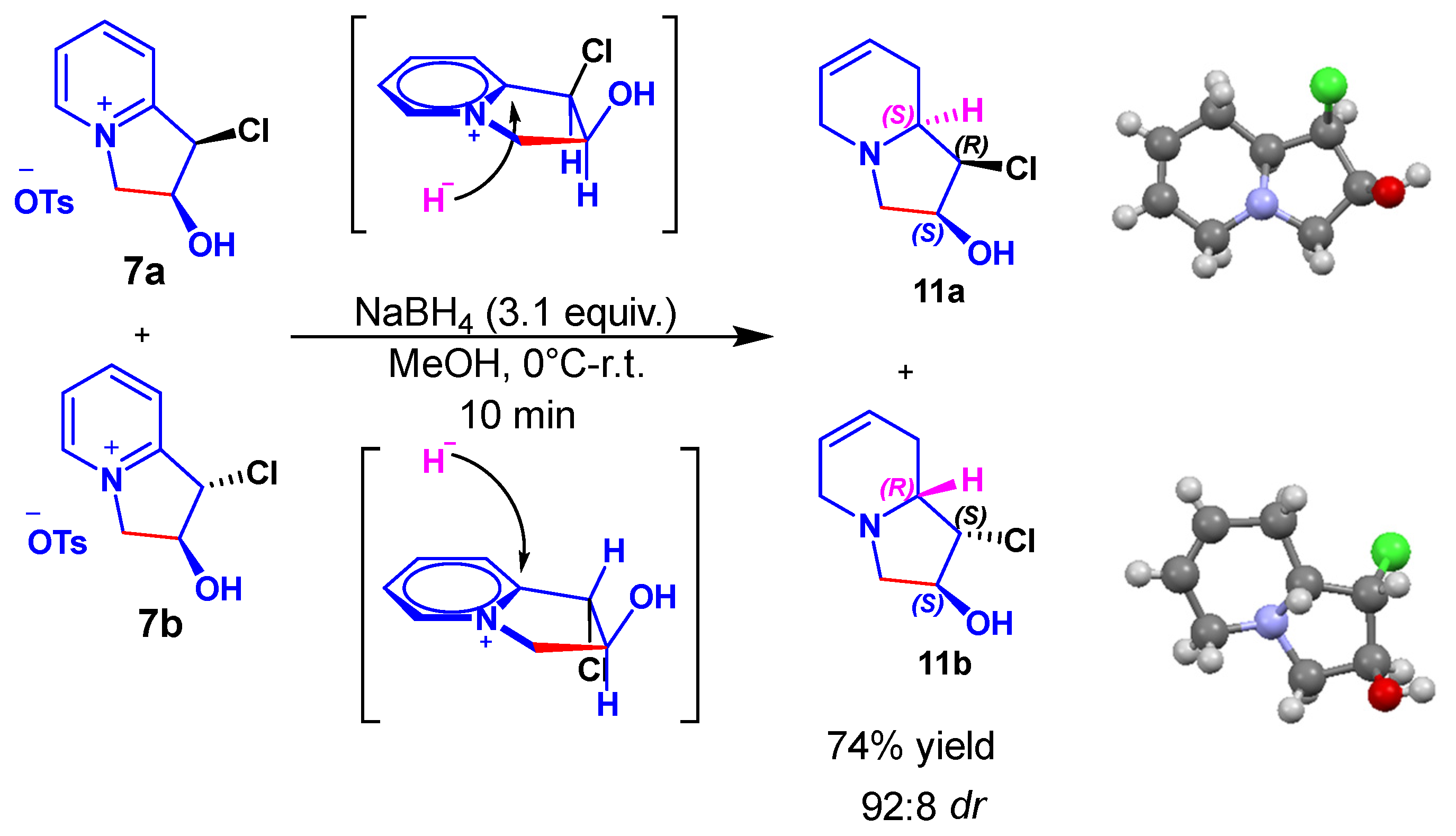
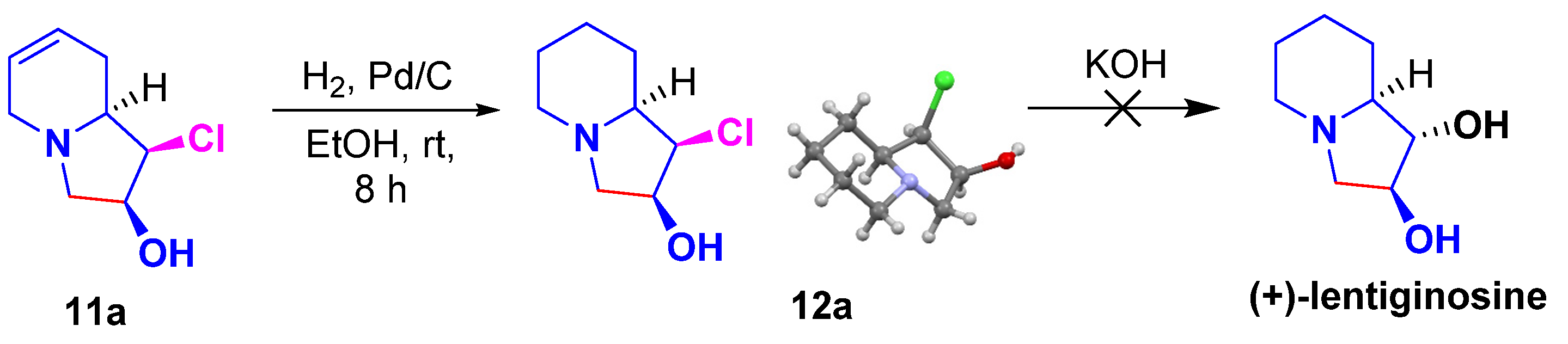
2. Results
3. Materials and Methods
3.1. General Information
3.2. General Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Macchi, B.; Minutolo, A.; Grelli, S.; Cardona, F.; Cordero, F.M.; Mastino, A.; Brandi, A. The novel proapoptotic activity of nonnatural enantiomer of Lentiginosine. Grycobiology 2010, 20, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Asano, N. Glycosidase-Inhibiting Alkaloids: Isolation, Structure, and Application. In Modern Alkaloids: Structure, Isolation, Synthesis and Biology; Fattorusso, E., Taglialatela-Scafati, O., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 111–138. [Google Scholar]
- Pastuszak, I.; Molyneux Russell, J.; James, L.F.; Elbein, A.D. Lentiginosine, a dihydroxyindolizidine alkaloid that inhibits amyloglucosidase. Biochemistry 1990, 29, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Brandi, A.; Cicchi, S.; Cordero, F.M.; Fringnoli, R.; Goti, A.; Picasso, S.; Vogel, P. Assignment of the Absolute Configuration of Natural Lentiginosine by Synthesis and Enzymatic Assays of Optically Pure (+) and (−)-Enantiomers. J. Org. Chem. 1995, 60, 6806–6812. [Google Scholar] [CrossRef]
- Dal Piaz, F.; Vassallo, A.; Chini, M.G.; Cordero, F.M.; Cardona, F.; Pisano, C.; Bifulco, G.; Detommasi, N.; Brandi, A. Natural Iminosugar (+)-Lentiginosine Inhibits ATPase and Chaperone Activity of Hsp90. PLoS ONE 2012, 7, e43316. [Google Scholar] [CrossRef]
- Minutolo, A.; Grelli, S.; Marino-Merlo, F.; Cordero, F.M.; Brandi, A.; Macchi, B.; Mastino, A. D(−)lentiginosine-induced apoptosis involves the intrinsic pathway and is p53-independent. Cell Death Dis. 2012, 3, e358. [Google Scholar] [CrossRef] [PubMed]
- Yoda, H.; Kitayama, H.; Katagiri, T.; Takabe, K. Synthesis of natural lentiginosine employing a cyclic imide with C2-symmetry derived from L-tartaric acid. Tetrahedron Asymmetry 1993, 4, 1455–1456. [Google Scholar] [CrossRef]
- Ha, D.C.; Yun, C.S.; Lee, Y. Samarium Diiodide-Promoted Cyclization of N-(ω-Iodoalkyl)imides to Polyhydroxylated Indolizidinones and Pyrrolizidinones: Synthesis of (+)-Lentiginosine. J. Org. Chem. 2000, 65, 621–623. [Google Scholar] [CrossRef]
- Yoda, H.; Katoh, H.; Ujihara, Y.; Takabe, K. SmI2-mediated hetero-coupling reaction of lactams with aldehydes; synthesis of indolizidine alkaloids, (−)-δ-coniceine, (+)-5-epiindolizidine 167B and (+)-lentiginosine. Tetrahedron Lett. 2001, 42, 2509–2512. [Google Scholar] [CrossRef]
- Klitzke, C.F.; Pilli, R.A. Enhanced trans diastereoselection in the allylation of cyclic chiral N-acyliminium ions. Synthesis of hydroxylated indolizidines. Tetrahedron Lett. 2001, 42, 5605. [Google Scholar] [CrossRef]
- El-Nezhawy, A.O.H.; El-Diwani, H.I.; Schmidt, R.R. O-(2-Oxopyrrolidin-5-yl)trichloroacetimidates as Amidoalkylating Agents—Synthesis of (+)-Lentiginosine. Eur. J. Org. Chem. 2002, 2002, 4137–4142. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ito, Y.; Nishiyama, T.; Isobe, M. Stereoselective Allyl Amine Synthesis through Enantioselective Addition of Diethylzinc and [1,3]-Chirality Transfer: Synthesis of Lentiginosine and Polyoxamic Acid Derivative. Chem. Eur. J. 2005, 11, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, Q.; Zhang, H.-K.; Chen, A. Practical synthesis of trans-dihydroxybutyrolactols as chiral C4 building blocks and their application to the synthesis of polyhydroxylated alkaloids. RSC Adv. 2013, 3, 20298–20307. [Google Scholar] [CrossRef]
- Cordero, L.F.; Cicchi, S.; Goti, A.; Brandi, A. Synthesis of lentiginosine by stereoselective chiral nitrone cycloaddition and thermal rearrangement of strained spiroisoxazolidine. Tetrahedron Lett. 1994, 35, 949–952. [Google Scholar] [CrossRef]
- Socha, D.; Jurczak, M.; Chmielewski, M. Synthesis of polyhydroxyindolizidines from 5,6-dihydro-2H-pyran-2-one. Carbohydr. Res. 2001, 336, 315–318. [Google Scholar] [CrossRef]
- McCaig, A.E.; Meldrum, K.P.; Wightman, R.H. Synthesis of trihydroxylated pyrrolizidines and indolizidines using cycloaddition reactions of functionalized cyclic nitrones, and the synthesis of (+)- and (−)-lentiginosine. Tetrahedron 1998, 54, 9429–9446. [Google Scholar] [CrossRef]
- Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. New Concise Total Synthesis of (+)-Lentiginosine and Some Structural Analogues. J. Org. Chem. 2005, 70, 6552–6555. [Google Scholar] [CrossRef]
- Yoda, H.; Kawauchi, M.; Takabe, K. Novel Asymmetric Synthesis of an Indolizidine Alkaloid, (+)-Lentiginosine Employing Highly Stereoselective Hydrogenation of α-Hydroxypyrrolidine. Synlett 1998, 1998, 137–138. [Google Scholar] [CrossRef]
- Chandra, K.L.; Chandrasekhar, M.; Singh, V.K. Total Synthesis of (−)- and (+)-Lentiginosine. J. Org. Chem. 2002, 67, 4630–4633. [Google Scholar] [CrossRef]
- Casiraghi, G.; Spanu, P.; Rassu, G.; Pinna, L.; Ulgheri, F. Total Synthesis of All Four Isomers of cis-1,2-Dihydroxypyrrolizidine. J. Org. Chem. 1994, 59, 2906–2909. [Google Scholar] [CrossRef]
- Chaudhari, V.D.; Ajish Kumar, K.S.; Dhavale, D.D. Synthesis of (−)-lentiginosine, its 8a-epimer and dehydroxylated pyrrolizidine alkaloid from d-glucose. Tetrahedron 2006, 62, 4349–4354. [Google Scholar] [CrossRef]
- Kim, I.S.; Zee, O.P.; Jung, Y.H. Regioselective and Diastereoselective Amination of Polybenzyl Ethers Using Chlorosulfonyl Isocyanate: Total Syntheses of 1,4-Dideoxy-1,4-imino-D-arabinitol and (−)-Lentiginosine. Org. Lett. 2006, 8, 4101–4104. [Google Scholar] [CrossRef]
- Angle, S.R.; Bensa, D.; Belanger, D.S. New Access to Indolizidine and Pyrrolizidine Alkaloids from and Enantiopure Proline: Total Syntheses of (−)-Lentiginosine and (1R,2R,7aR)-Dihydroxypyrrolizidine. J. Org. Chem. 2007, 72, 5592–5597. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, R.; Kokatla, H.P.; Vankar, Y.D. An improved method of ring closing metathesis in the presence of basic amines: Application to the formal synthesis of (+)-lentiginosine and other piperidines and carbamino sugar analogs. Tetrahedron Lett. 2011, 52, 781–786. [Google Scholar] [CrossRef]
- Kamal, A.; Vangala, S.R. An expedient total synthesis of optically active piperidine and indolizidine alkaloids (–)-β-conhydrine and (−)-lentiginosine. Tetrahedron 2011, 67, 1341–1347. [Google Scholar] [CrossRef]
- Kim, I.S.; Li, Q.R.; Dong, G.R.; Kim, Y.C.; Hong, Y.J.; Lee, M.; Chi, K.-W.; Oh, J.S.; Jung, Y.H. A Facile Synthesis of Lentiginosine Analogues Based on a Highly Regio- and Diastereoselective Allylic Amination Using Chlorosulfonyl Isocyanate. Eur. J. Org. Chem. 2010, 2010, 1569–1573. [Google Scholar] [CrossRef]
- Gurjar, M.K.; Ghosh, L.; Syamala, M.; Jayasree, V. Stereoselective synthesis of (+)- and (−)-lentiginosine. Tetrahedron Lett. 1994, 35, 8871–8872. [Google Scholar] [CrossRef]
- Nukui, S.; Sodeoka, M.; Sasai, H.; Shibasaki, M. Regio- and Stereoselective Functionalization of Optically Active Tetrahydroindolizidine Derivative. Catalytic Asymmetric Syntheses of Lentiginosine, 1,2-Diepilentiginosine, and Gephyrotoxin 209D. J. Org. Chem. 1995, 60, 398–404. [Google Scholar] [CrossRef]
- Rasmussen, M.O.; Delair, P.; Greene, A.E. Enantiocontrolled Preparation of Indolizidines: Synthesis of (−)-2-Epilentiginosine and (+)-Lentiginosine. J. Org. Chem. 2001, 66, 5438–5443. [Google Scholar] [CrossRef]
- Lim, S.H.; Ma, S.; Beak, P. Asymmetric Syntheses of Fused Bicyclic Lactams. J. Org. Chem. 2001, 66, 9056–9062. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-X.; Zhou, W.-S. A novel concise total synthesis of (+)-lentiginosine. Tetrahedron Lett. 2003, 44, 497–498. [Google Scholar] [CrossRef]
- Ayad, T.; Genisson, Y.; Baltas, M. Asymmetric syntheses of (−)-lentiginosine and an original pyrrolizidinic analogue thereof from a versatile epoxyamine intermediate. Org. Biomol. Chem. 2005, 3, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-W.; Hsu, H.-C.; Chang, C.-H.; Tsai, H.-H.T.; Hou, D.-R. Asymmetric Synthesis of (−)-Lentiginosine by Double Aza-Michael Reaction. Eur. J. Org. Chem. 2010, 2010, 4771–4773. [Google Scholar] [CrossRef]
- Shao, J.; Yang, J.-S. A Diastereoselective Cyclic Imine Cycloaddition Strategy To Access Polyhydroxylated Indolizidine Skeleton: Concise Syntheses of (+)-/(−)-Lentiginosine and (−)-2-epi-Steviamine. J. Org. Chem. 2012, 77, 7891–7900. [Google Scholar] [CrossRef]
- Azzouz, R.; Fruit, C.; Bischoff, L.; Marsais, F. A Concise Synthesis of Lentiginosine Derivatives Using a Pyridinium Formation via the Mitsunobu Reaction. J. Org. Chem. 2008, 73, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Giomi, D.; Alfini, R.; Micoli, A.; Calamai, E.; Faggi, C.; Brandi, A. Synthesis of 1,2-Dihydroxyindolizidines from 1-(2-Pyridyl)-2-propen-1-ol. J. Org. Chem. 2011, 76, 9536–9541. [Google Scholar] [CrossRef] [PubMed]
- Gordillo, P.G.; Aparicio, D.M.; Flores, M.; Mendoza, A.; Orea, L.; Juárez, J.R.; Huelgas, G.; Gnecco, D.; Terán, J.L. Oxazolidine Sulfur Ylides Derived from Phenylglycinol for the Specific and Highly Diastereoselective Synthesis of Aryl and Alkyl trans-Epoxyamides. Eur. J. Org. Chem. 2013, 2013, 5561–5565. [Google Scholar] [CrossRef]
- CCDC: 2181510 (3), 2181511 (11a), 2181512 (11b) and 2181513 (12a). See the ESI for Details. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 5 April 2023).
- Zapata-Machin, E.; Castellan, T.; Baudoin-Dehoux, C.; Génisson, Y. Asymmetric Synthesis of Two Hydroxylated Pyrrolizidines From a C-Allyl Epoxypyrrolidine. Synth. Commun. 2015, 45, 645–652. [Google Scholar] [CrossRef]
- Stoe & Cie. X-AREA and X-RED32; Stoe & Cie: Darmstadt, Germany, 2019. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, C71, 3–8. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
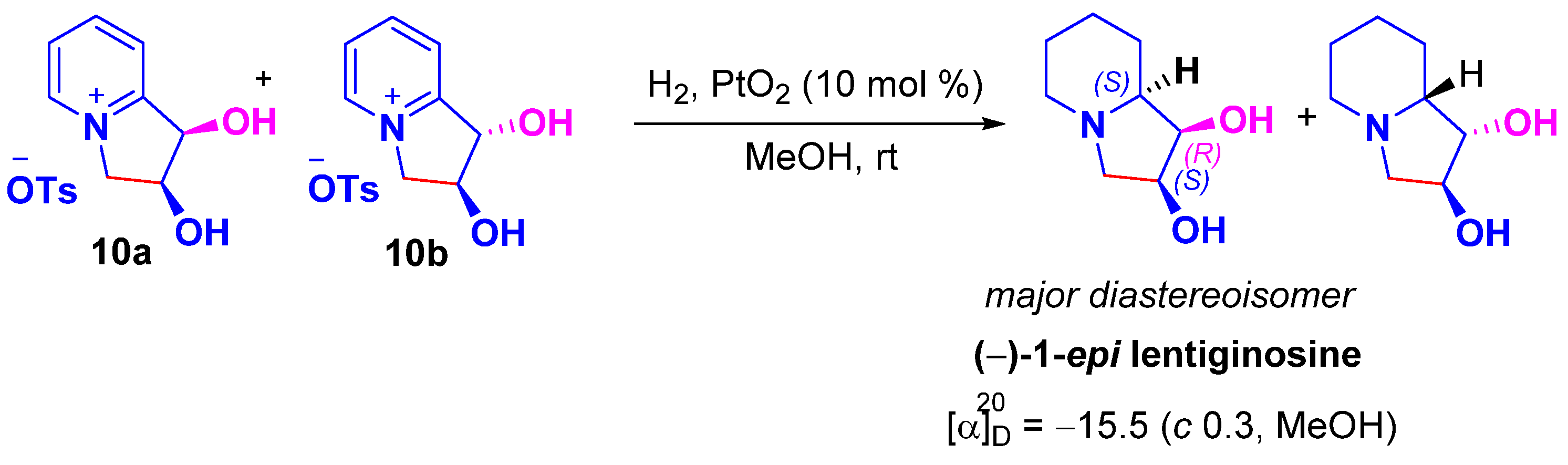{kind=link}
 | |||
|---|---|---|---|
| Entry | Solvent | Time | Yield [%] |
| 1 | THF | 3.5 h | 95 |
| 2 | CHCl3 | 1 h | 98 |
| 3 | Toluene | 3 h | 95 |
| 4 | CCl4 | 40 min | 80 |
| 5 | MeOH | 72 h | NR |
| 6 | CH3CN | 20 h | 80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Matsui, H.; Aparicio, D.M.; Orea, M.L.; Juárez, J.R.; Gómez-Calvario, V.; Gnecco, D.; Carrasco-Carballo, A.; Terán, J.L. Chiral Indolizinium Salts Derived from 2-Pyridinecarbaldehyde—First Diastereoselective Syntheses of (−)-1-epi-lentiginosine. Molecules 2023, 28, 3719. https://doi.org/10.3390/molecules28093719
Rodriguez-Matsui H, Aparicio DM, Orea ML, Juárez JR, Gómez-Calvario V, Gnecco D, Carrasco-Carballo A, Terán JL. Chiral Indolizinium Salts Derived from 2-Pyridinecarbaldehyde—First Diastereoselective Syntheses of (−)-1-epi-lentiginosine. Molecules. 2023; 28(9):3719. https://doi.org/10.3390/molecules28093719
Chicago/Turabian StyleRodriguez-Matsui, Hisami, David M. Aparicio, María L. Orea, Jorge R. Juárez, Victor Gómez-Calvario, Dino Gnecco, Alan Carrasco-Carballo, and Joel L. Terán. 2023. "Chiral Indolizinium Salts Derived from 2-Pyridinecarbaldehyde—First Diastereoselective Syntheses of (−)-1-epi-lentiginosine" Molecules 28, no. 9: 3719. https://doi.org/10.3390/molecules28093719
APA StyleRodriguez-Matsui, H., Aparicio, D. M., Orea, M. L., Juárez, J. R., Gómez-Calvario, V., Gnecco, D., Carrasco-Carballo, A., & Terán, J. L. (2023). Chiral Indolizinium Salts Derived from 2-Pyridinecarbaldehyde—First Diastereoselective Syntheses of (−)-1-epi-lentiginosine. Molecules, 28(9), 3719. https://doi.org/10.3390/molecules28093719