
Tuning the Biological Activity of PI3Kδ Inhibitor by the Introduction of a Fluorine Atom Using the Computational Workflow

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
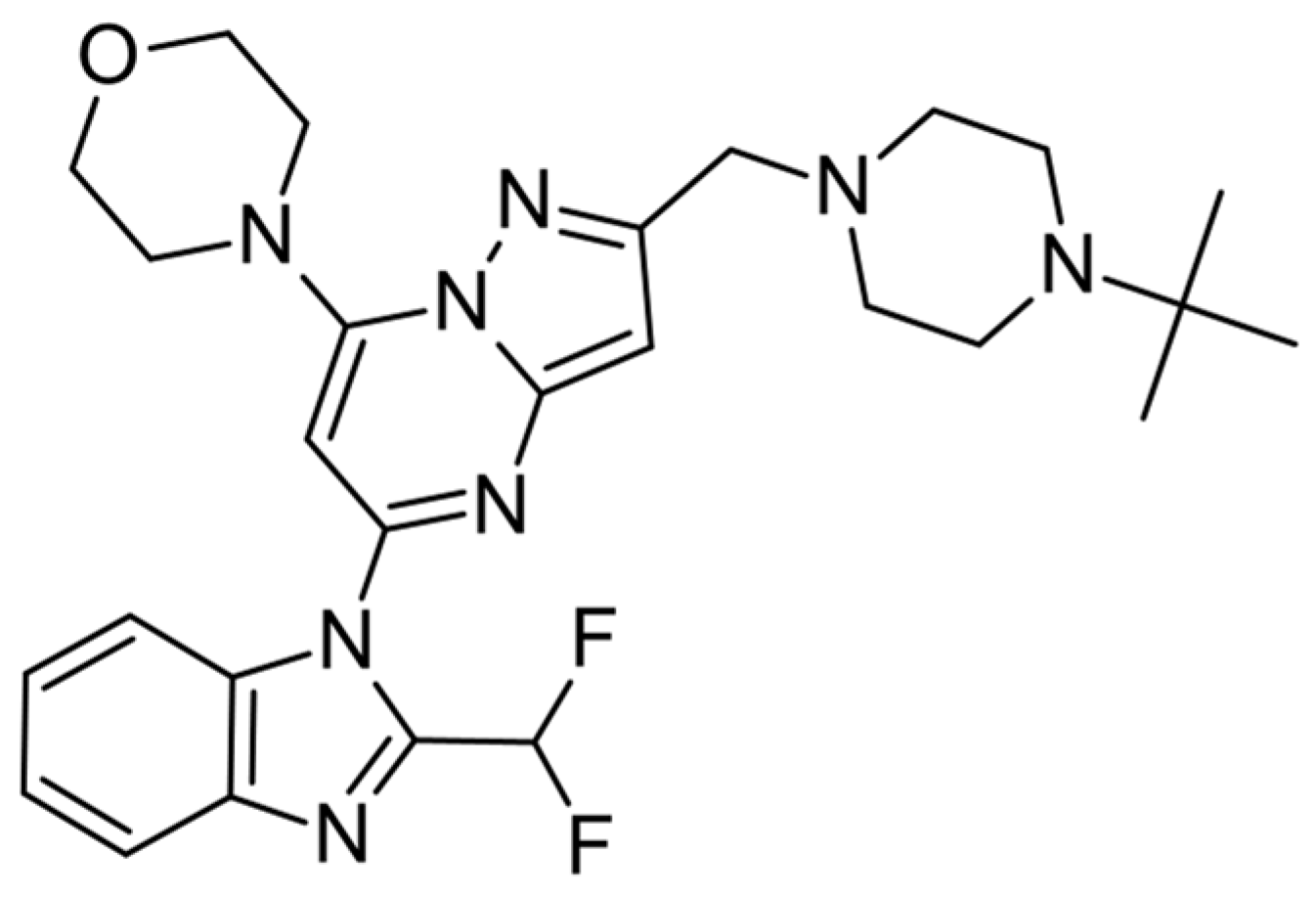
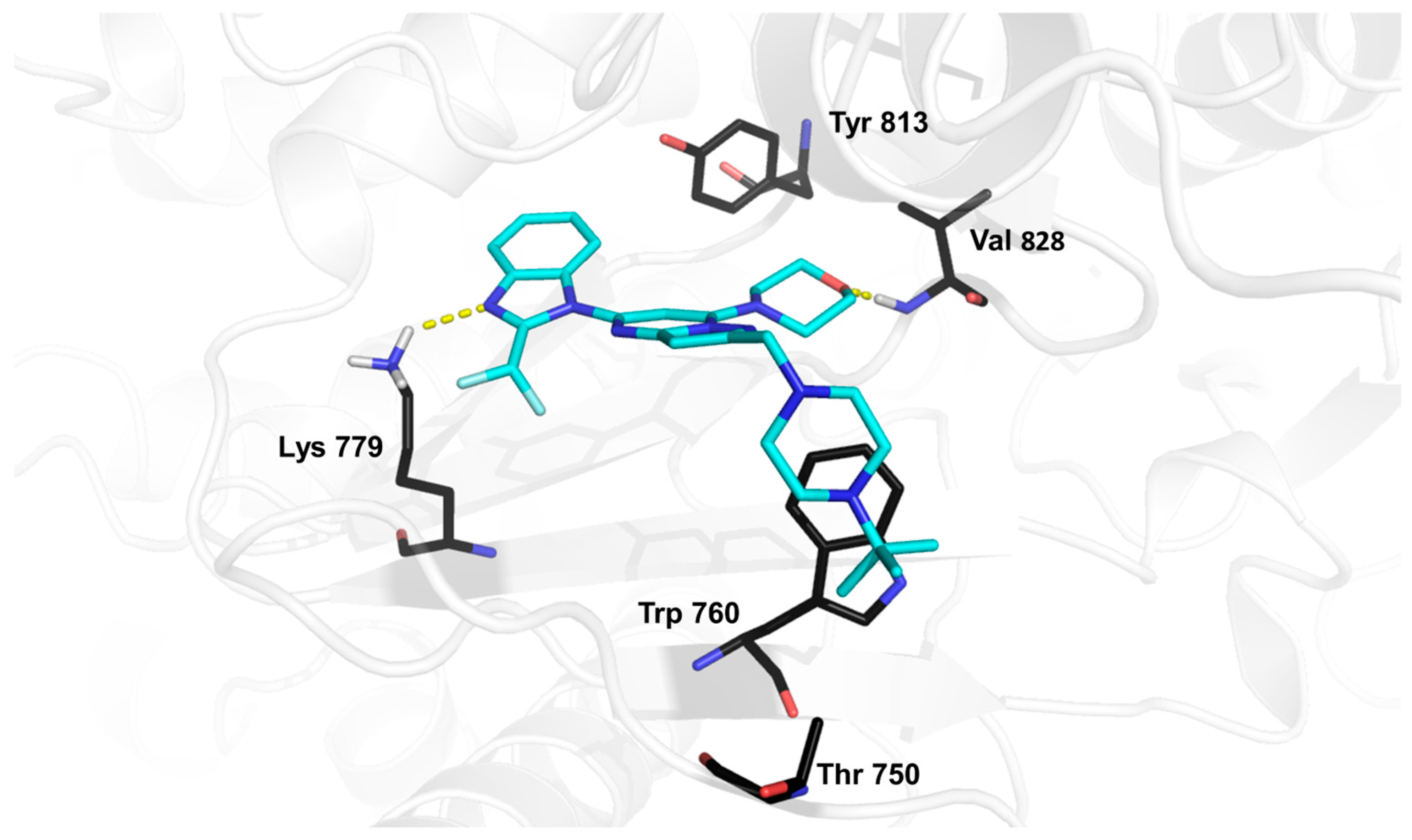
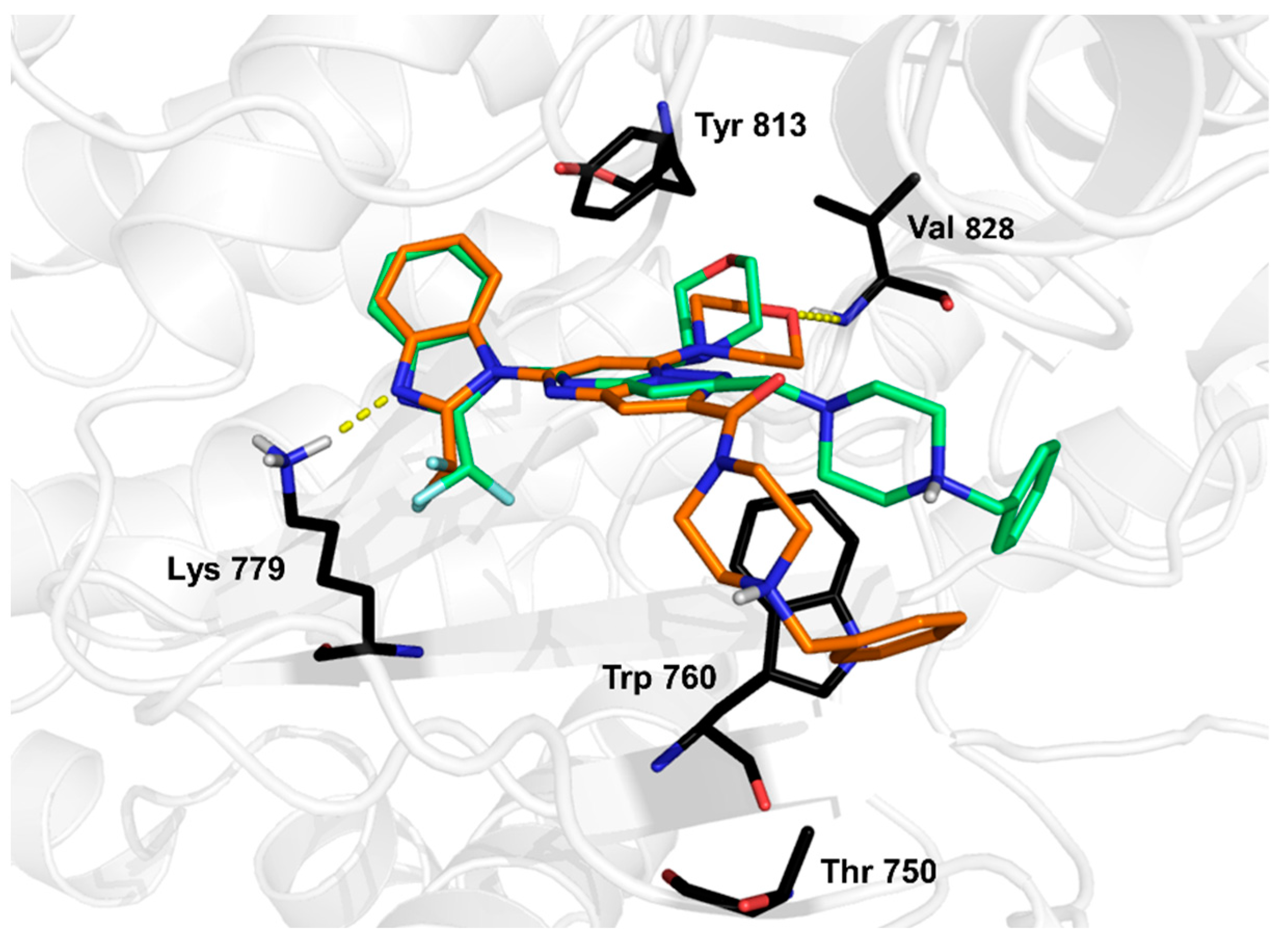
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.2. General Information
3.3. Synthesis
3.3.1. General Procedure for the Buchwald–Hartwig Reaction
- 1-{2-[(4-Tert-butylpiperazin-1-yl)methyl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl}-2-(trifluoromethyl)-1H-benzimidazole (3).
- Compound 3 was synthesized from 4-{2-[(4-tert-butylpiperazin-1-yl)methyl]-5-chloropyrazolo[1,5-a]pyrimidin-7-yl}morpholine (0.88 g, 2.04 mmol), 2-(trifluoromethyl)-benzimidazole (0.57 g, 3.06 mmol), tris(dibenzylideneacetone)dipalladium (93.3 mg, 0.102 mmol), 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene (124.0 mg, 0.204 mmol), cesium carbonate (1.34 g, 4.08 mmol), and toluene (8.0 mL), according to the general procedure for the Buchwald–Hartwig reaction. The resulting crude product was purified by flash chromatography (0–100% AcOEt gradient in heptane, amine-functionalized gel column) to obtain compound 3 as a white solid (38.0 mg, 0.07 mmol) with a 3% yield.
- 1H NMR (600 MHz, CDCl3) δ 7.94–7.93 (m, 1H, Ar-H), 7.55–7.53 (m, 1H, Ar-H), 7.46–7.42 (m, 2H, Ar-H), 6.63 (s, 1H, Ar-H), 6.16 (s, 1H, Ar-H), 3.98–3.97 (m, 4H, morph.), 3.90–3.89 (m, 4H, morph.), 3.82 (s, 2H, CH2), 2.67 (d, J = 2.1 Hz, 8H, piperaz.), and 1.13–1.06 (m, 9H, t-Bu.).
- 13C{1H, 19F}NMR (151 MHz, CDCl3) δ 151.2, 150.2, 147.0, 141.0, 139.9, 135.4, 126.3, 124.5, 121.7, 119.7, 118.0, 111.9, 97.2, 88.1, 66.2, 56.3, 53.8, 48.6, 45.7, 45.0, 29.7, 25.9, and 25.8.
- HRMS (ESI/MS): m/z calculated for C27H33F3N8O [M + H]+ 543.2802, found 543.2806.
- 1-{2-[(4-Tert-butylpiperazin-1-yl)methyl]-3-chloro-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl}-2-(difluoromethyl)-1H-benzimidazole (4).
- To the solution of 1-{2-[(4-tert-butylpiperazin-1-yl)methyl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl}-2-(difluoromethyl)-1H-benzimidazole (230.0 mg, 0.44 mol, 1.1 eq) in dichloromethane (DCM) (4 mL), N-chlorosuccinimide (64.4 mg, 0.48 mmol) was added. The reaction mixture was stirred for 1 h at room temperature. Then, sodium metabisulfite (3 mL) and water (5 mL) were added and the aqueous mixture was extracted with DCM (3 × 5 mL). The combined organic extracts were washed with water, dried over Na2SO4, filtered, and concentrated. The resulting crude product was purified by flash chromatography (0–100% ethyl acetate gradient in heptane, amine-functionalized gel column) and crystallization (AcOEt) to obtain compound 4 (134.0 mg; 0.24 mmol) as a white solid with a 55% yield.
- 1H NMR (600 MHz, CDCl3) δ 7.92–7.91 (m, 1H, Ar-H), 7.70–7.69 (m, 1H, Ar-H), 7.46–7.40 (m, 2H, Ar-H), 7.31 (t, J = 53.6 Hz, 1H, CHF2), 6.35 (s, 1H, Ar-H), 3.98–3.96 (m, 4H, morph.), 3.94–3.91 (m, 6H, morph.), 2.71–2.65 (m, 8H), and 1.10 (s, 9H, t-Bu.).
- 13C{1H, 19F}NMR (151 MHz, CDCl3) δ 151.3, 150.4, 148.1, 145.2, 144.7, 141.9, 134.5, 125.9, 124.3, 121.6, 111.8, 109.4 (CF2), 87.7, 66.2, 53.3, 48.7, 45.7, and 25.9 (t-Bu.).
- HRMS (ESI/MS): m/z calculated for C27H33ClF2N8O [M + H]+ 558.2434, found 538.2442.
- 1-{3-Bromo-2-[(4-tert-butylpiperazin-1-yl)methyl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl}-2-(difluoromethyl)-1H-benzimidazole (5).
- To the solution of 1-{2-[(4-tert-butylpiperazin-1-yl)methyl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl}-2-(difluoromethyl)-1H-benzimidazole (500.0 mg, 0.94 mol) in DCM (7 mL), N-bromosuccinimide (204.0 mg, 1.13 mmol, and 1.2 eq) was added. The reaction mixture was stirred for 1 h at room temperature. Then, sodium metabisulfite (5 mL) and water (10 mL) were added and the aqueous mixture was extracted with DCM (3 × 10 mL). The combined organic extracts were washed with water, dried over Na2SO4, filtered, and concentrated. The resulting crude product was purified by flash chromatography (0–100% ethyl acetate gradient in heptane, amine-functionalized gel column) and crystallization (AcOEt) to obtain compound 5 (370.0 mg; 0.61 mmol) as a white solid with a 65% yield.
- 1H NMR (600 MHz, CDCl3) δ 7.92–7.91 (m, 1H, Ar-H), 7.71–7.70 (m, 1H, Ar-H), 7.46–7.40 (m, 2H, Ar-H), 7.39 (t, J = 53.5 Hz, 1H, CHF2), 6.37 (s, 1H, Ar-H), 3.98–3.96 (m, 4H, morph.), 3.94–3.92 (m, 4H, morph.), 3.90 (s, 2H, CH2), 2.81–2.57 (m, 8H), and 1.08 (s, 9H, t-Bu.).
- 13C{1H, 19F}NMR (151 MHz, CDCl3) δ 151.9, 151.4, 148.4, 146.6, 144.8, 141.9, 134.5, 125.9, 124.3, 121.6, 111.8, 109.4 (CF2), 87.7, 66.2, 53.6, 48.7, 45.7, and 25.8 (t-Bu.).
- HRMS (ESI/MS): m/z calculated for C27H33BrF2N8O [M + H]+ 602.1929, found 602.1936.
3.3.2. General Procedure for the Reductive Amination Reaction
- 1-{2-[(4-Benzylpiperazin-1-yl)methyl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl}-2-(difluoromethyl)-1H-benzimidazole (6).
- Compound 6 was prepared from aldehyde 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (0.15 g, 0.37 mmol), 1-benzylpiperazine (0.80 g, 0.45 mmol) as an amine, DCM (3.0 mL), and sodium triacetoxyborohydride (0.12 g, 0.56 mmol), according to the general procedure for reductive amination reaction. The resulting crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to obtain compound 6 (0.1 g, 0.18 mmol) with a 47% yield.
- 1H NMR (600 MHz, DMSO-d6) δ 7.88–7.87 (m, 1H, Ar-H), 7.81 (d, J = 7.6 Hz, 1H, Ar-H), 7.58 (t, J = 52.5 Hz, 1H, CHF2), 7.47–7.41 (m, 2H, Ar-H), 7.31–7.26 (m, 4H, Ar-H), 7.24–7.21 (m, 1H, Ar-H), 6.65 (s, 1H, Ar-H), 6.52 (s, 1H, Ar-H), 3.93–3.91 (m, 4H, morph.), 3.83 (t, J = 4.6 Hz, 4H, morph.), 3.68 (s, 2H, CH2), 3.44 (s, 2H, CH2), 2.50–2.48 (m, 4H, piperaz.), and 2.39–2.37 (m, 4H, piperaz.).
- 13C NMR (151 MHz, DMSO-d6) δ 154.9, 150.8, 149.6, 147.0, 144.6, 141.2, 138.2, 134.0, 128.7, 128.1, 126.8, 125.4, 123.9, 120.6, 112.4, 108.5, 95.4, 87.8, 65.5, 62.0, 55.7, 52.6, 52.6, and 48.1.
- HRMS (ESI/MS): m/z calculated for C30H32F2N8O [M + H]+ 558.2667, found 558.2681.
- 1-[2-({4-[(2-Fluorophenyl)methyl]piperazin-1-yl}methyl)-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl]-2-(propan-2-yl)-1H-benzimidazole (7).
- Compound 7 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (0.20 g, 0.50 mmol), 1-[(2-fluorophenyl)methyl]piperazine (0.105 mL, 0.12 g, 0.60 mmol) as an amine, DCM (2.0 mL), and sodium triacetoxyborohydride (0.16 g, 0.75 mmol), according to the general procedure for reductive aminationreaction. The resulting crude product was purified by flash chromatography (0–100% AcOEt gradient in heptane) to obtain compound 7 (0.74 g, 0.13 mmol) with a 26% yield.
- 1H NMR (600 MHz, DMSO-d6) δ 7.88–7.87 (m, 1H, Ar-H), 7.82 (d, J = 7.6 Hz, 1H, Ar-H), 7.57 (t, J = 52.5 Hz, 1H, CHF2), 7.46–7.41 (m, 2H, Ar-H), 7.32–7.26 (m, 2H, Ar-H), 7.13–7.10 (m, 2H, Ar-H), 6.65 (s, 1H, Ar-H), 6.56 (s, 1H, Ar-H), 4.37 (dd, J = 8.4, 6.5 Hz, 1H), 3.93–3.99 (1H), 3.91–3.90 (m, 4H, morph.), 3.86–3.81 (m, 4H, morph.), 3.45 (s, 2H, CH2), 3.13 (dd, J = 16.2, 8.6 Hz, 1H), 2.99 (dd, J = 16.3, 6.4 Hz, 1H), 2.54 (d, J = 8.5 Hz, 2H, CH2), and 2.40–2.32 (m, 4H, piepraz.).
- 13C NMR (151 MHz, DMSO-d6) δ 206.6, 160.7, 155.0, 150.8, 149.4, 147.0, 144.7, 141.2, 134.0, 131.5, 128.9, 125.4, 124.5, 124.0, 123.9, 120.6, 115.0, 112.4, 108.4, 95.2, 87.8, 65.6, 58.2, 54.5, 52.8, 48.1, 43.6, and 30.0.
- HRMS (ESI/MS): m/z calculated for C30H31F3N8O [M + H]+ 576.2564, found 576.2573.
- 1-[2-({4-[(3-Fluorophenyl)methyl]piperazin-1-yl}methyl)-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl]-2-(propan-2-yl)-1H-benzimidazole (8).
- Compound 10 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (0.25 g, 0.63 mmol), 1-[(3-fluorophenyl)methyl]piperazine (0.15 g, 0.75 mmol) as an amine, DCM (2.5 mL), and sodium triacetoxyborohydride (0.20 g, 0.94 mmol), according to the general procedure for reductive amination reaction. The resulting crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to obtain compound 8 (0.24 g, 0.41 mmol) with a 65% yield.
- 1H NMR (600 MHz, DMSO-d6) δ 7.84 (dd, J = 40.8, 7.5 Hz, 2H, Ar-H), 7.58 (t, J = 52.4 Hz, 1H, CHF2), 7.47–7.41 (m, 2H, Ar-H), 7.34 (dd, J = 14.0, 7.8 Hz, 1H, Ar-H), 7.12–7.03 (m, 3H, Ar-H), 6.65 (s, 1H, Ar-H), 6.52 (d, J = 3.5 Hz, 1H, Ar-H), 3.92 (s, 4H, morph.), 3.83 (d, J = 4.3 Hz, 4H, morph.), 3.69 (s, 2H, CH2), 3.47 (s, 4H, piperaz.), and 2.40 (s, 4H, piperaz.).
- 13C NMR (151 MHz, DMSO-d6) δ 163.0, 161.4, 154.9, 150.9, 149.7, 147.0, 141.5, 141.2, 134.0, 130.0, 125.4, 124.6, 123.9, 120.7, 115.0, 113.7, 112.4, 108.5, 95.4, 87.8, 65.5, 61.2, 55.7, 52.6, and 48.2.
- HRMS (ESI/MS): m/z calculated for C30H31F3N8O [M + H]+ 576.2573, found 576.2590.
- 1-[2-({4-[(4-Fluorophenyl)methyl]piperazin-1-yl}methyl)-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl]-2-(propan-2-yl)-1H-benzimidazole (9).
- Compound 9 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (0.20 g, 0.50 mmol), 1-[(4-fluorophenyl)methyl]piperazine (0.12 g, 0.60 mmol) as an amine, DCM (2.0 mL), and sodium triacetoxyborohydride (0.16 g, 0.75 mmol), according to the general procedure for reductive amination reaction. The resulting crude product was purified by flash chromatography (0–100% AcOEt gradient in heptane) to obtain compound 9 (0.25 g, 0.44 mmol) with a 88% yield.
- 1H NMR (400 MHz, CDCl3) δ 7.93–7.91 (m, 1H, Ar-H), 7.66–7.64 (m, 1H, Ar-H), 7.45–7.39 (m, 2H, Ar-H), 7.30–7.26 (m, 3H, Ar-H), 7.16 (s, 1H, Ar-H), 7.01–6.97 (m, 2H, Ar-H), 6.59 (s, 1H, Ar-H), 6.30 (s, 1H, Ar-H), 3.98 (q, J = 3.1 Hz, 4H, morph.), 3.92–3.88 (m, 4H, morph.), 3.80 (s, 2H, CH2), 3.48 (s, 2H, CH2), 2.63 (s, 4H, piperaz.), and 2.51 (s, 4H, piperaz.).
- 13C NMR (101 MHz, CDCl3) δ 163.2, 160.8, 155.6, 151.3, 150.1, 147.5, 144.7, 141.8, 134.6, 130.7, 130.6, 125.7, 124.2, 121.6, 115.1, 114.9, 111.7, 111.7, 109.3, 96.6, 87.3, 66.2, 62.2, 56.4, 53.1, 48.5, 31.6, 22.6, and 14.1.
- HRMS (ESI/MS): m/z calculated for C30H31F3N8O [M + H]+ 576.2573, found 576.2588.
3.3.3. General Procedure for the Amidation Reaction
- 1-[2-(4-Benzylpiperazine-1-carbonyl)-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl]-2-(difluoromethyl)-1H-benzimidazole (10).
- Compound 10 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carboxylic acid (0.45 g, 1.09 mmol), 1-benzylpiperazine (0.20 g, 1.14 mmol), HATU (0.46 g, 1.19 mmol), TEA (0.23 mL, 0.16 g, and 1.63 mmol), and DCM (4.0 mL), according to the general procedure for amidation reaction. The resulting crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to obtain compound 10 (0.32 g, 0.56 mmol) as a light yellow solid with a 51% yield.
- 1H NMR (600 MHz, DMSO-d6) δ 7.89 (d, J = 7.6 Hz, 1H, Ar-H), 7.82 (d, J = 7.8 Hz, 1H, Ar-H), 7.58 (t, J = 52.4 Hz, 1H, CHF2), 7.48–7.42 (m, 2H, Ar-H), 7.35–7.31 (m, 4H, Ar-H), 7.26 (td, J = 5.9, 2.6 Hz, 1H, Ar-H), 6.83 (s, 1H, Ar-H), 6.81 (s, 1H, Ar-H), 3.91–3.90 (m, 4H, morph.), 3.83 (t, J = 4.5 Hz, 4H, morph.), 3.72 (d, J = 41.1 Hz, 4H, piperaz.), 3.53 (s, 2H, CH2), and 2.44 (dt, J = 23.1, 4.6 Hz, 4H, piperaz.).
- 13C NMR (151 MHz, DMSO-d6) δ 161.6, 151.1, 150.3, 149.2, 147.7, 144.6, 141.2, 137.7, 134.0, 128.9, 127.0, 125.5, 124.0, 120.7, 108.5, 65.5, 52.9, 48.4, 30.9, and 26.8.
- HRMS (ESI/MS): m/z calculated for C30H30F2N8O2 [M + H]+ 572.2460, found 572.2477.
- 2-(Difluoromethyl)-1-(2-{4-[(2-fluorophenyl)methyl]piperazine-1-carbonyl}-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl)-1H-benzimidazole (11).
- Compound 11 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carboxylic acid (0.20 g, 0.48 mmol), 1-(2-fluorobenzyl)piperazine (0.89 mL, 0.10 g, and 0.51 mmol), HATU (0.20 g, 0.53 mmol), TEA (0.10 mL, 0.073 g, and 0.72 mmol), and DCM (2.0 mL), according to the general procedure for amidationreaction. The resulting crude product was purified by flash chromatography (0–5% MeOH gradient in AcOEt) to obtain compound 11 (0.17 g, 0.29 mmol) as a white solid with a 60% yield.
- 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, J = 7.3 Hz, 1H, Ar-H), 7.82 (d, J = 7.3 Hz, 1H, Ar-H), 7.61 (t, J = 52.6 Hz, 1H, CHF2), 7.47–7.41 (m, 3H, Ar-H), 7.33 (d, J = 7.7 Hz, 1H, Ar-H), 7.20–7.15 (m, 2H, Ar-H), 6.83 (s, 1H, Ar-H), 6.80 (s, 1H, Ar-H), 3.83 (d, J = 3.8 Hz, 4H, morph.), 3.72 (d, J = 27.4 Hz, 4H, morph.), 3.60 (s, 2H,), and 2.49–2.46 (m, 4H).
- 13C NMR (101 MHz, DMSO-d6) δ 161.7, 159.6, 151.2, 150.2, 149.2, 147.8, 144.7, 141.2, 134.0, 131.7, 129.2, 125.5, 124.2, 124.0, 120.7, 115.3, 112.4, 108.5, 96.7, 89.3, 65.6, 54.4, 52.7, 52.1, 48.4, and 41.8.
- HRMS (ESI/MS): m/z calculated for C30H29F3N8O2 [M + H]+ 590.2366, found 590.2381.
- 2-(Difluoromethyl)-1-(2-{4-[(3-fluorophenyl)methyl]piperazine-1-carbonyl}-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl)-1H-benzimidazole (12).
- Compound 12 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carboxylic acid (0.24 g, 0.59 mmol), 1-(3-fluorobenzyl)piperazine (0.12 g, 0.62 mmol), HATU (0.25 g, 0.65 mmol), TEA (0.12 mL, 0.09 g, and 0.88 mmol), and DCM (2.0 mL), according to the general procedure for amidation reaction. The resulting crude product was purified by flash chromatography (0–15% MeOH gradient in AcOEt) to obtain compound 12 (0.13 g, 0.21 mmol) as a white solid with a 36% yield.
- 1H NMR (600 MHz, DMSO-d6) δ 7.89 (d, J = 7.6 Hz, 1H, Ar-H), 7.82 (d, J = 8.1 Hz, 1H, Ar-H), 7.58 (t, J = 52.4 Hz, 1H, CHF2), 7.48–7.42 (m, 2H, Ar-H), 7.37 (dd, J = 14.1, 7.9 Hz, 1H, Ar-H), 7.17–7.15 (m, 2H, Ar-H), 7.10–7.07 (m, 1H, Ar-H), 6.84 (s, 1H, Ar-H), 6.81 (s, 1H, Ar-H), 3.91–3.90 (m, 4H, morph.), 3.84–3.82 (m, 4H, morph.), 3.73 (d, J = 41.5 Hz, 4H, piperaz.), 3.56 (s, 2H), 2.68 (s, 3H), 2.44 (d, J = 4.5 Hz, 4H, piperaz.), and 2.43 (t, J = 4.5 Hz, 2H).
- 13C NMR (151 MHz, DMSO-d6) δ 162.2, 161.7, 151.1, 150.3, 149.2, 147.7, 144.6, 141.2, 140.9, 134.0, 130.1, 125.5, 124.7, 124.0, 120.7, 115.2, 113.8, 112.4, 108.5, 96.7, 89.2, 65.5, 48.4, and 38.2.
- HRMS (ESI/MS): m/z calculated for C30H29F3N8O2 [M + H]+ 590.2366, found 590.2385.
- 2-(Difluoromethyl)-1-(2-{4-[(4-fluorophenyl)methyl]piperazine-1-carbonyl}-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidin-5-yl)-1H-benzimidazole (13).
- Compound 13 was prepared from 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carboxylic acid (0.15 g, 0.36 mmol), 1-(4-fluorobenzyl)piperazine (0.075 g, 0.38 mmol), HATU (0.15 g, 0.40 mmol), TEA (0.076 mL, 0.05 g, and 0.54 mmol), and DCM (1.5 mL), according to the general procedure for amidationreaction. The resulting crude product was purified by flash chromatography (0–5% MeOH gradient in AcOEt) to obtain compound 13 (0.13 g, 0.22 mmol) as a white solid with a 62% yield.
- 1H NMR (600 MHz, DMSO-d6) δ 7.89 (d, J = 7.4 Hz, 1H, Ar-H), 7.82 (d, J = 7.8 Hz, 1H, Ar-H), 7.58 (t, J = 52.6 Hz, 1H, CHF2), 7.48–7.42 (m, 2H, Ar-H), 7.37–7.35 (m, 2H, Ar-H), 7.15 (t, J = 8.7 Hz, 2H, Ar-H), 6.83 (s, 1H, Ar-H), 6.81 (s, 1H, Ar-H), 3.91 (t, J = 4.6 Hz, 4H, morph.), 3.83 (t, J = 4.6 Hz, 4H, morph.), 3.72 (d, J = 42.1 Hz, 4H, piperaz.), 3.51 (s, 2H, CH2), and 2.43 (d, J = 23.5 Hz, 3H).
- 13C NMR (151 MHz, DMSO-d6) δ 161.7, 151.1, 150.3, 149.2, 147.8, 144.7, 141.2, 134.0, 130.7, 130.7, 125.5, 124.0, 120.7, 115.0, 114.8, 112.4, 108.5, 96.7, 89.3, 65.5, 60.8, and 48.4.
- HRMS (ESI/MS): m/z calculated for C30H29F3N8O2 [M + H]+ 590.2366, found 590.2380.
3.4. In Vitro PI3Kδ Inhibition Assay
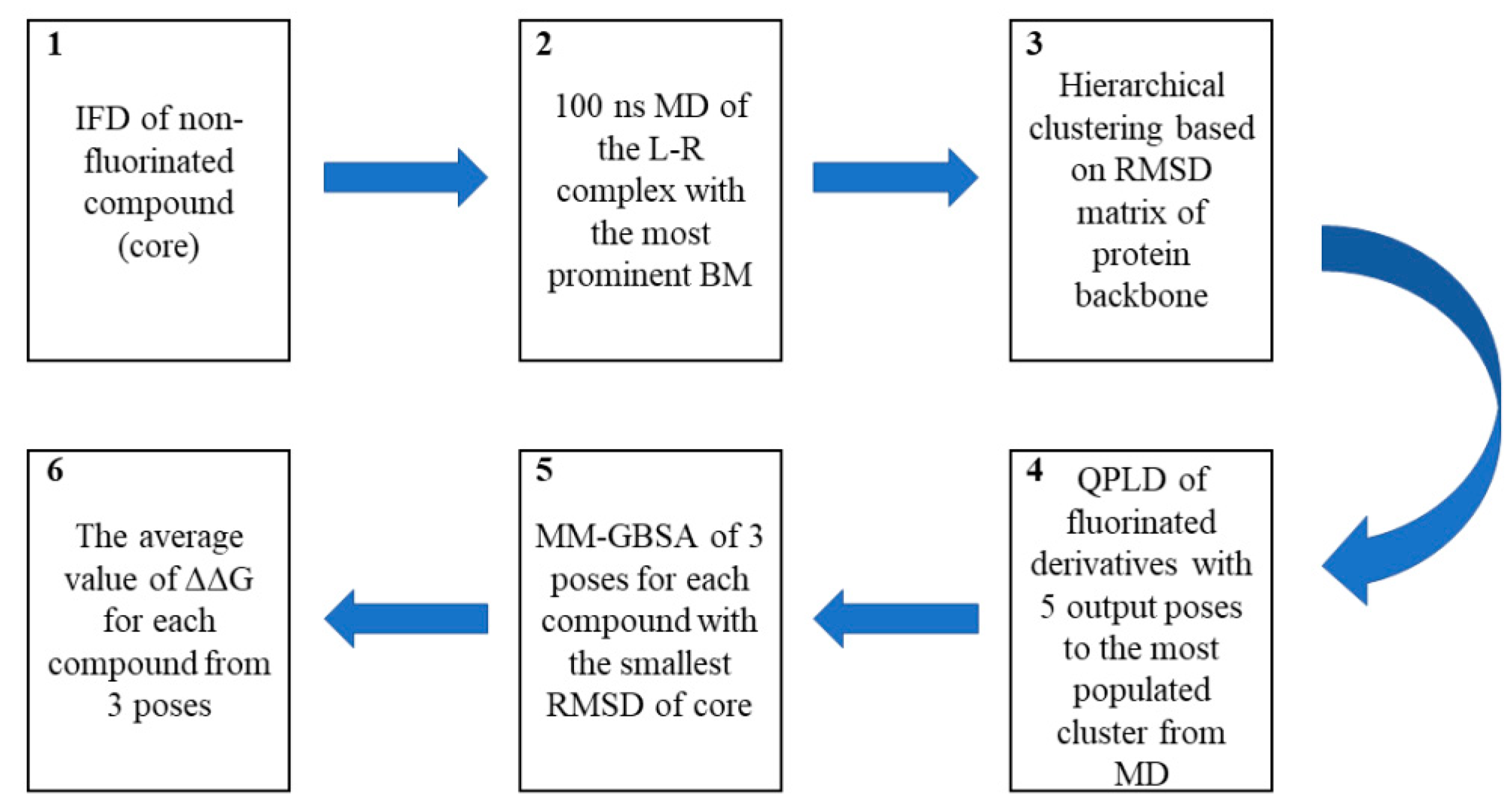
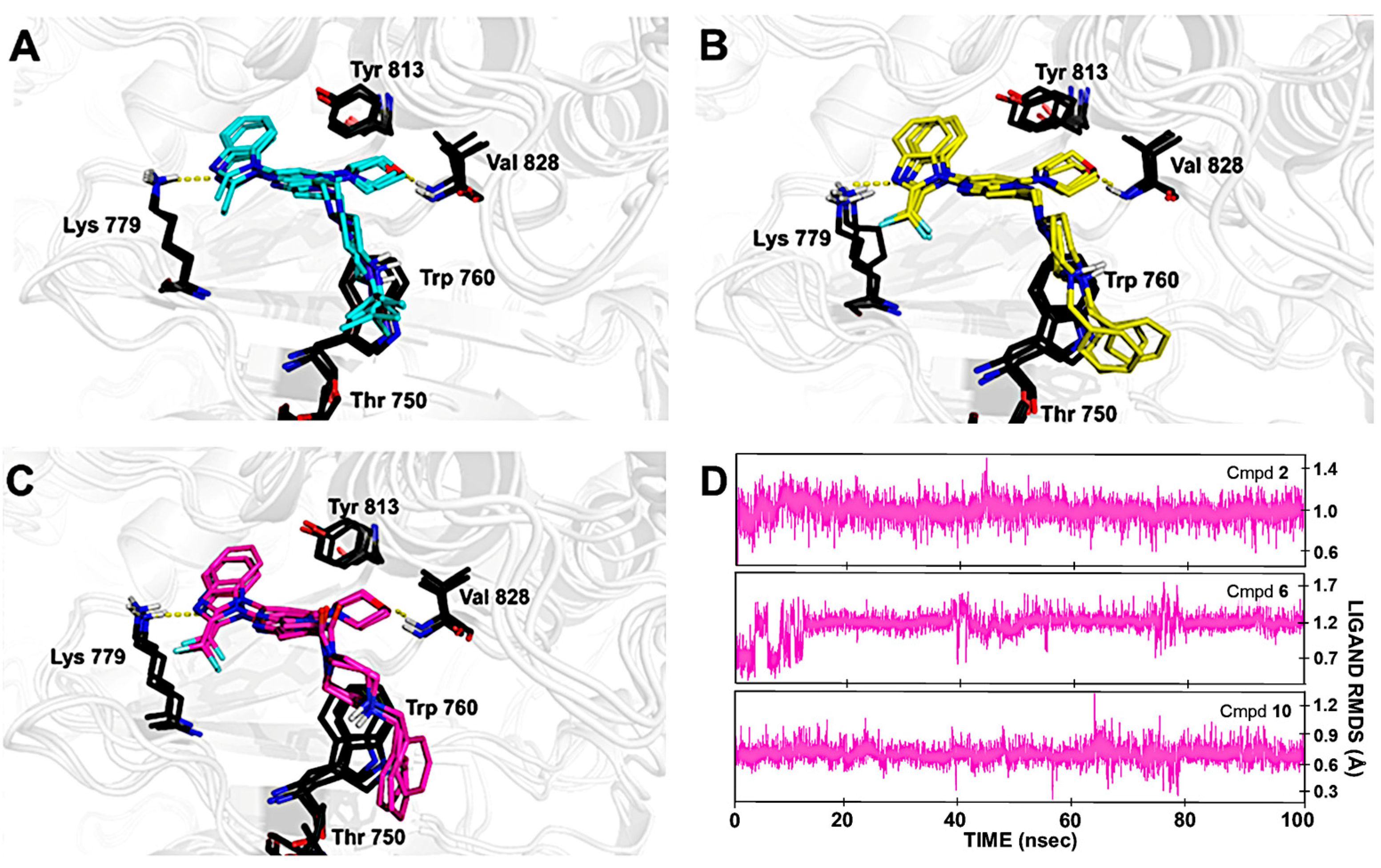
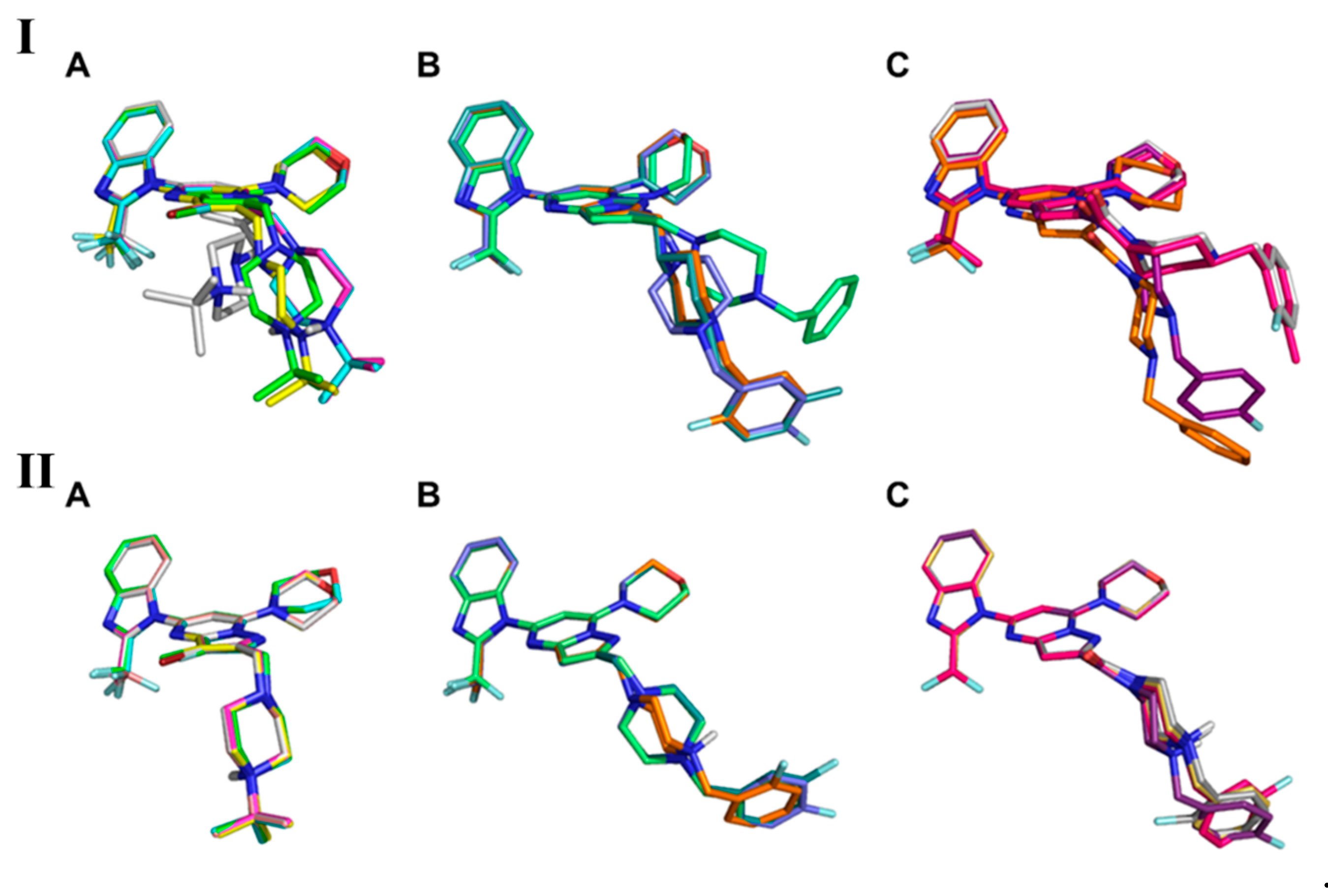
3.5. Computational Workflow Used to Predict the Most Potent Fluorine Derivative
3.5.1. IFD
3.5.2. MD
3.5.3. QPLD
3.5.4. Binding-Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Foster, J.G.; Blunt, M.D.; Carter, E.; Ward, S.G. Inhibition of PI3K Signaling Spurs New Therapeutic Opportunities in Inflammatory/Autoimmune Diseases and Hematological Malignancies. Pharmacol. Rev. 2012, 64, 1027–1054. [Google Scholar] [CrossRef]
- Banham-Hall, E. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 245–258. [Google Scholar] [CrossRef]
- Stark, A.-K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef]
- Puri, K.D.; Gold, M.R. Selective inhibitors of phosphoinositide 3-kinase delta: Modulators of B-cell function with potential for treating autoimmune inflammatory diseases and B-cell malignancies. Front. Immunol. 2012, 3, 256. [Google Scholar] [CrossRef]
- Haselmayer, P.; Camps, M.; Muzerelle, M.; El Bawab, S.; Waltzinger, C.; Bruns, L.; Abla, N.; Polokoff, M.A.; Jond-Necand, C.; Gaudet, M.; et al. Characterization of Novel PI3Kδ Inhibitors as Potential Therapeutics for SLE and Lupus Nephritis in Pre-Clinical Studies. Front. Immunol. 2014, 5, 233. [Google Scholar] [CrossRef]
- Pietruś, W.; Kafel, R.; Bojarski, A.J.; Kurczab, R. Hydrogen Bonds with Fluorine in Ligand–Protein Complexes-the PDB Analysis and Energy Calculations. Molecules 2022, 27, 1005. [Google Scholar] [CrossRef]
- Grychowska, K.; Olejarz-Maciej, A.; Blicharz, K.; Pietruś, W.; Karcz, T.; Kurczab, R.; Koczurkiewicz, P.; Doroz-Płonka, A.; Latacz, G.; Keeri, A.R.; et al. Overcoming undesirable hERG affinity by incorporating fluorine atoms: A case of MAO-B inhibitors derived from 1 H-pyrrolo-[3,2-c]quinolines. Eur. J. Med. Chem. 2022, 236, 114329. [Google Scholar] [CrossRef]
- Pietruś, W.; Kurczab, R.; Stumpfe, D.; Bojarski, A.J.; Bajorath, J. Data-driven analysis of fluorination of ligands of aminergic g protein coupled receptors. Biomolecules 2021, 11, 1647. [Google Scholar] [CrossRef]
- Pietruś, W.; Kurczab, R.; Warszycki, D.; Bojarski, A.J.; Bajorath, J. Isomeric Activity Cliffs—A Case Study for Fluorine Substitution of Aminergic G Protein-Coupled Receptor Ligands. Molecules 2023, 28, 490. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Swallow, S. Fluorine in Medicinal Chemistry. In Progress in Medicinal Chemistry; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 54, pp. 65–133. ISBN 0306-0012. [Google Scholar]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Batta, A.; Kalra, B.S.; Khirasaria, R. Trends in FDA drug approvals over last 2 decades: An observational study. J. Fam. Med. Prim. Care 2020, 9, 105. [Google Scholar] [CrossRef]
- Rojas, S.; Parravicini, O.; Vettorazzi, M.; Tosso, R.; Garro, A.; Gutiérrez, L.; Andújar, S.; Enriz, R. Combined MD/QTAIM techniques to evaluate ligand-receptor interactions. Scope and limitations. Eur. J. Med. Chem. 2020, 208, 112792. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. [Google Scholar] [CrossRef]
- Xu, M.; Lill, M.A. Induced fit docking, and the use of QM/MM methods in docking. Drug Discov. Today Technol. 2013, 10, e411–e418. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Stypik, M.; Michałek, S.; Orłowska, N.; Zagozda, M.; Dziachan, M.; Banach, M.; Turowski, P.; Gunerka, P.; Zdżalik-Bielecka, D.; Stańczak, A.; et al. Design, Synthesis, and Development of Pyrazolo[1,5-a]pyrimidine Derivatives as a Novel Series of Selective PI3Kδ Inhibitors: Part II—Benzimidazole Derivatives. Pharmaceuticals 2022, 15, 927. [Google Scholar] [CrossRef]
- Berry, M.; Fielding, B.; Gamieldien, J. Practical Considerations in Virtual Screening and Molecular Docking. In Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 2, pp. 487–502. ISBN 9788578110796. [Google Scholar]
- Pietruś, W.; Kurczab, R.; Kalinowska-Tłuścik, J.; Machalska, E.; Golonka, D.; Barańska, M.; Bojarski, A.J. Influence of Fluorine Substitution on Nonbonding Interactions in Selected Para-Halogeno Anilines. ChemPhysChem 2021, 22, 2115–2127. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R. The evaluation of QM/MM-driven molecular docking combined with MM/GBSA calculations as a halogen-bond scoring strategy. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 188–194. [Google Scholar] [CrossRef]
- Stypik, M.; Zagozda, M.; Michałek, S.; Dymek, B.; Zdżalik-Bielecka, D.; Dziachan, M.; Orłowska, N.; Gunerka, P.; Turowski, P.; Hucz-Kalitowska, J.; et al. Design, Synthesis, and Development of pyrazolo[1,5-a]pyrimidine Derivatives as a Novel Series of Selective PI3Kδ Inhibitors: Part I—Indole Derivatives. Pharmaceuticals 2022, 15, 949. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, D.; López, C.; Claramunt, R.; Alkorta, I.; Elguero, J. 19F-NMR Diastereotopic Signals in Two N-CHF2 Derivatives of (4S,7R)-7,8,8-Trimethyl-4,5,6,7-tetrahydro-4,7-methano-2H-indazole. Molecules 2017, 22, 2003. [Google Scholar] [CrossRef] [PubMed]
- Berndt, A.; Miller, S.; Williams, O.; Le, D.D.; Houseman, B.T.; Pacold, J.I.; Gorrec, F.; Hon, W.-C.; Ren, P.; Liu, Y.; et al. The p110δ structure: Mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, USA, 2021.
- Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2021.
- Abascal, J.L.F.; Sanz, E.; García Fernández, R.; Vega, C. A potential model for the study of ices and amorphous water: TIP4P/Ice. J. Chem. Phys. 2005, 122, 234511. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. 1994, 100, 2975–2988. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Compound | R1 | R2 | IC50 PI3Kδ (nM) a | Docking Score | |
| First Pose | Average of Top Three Poses | ||||
| 1 | CH3 | H | 236 | −9.3 | −8.8 |
| 2 (CPL302415) | CHF2 | H | 18 | −9.9 | −9.8 |
| 3 | CF3 | H | 907 | −9.6 | −9.3 |
| 4 | CHF2 | Cl | 44 | −10.4 | −10.1 |
| 5 | CHF2 | Br | 50 | −10.5 | −10.1 |
 | |||||
| Compound | X | R3 | IC50 PI3Kδ (nM) a | Docking score | |
| First Pose | Average of Top Three Poses | ||||
| 6 | CH2 | - | 118 | −6.8 | −6.4 |
| 7 | CH2 | o-F | 640 | −10.8 | −10.4 |
| 8 | CH2 | m-F | 751 | −9.0 | −8.3 |
| 9 | CH2 | p-F | 489 | −9.2 | −8.1 |
| 10 | CO | - | 275 | −10.1 | −9.1 |
| 11 | CO | o-F | 212 | −10.3 | −9.3 |
| 12 | CO | m-F | 92 | −10.9 | −10.8 |
| 13 | CO | p-F | 181 | −10.5 | −10.1 |
| Compound | R1 | R2 | IC50 PI3Kδ (nM) b | ΔΔG (kcal/mol) | |
|---|---|---|---|---|---|
| 1 | CH3 | H | 236 | −75.0 | – |
| 2 (CPL302415) | CHF2 | H | 18 | −82.6 | −7.6 |
| 3 | CF3 | H | 907 | −69.6 | 5.4 |
| 4 | CHF2 | Cl | 44 | −81.3 | 1.3 c |
| 5 | CHF2 | Br | 50 | −81.2 | 1.4 c |
| Compound | X | R3 | IC50 PI3Kδ (nM) b | ΔΔG (kcal/mol) | |
|---|---|---|---|---|---|
| 6 | CH2 | - | 118 | −86.7 | – |
| 7 | CH2 | o-F | 640 | −83.4 | 3.3 |
| 8 | CH2 | m-F | 751 | −81.8 | 4.9 |
| 9 | CH2 | p-F | 489 | −84.1 | 2.6 |
| 10 | CO | - | 275 | −78.9 | – |
| 11 | CO | o-F | 212 | −83.8 | −4.9 |
| 12 | CO | m-F | 92 | −86.6 | −7.7 |
| 13 | CO | p-F | 181 | −83.8 | −4.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietruś, W.; Stypik, M.; Zagozda, M.; Banach, M.; Gurba-Bryśkiewicz, L.; Maruszak, W.; Leniak, A.; Kurczab, R.; Ochal, Z.; Dubiel, K.; et al. Tuning the Biological Activity of PI3Kδ Inhibitor by the Introduction of a Fluorine Atom Using the Computational Workflow. Molecules 2023, 28, 3531. https://doi.org/10.3390/molecules28083531
Pietruś W, Stypik M, Zagozda M, Banach M, Gurba-Bryśkiewicz L, Maruszak W, Leniak A, Kurczab R, Ochal Z, Dubiel K, et al. Tuning the Biological Activity of PI3Kδ Inhibitor by the Introduction of a Fluorine Atom Using the Computational Workflow. Molecules. 2023; 28(8):3531. https://doi.org/10.3390/molecules28083531
Chicago/Turabian StylePietruś, Wojciech, Mariola Stypik, Marcin Zagozda, Martyna Banach, Lidia Gurba-Bryśkiewicz, Wioleta Maruszak, Arkadiusz Leniak, Rafał Kurczab, Zbigniew Ochal, Krzysztof Dubiel, and et al. 2023. "Tuning the Biological Activity of PI3Kδ Inhibitor by the Introduction of a Fluorine Atom Using the Computational Workflow" Molecules 28, no. 8: 3531. https://doi.org/10.3390/molecules28083531
APA StylePietruś, W., Stypik, M., Zagozda, M., Banach, M., Gurba-Bryśkiewicz, L., Maruszak, W., Leniak, A., Kurczab, R., Ochal, Z., Dubiel, K., & Wieczorek, M. (2023). Tuning the Biological Activity of PI3Kδ Inhibitor by the Introduction of a Fluorine Atom Using the Computational Workflow. Molecules, 28(8), 3531. https://doi.org/10.3390/molecules28083531