

Recent Developments in Reactions and Catalysis of Protic Pyrazole Complexes


{kind=link}
{kind=link}
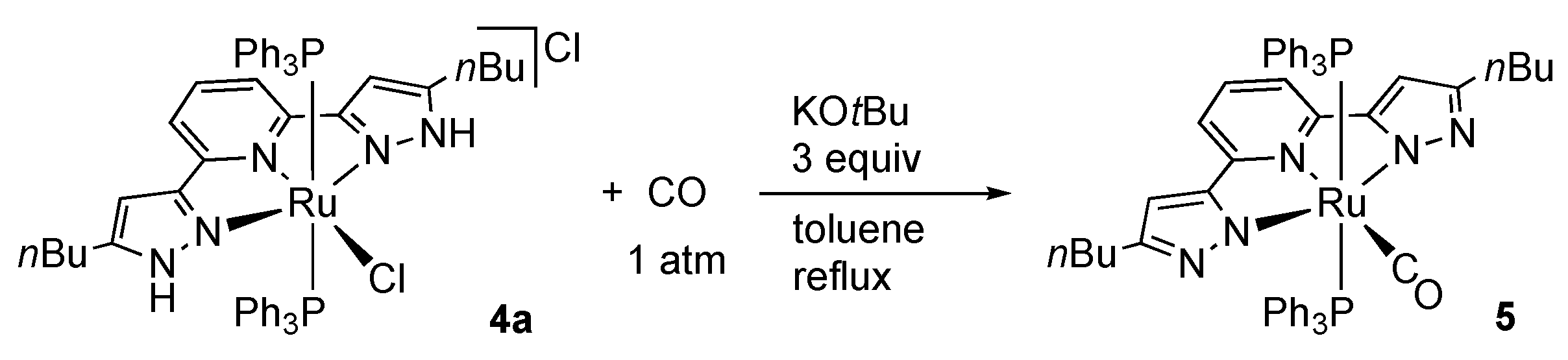{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
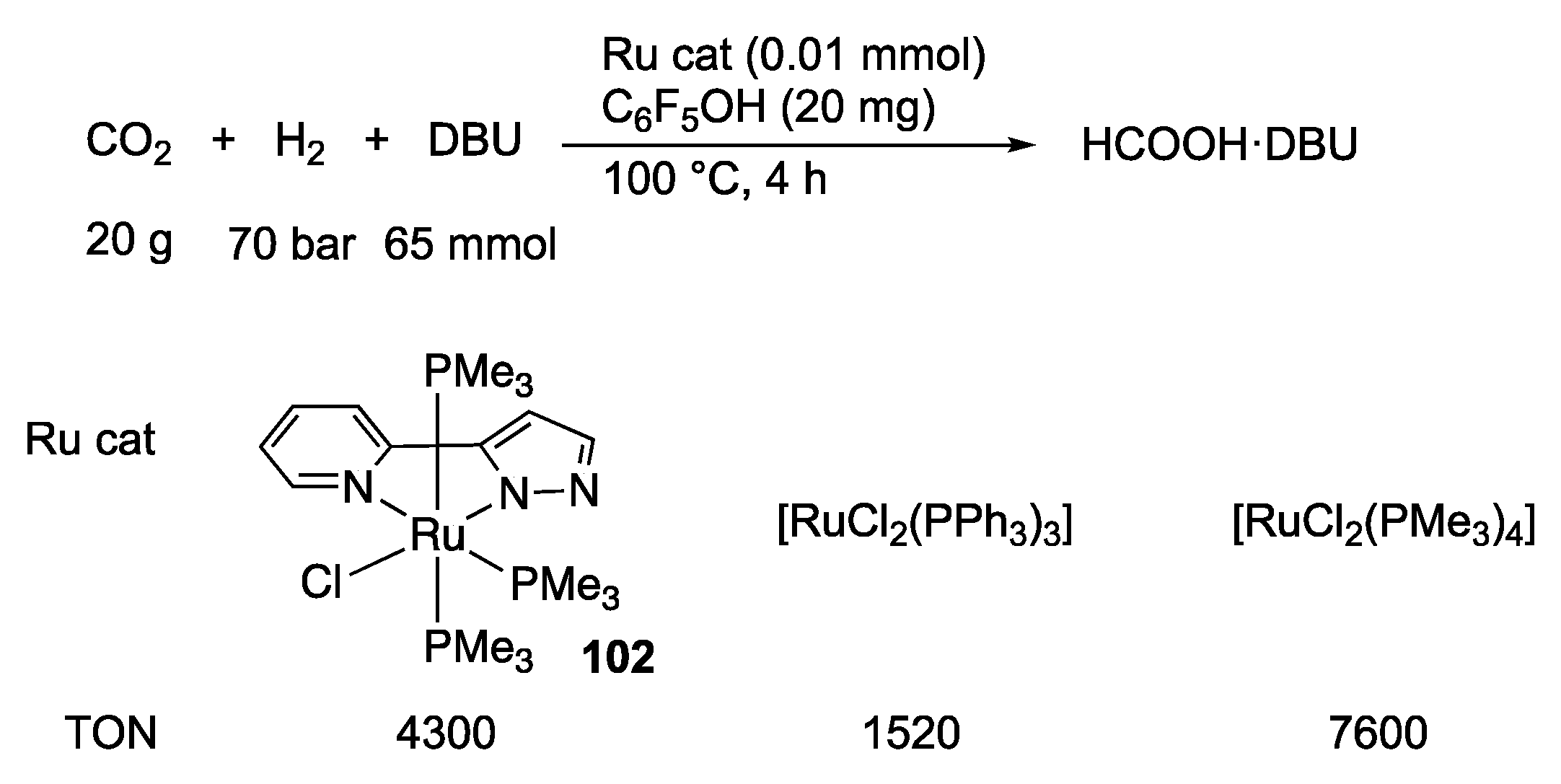{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
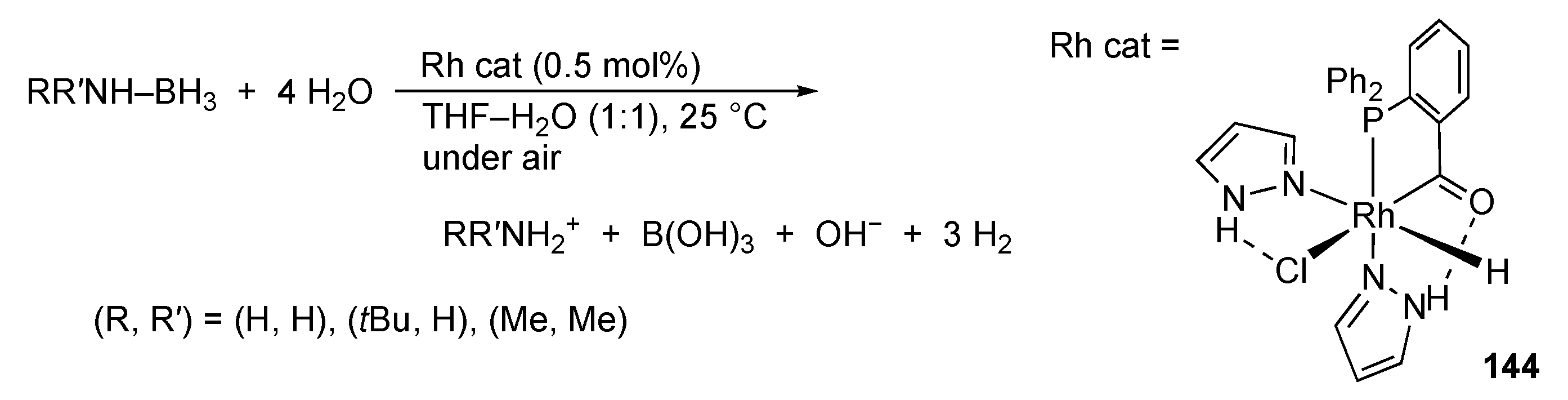{kind=link}
{kind=link}
Abstract
1. Introduction
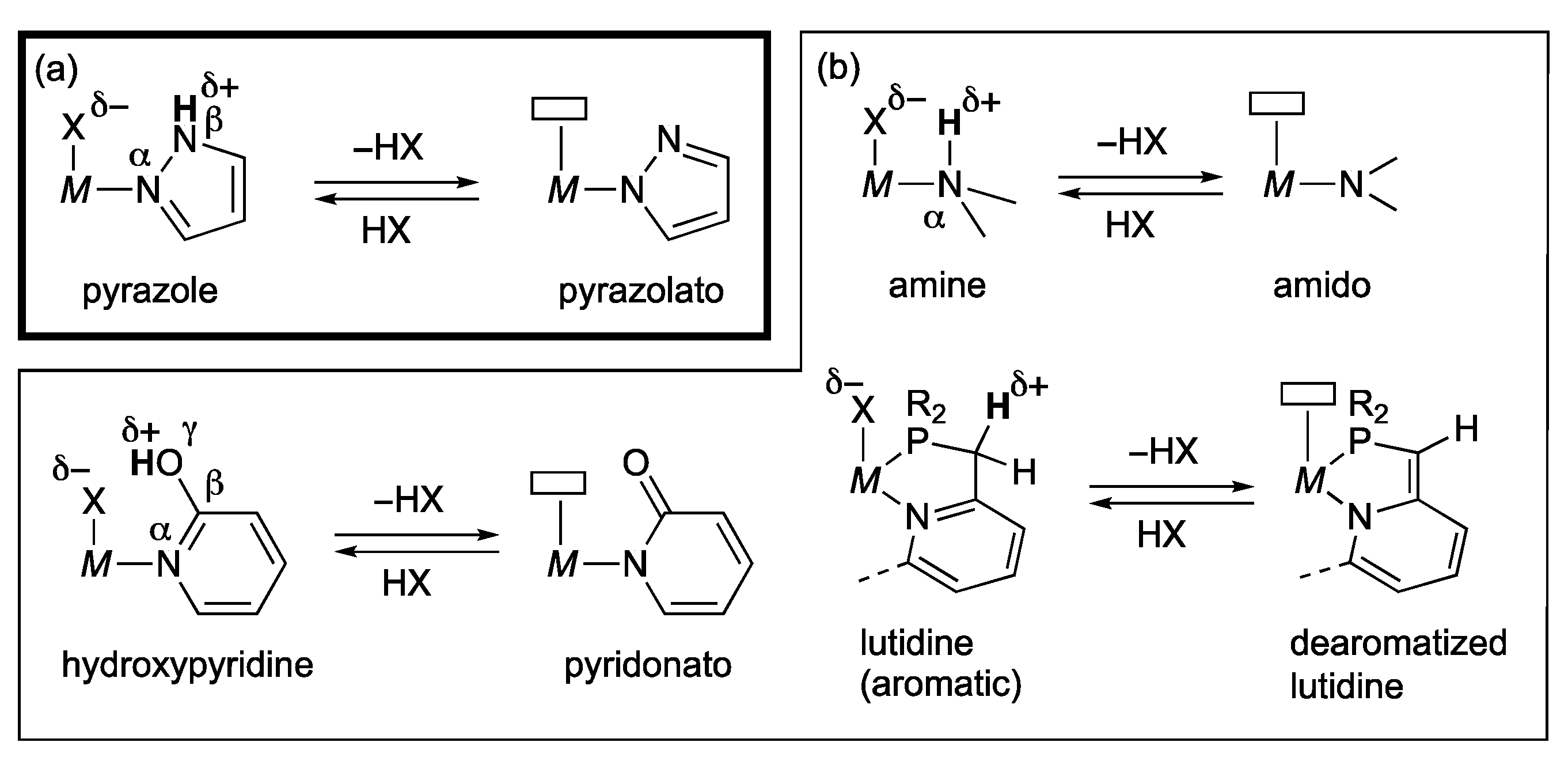
2. Reactions of Protic Pyrazole Complexes
2.1. Pincer-Type Complexes Bearing Protic Pyrazole Arms
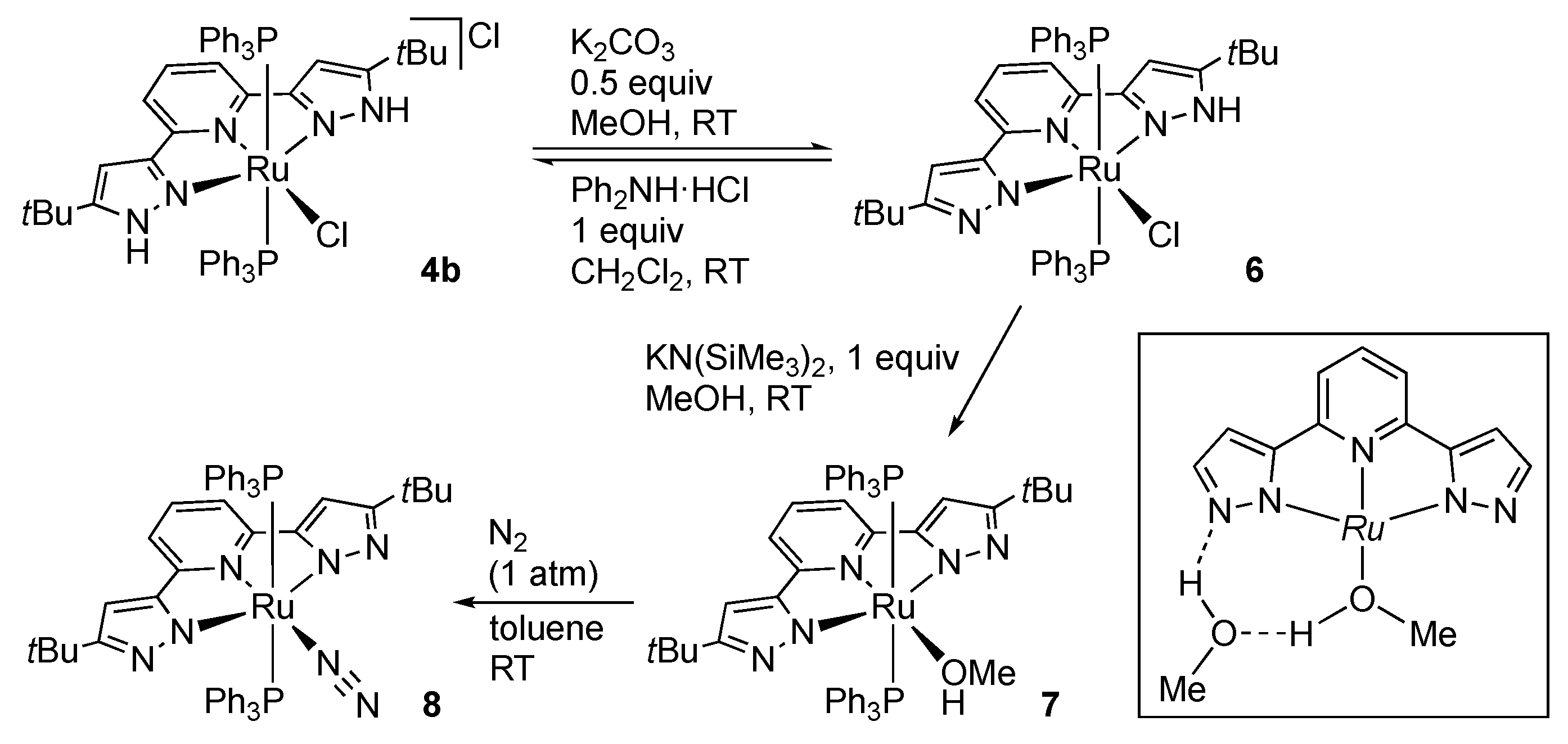
2.1.1. Bis(1H-pyrazol-3-yl)pyridine Complexes
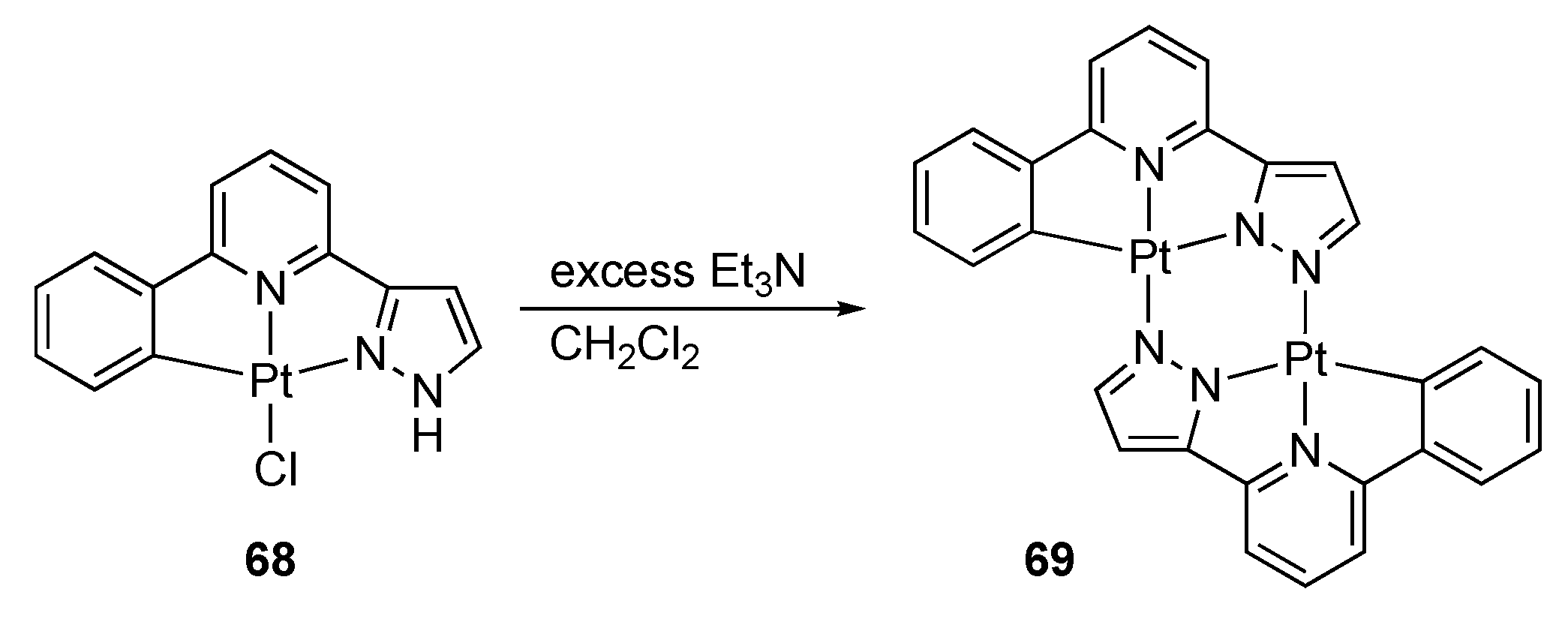
Ruthenium and Osmium
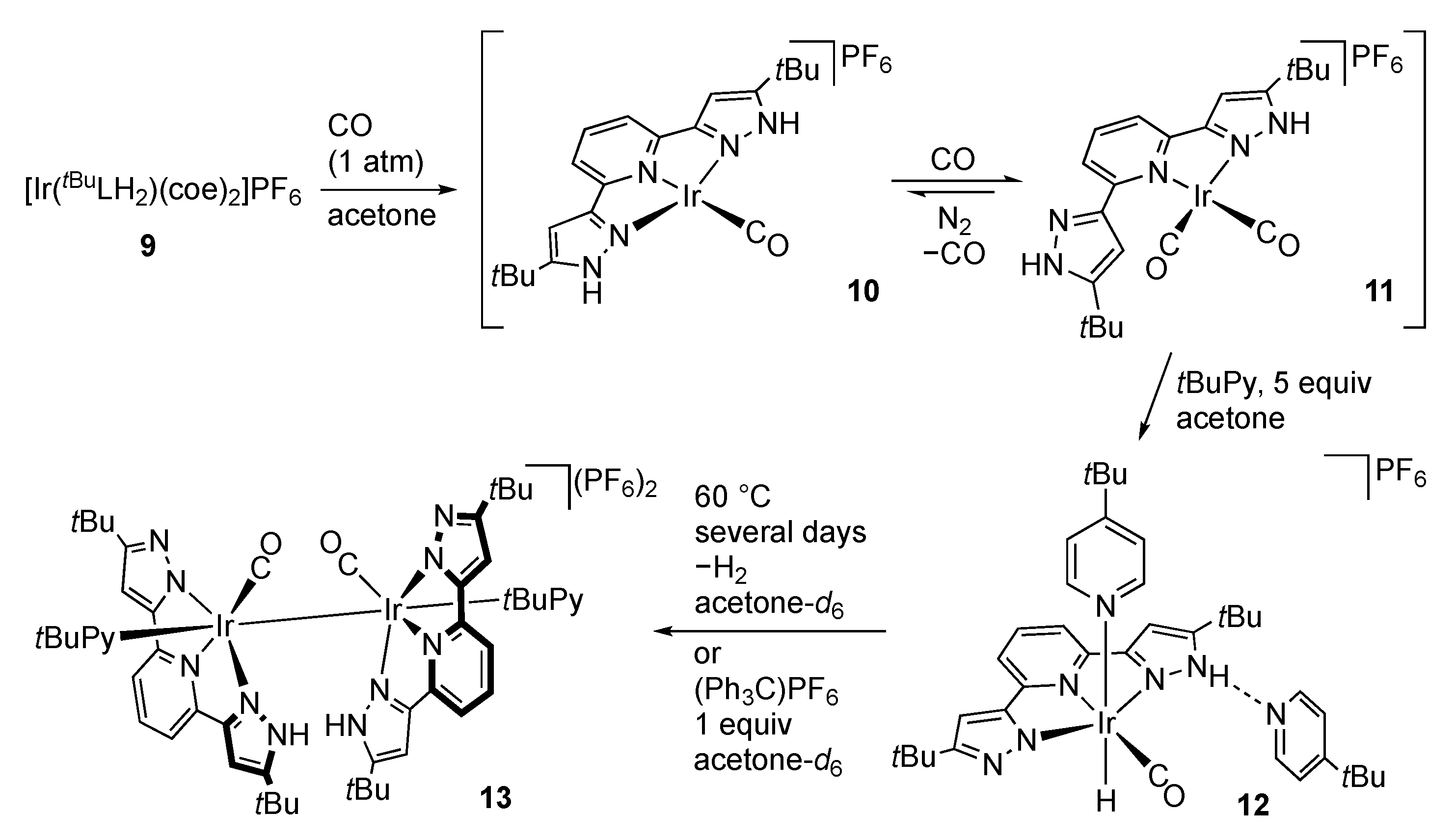
Rhodium and Iridium
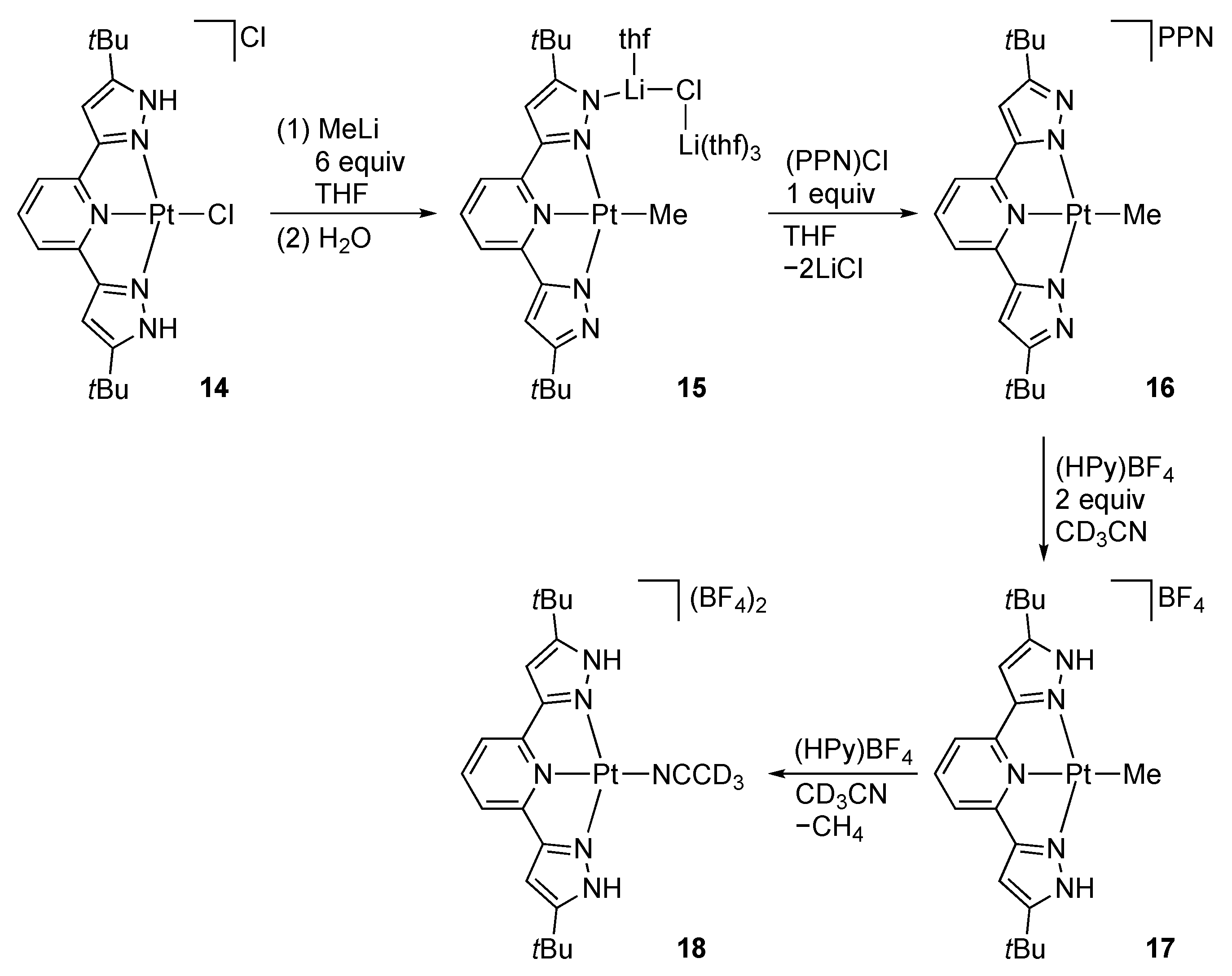
Platinum
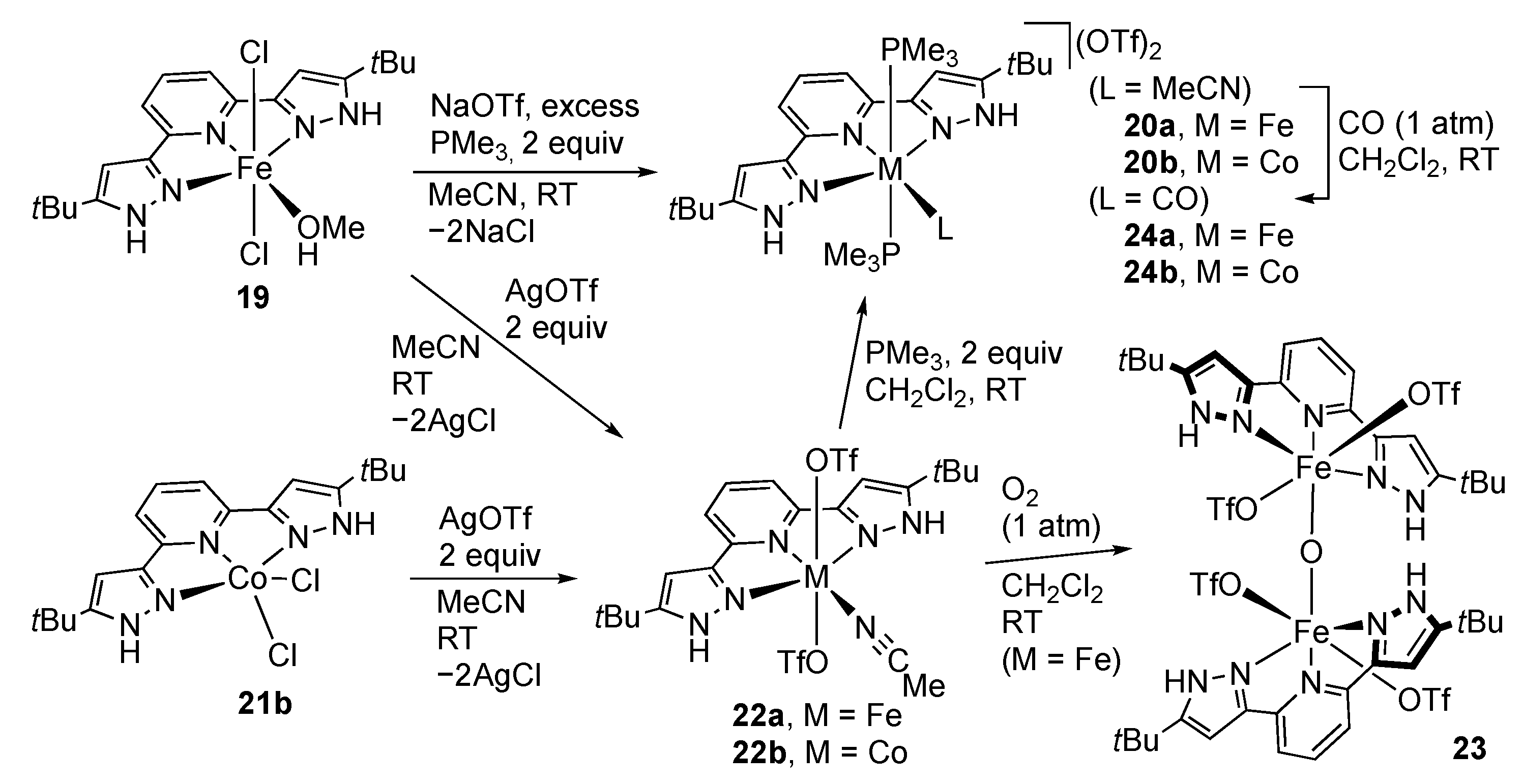
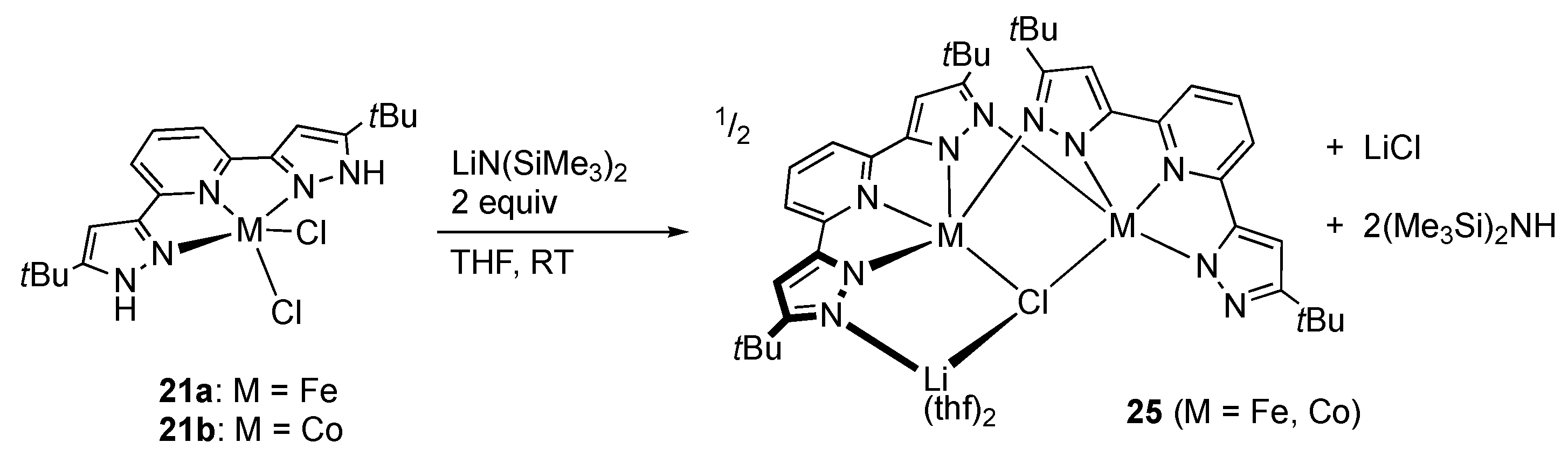
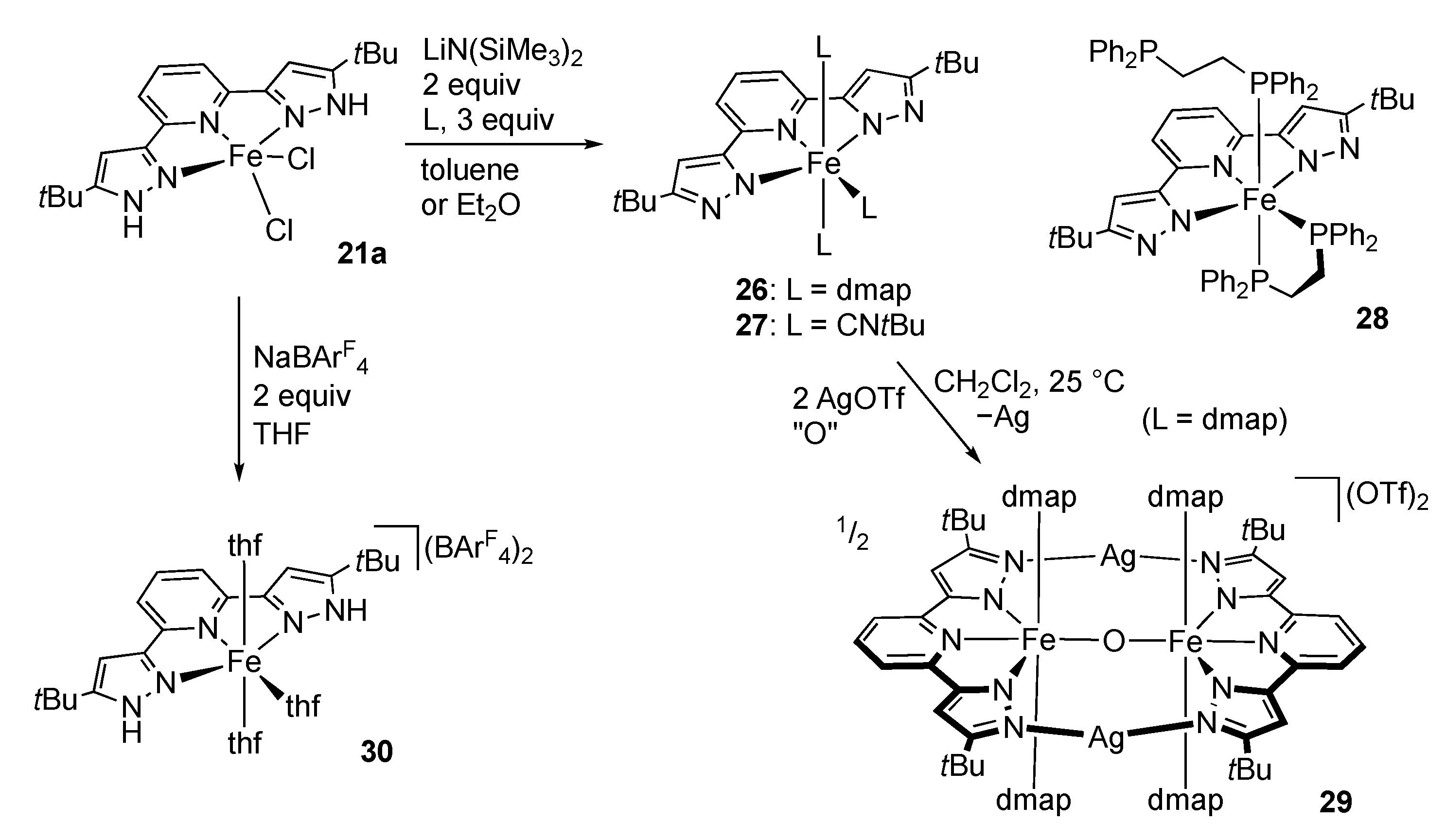
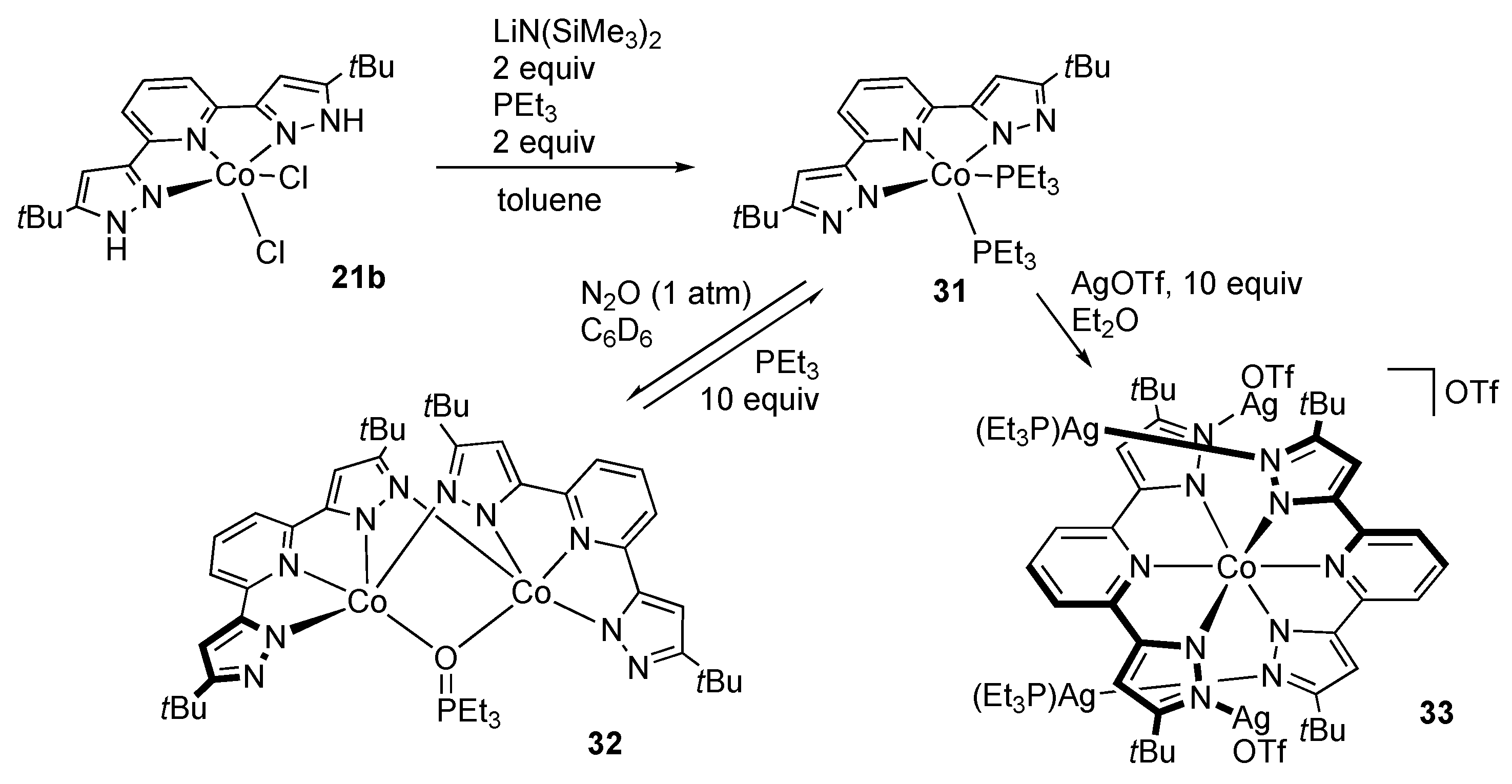
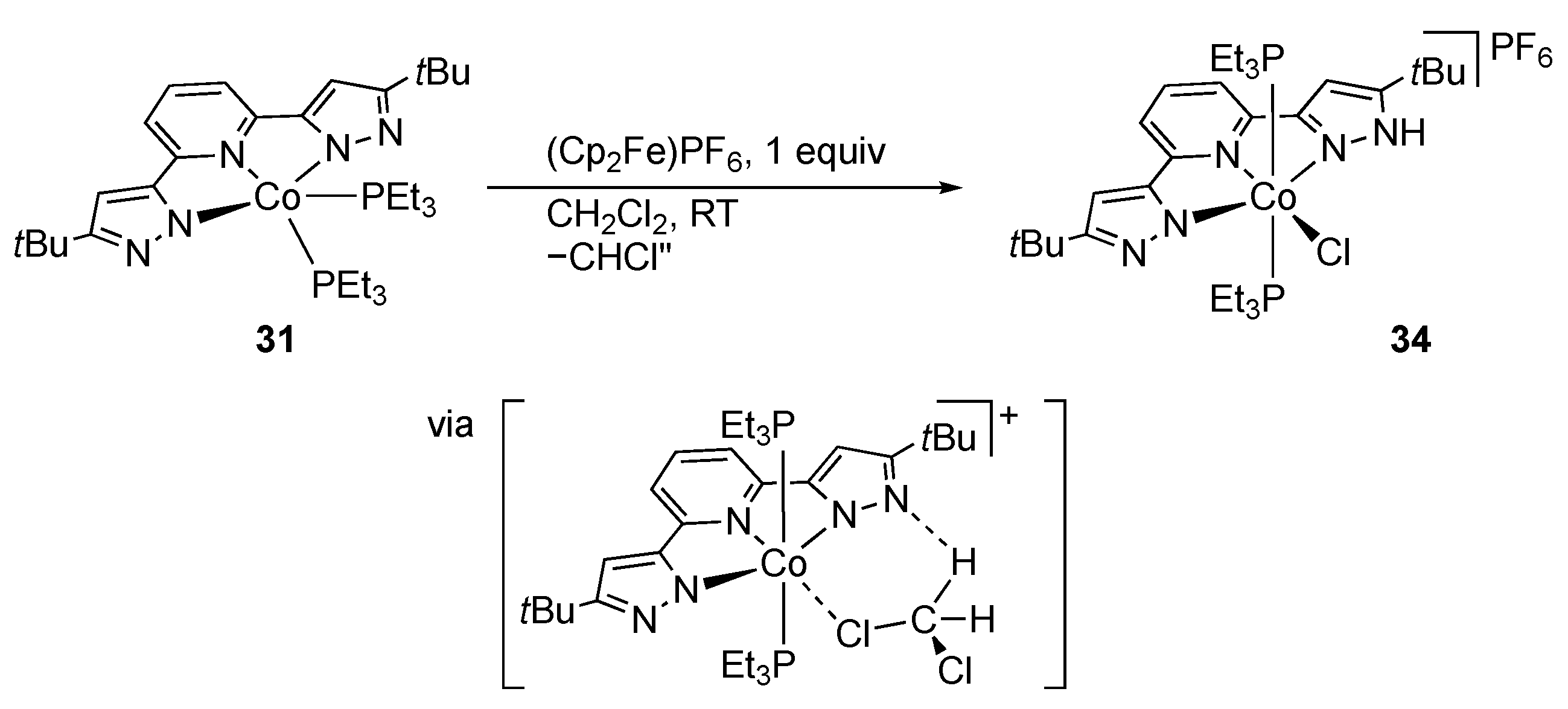
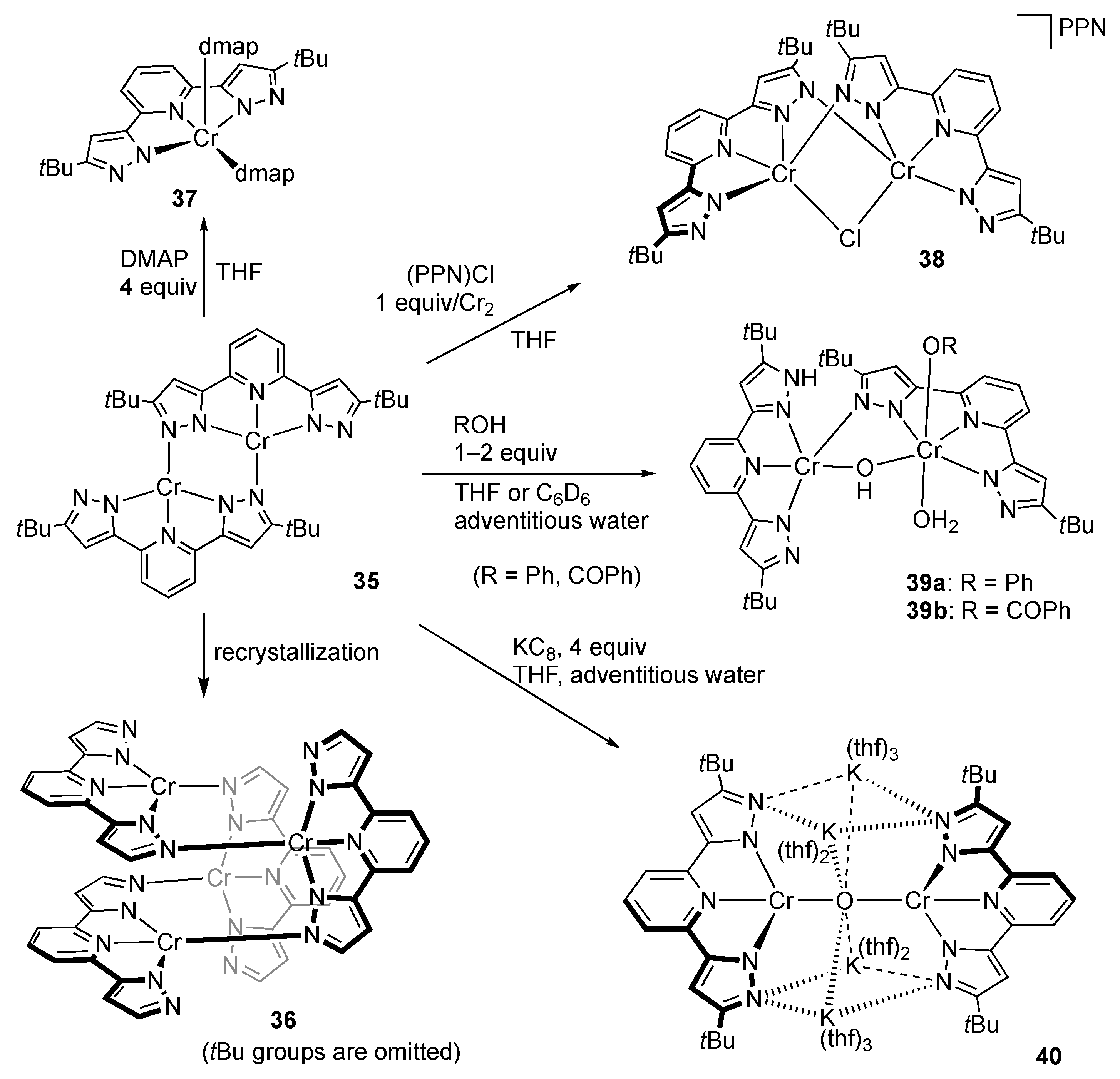
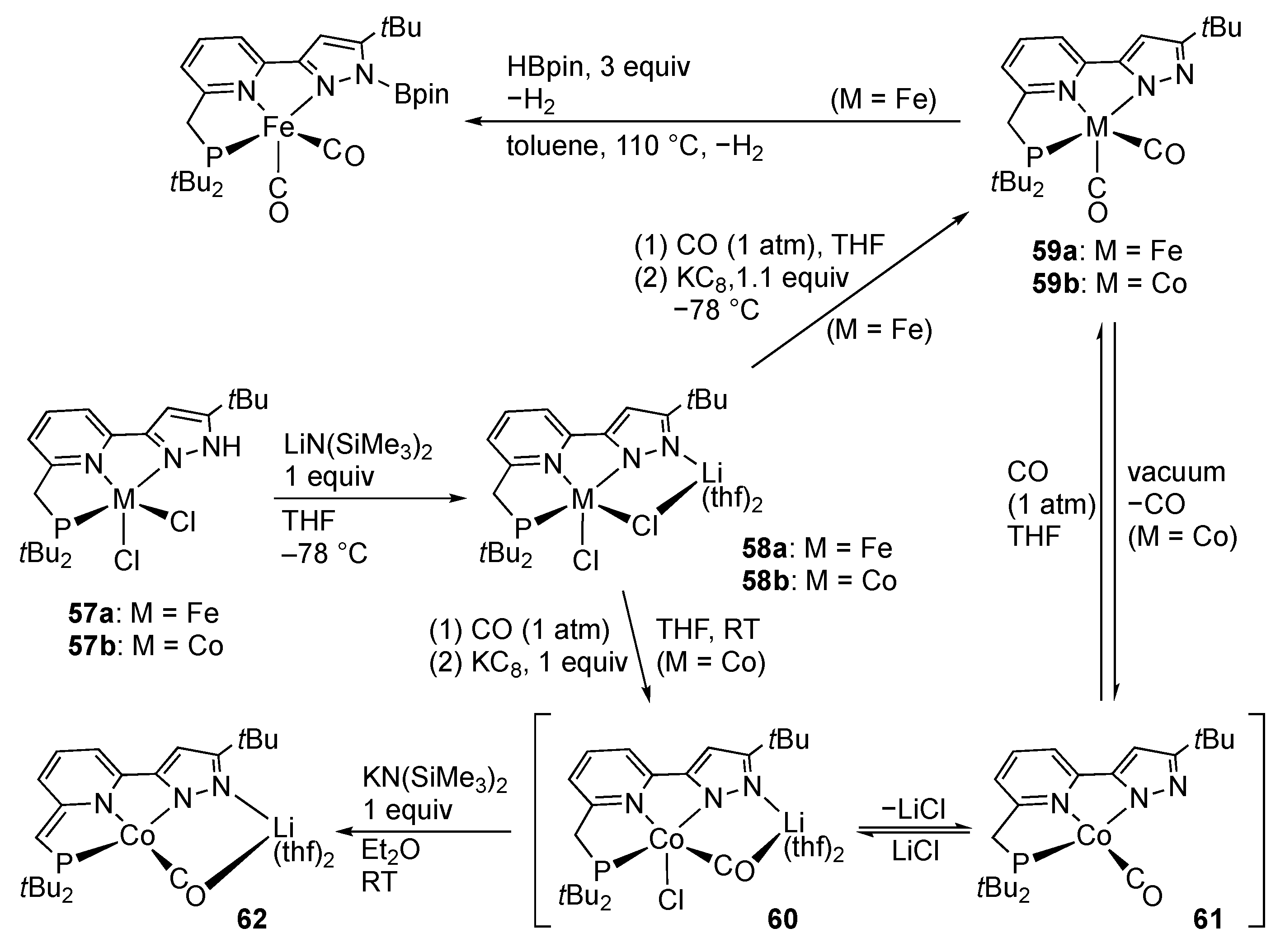
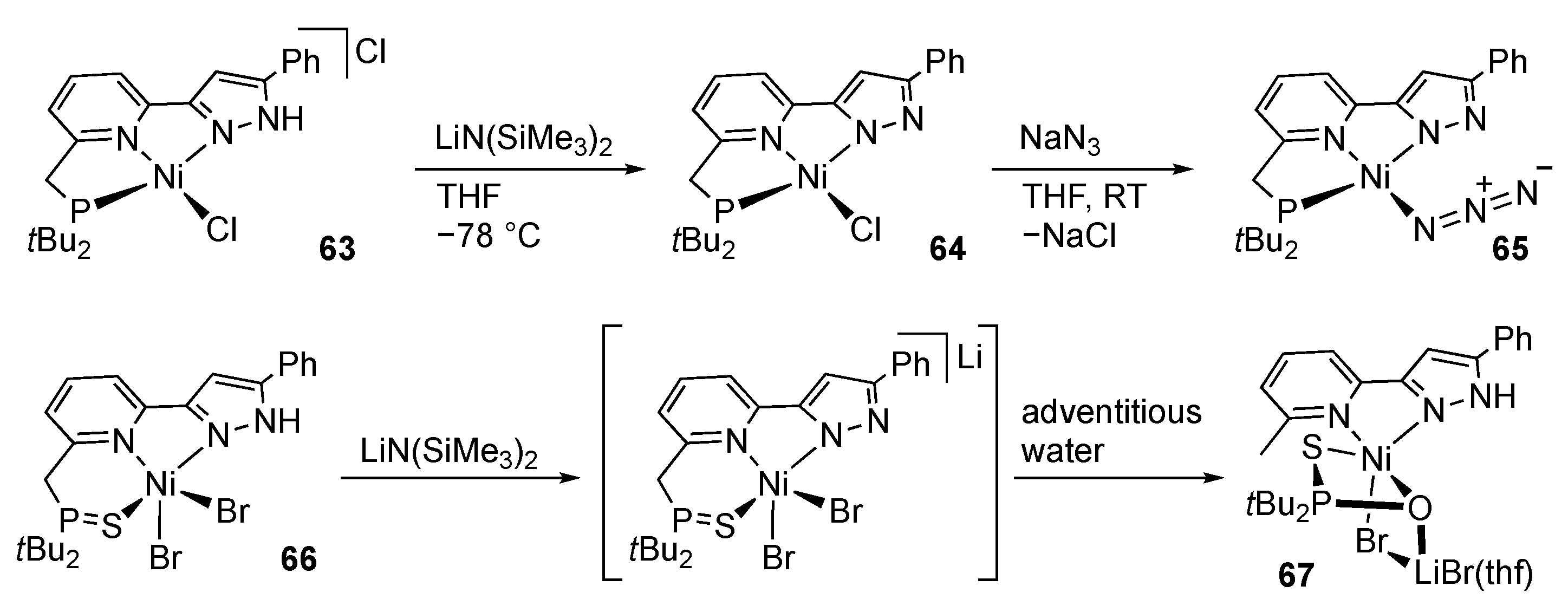
First-Row Transition Metals
2.1.2. Modified 1H-Pyrazol-3-yl Pincer Complexes
Modification at Pincer Center
Unsymmetrical Pincer-Type Complexes
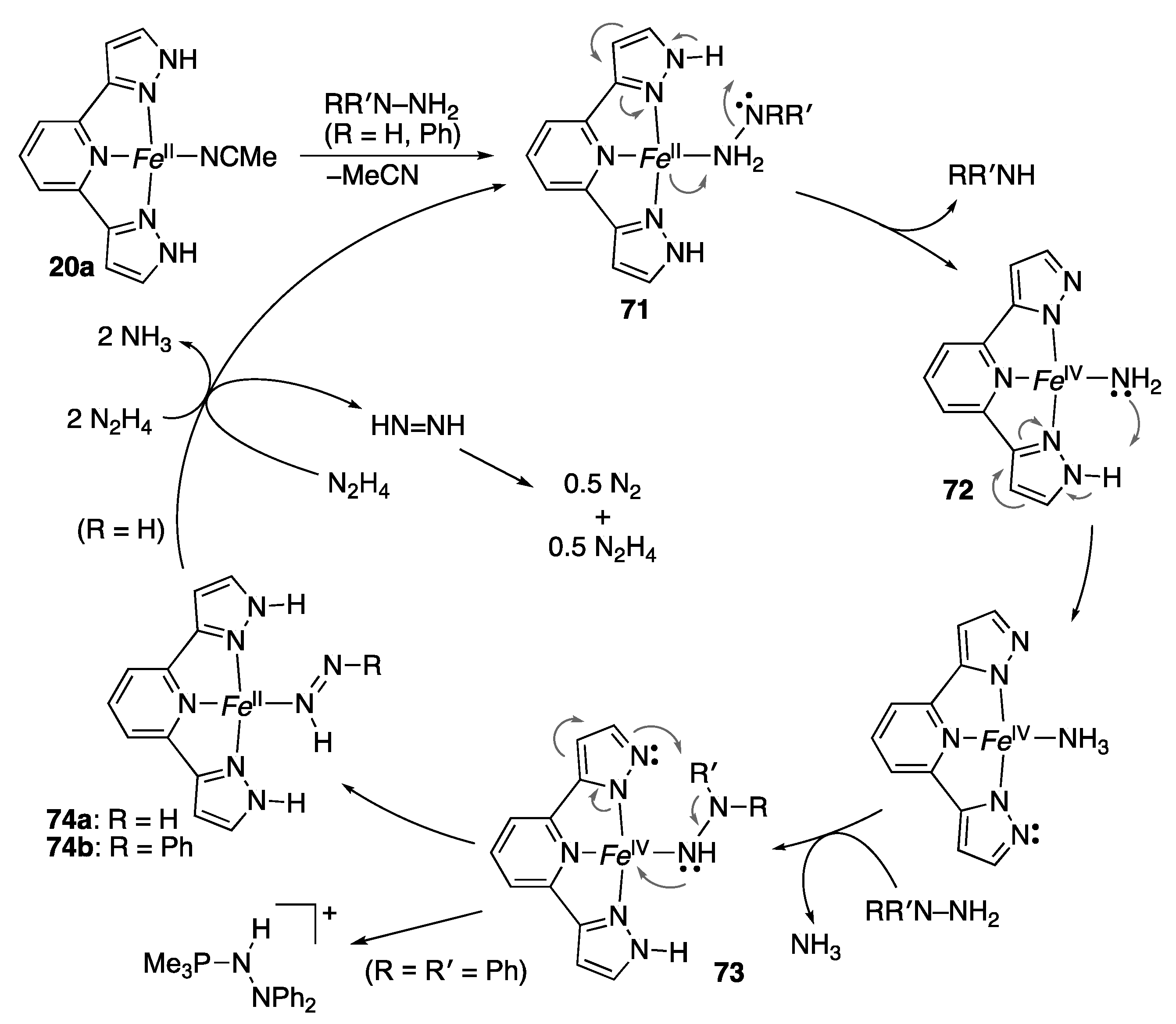
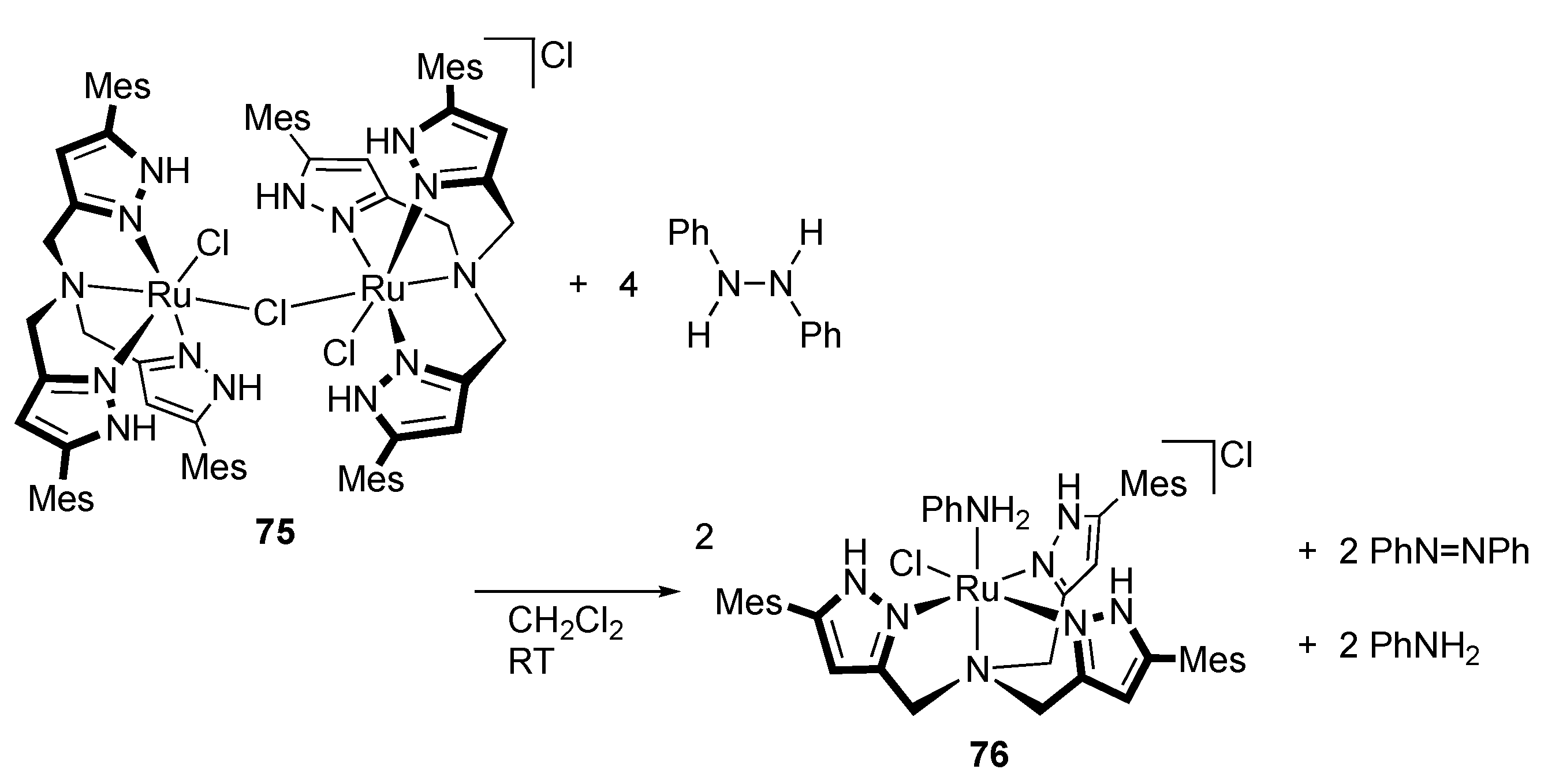
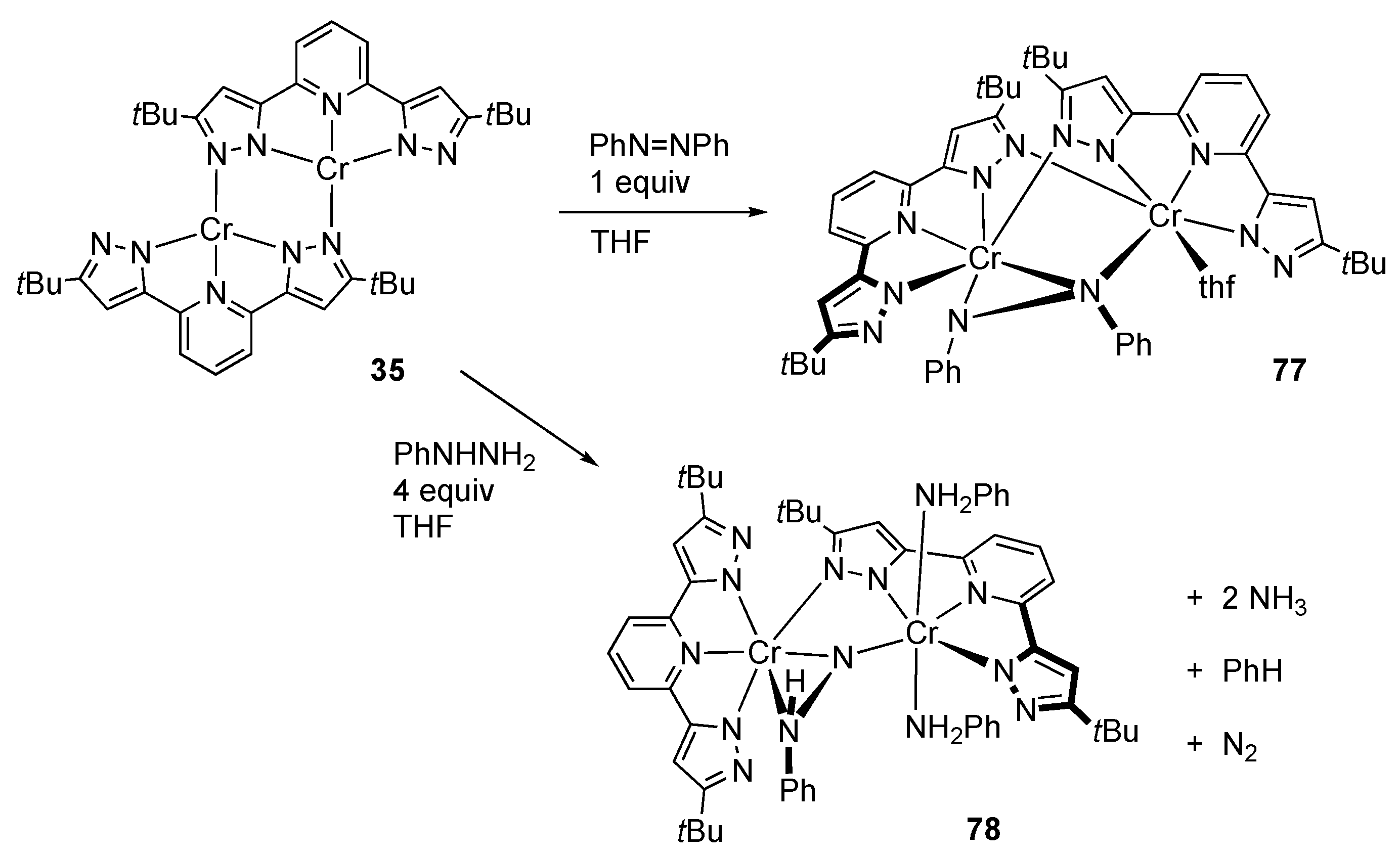
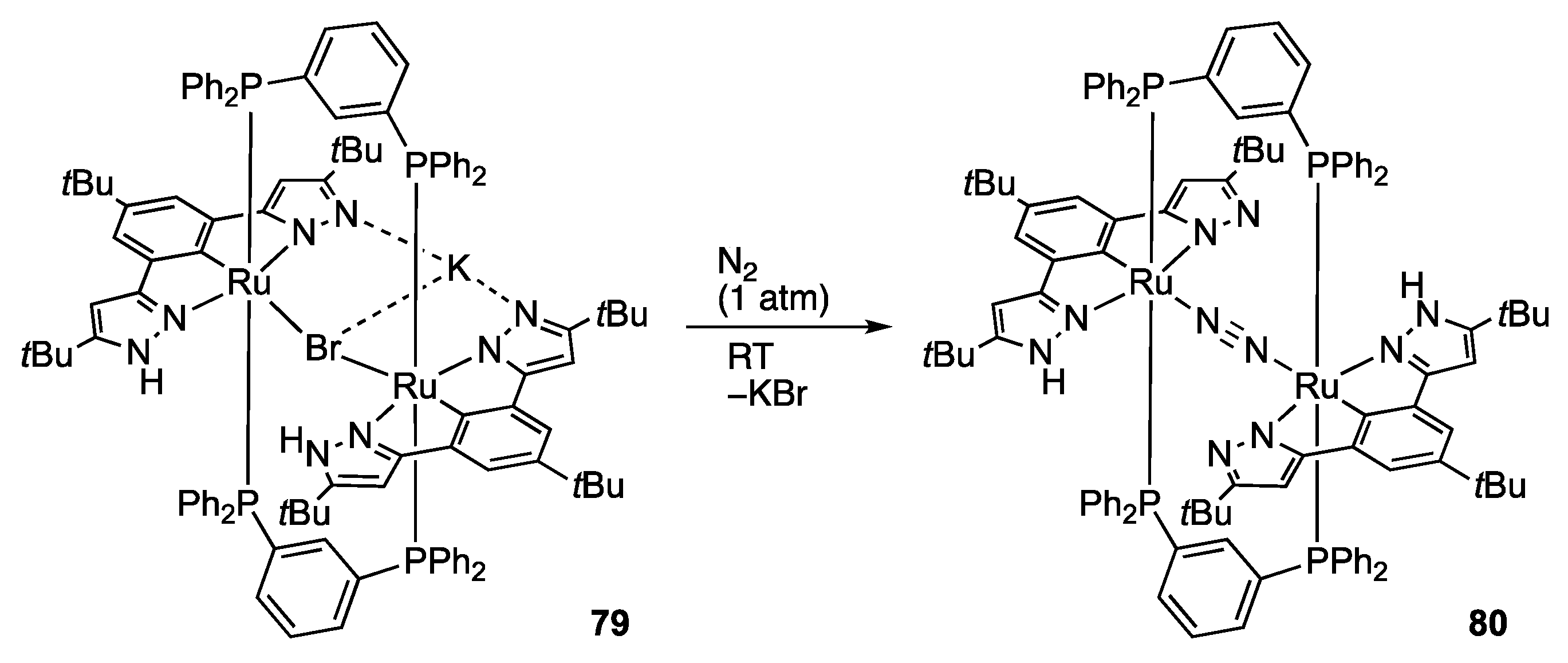
2.2. Redox Reactions of Hydrazines and Azobenzene
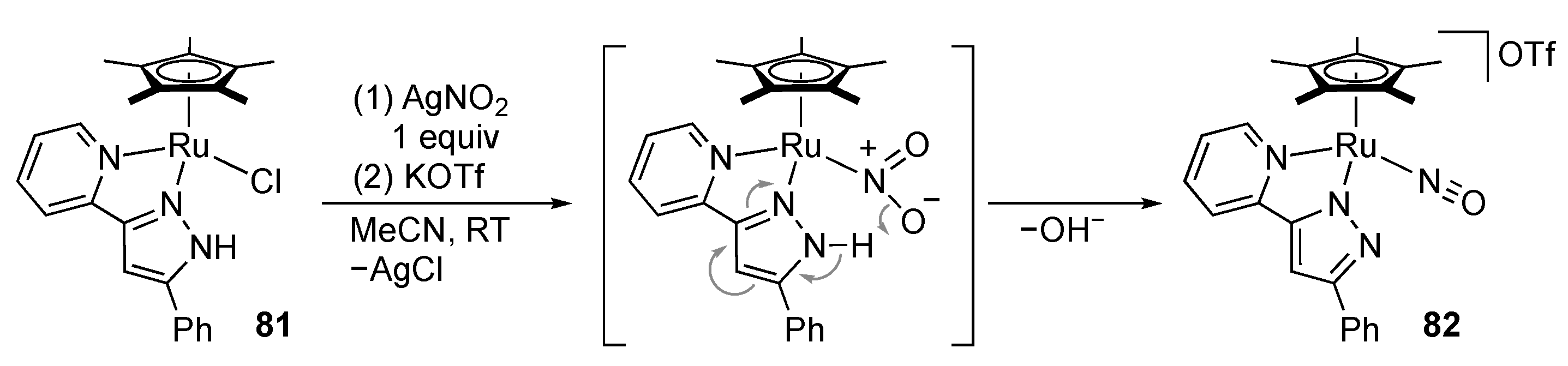
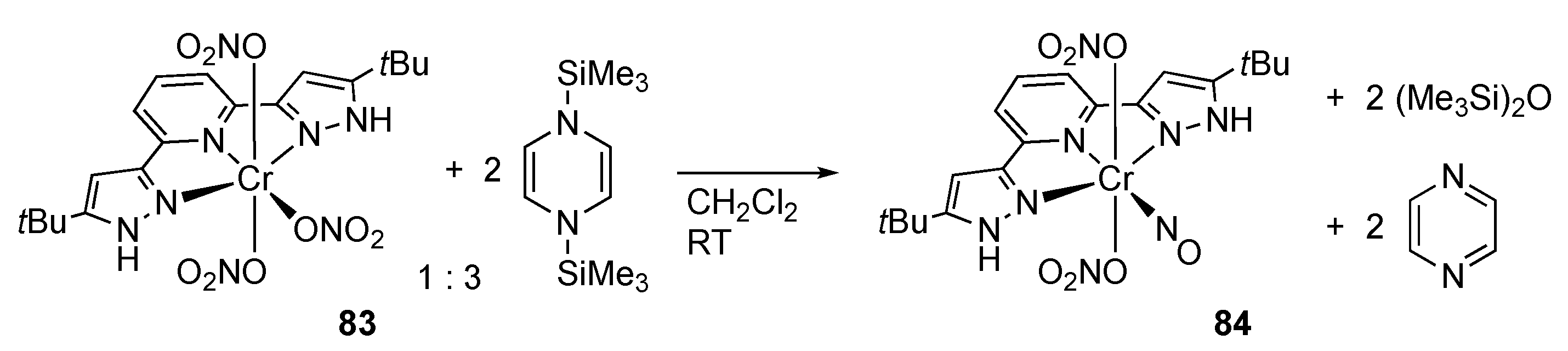
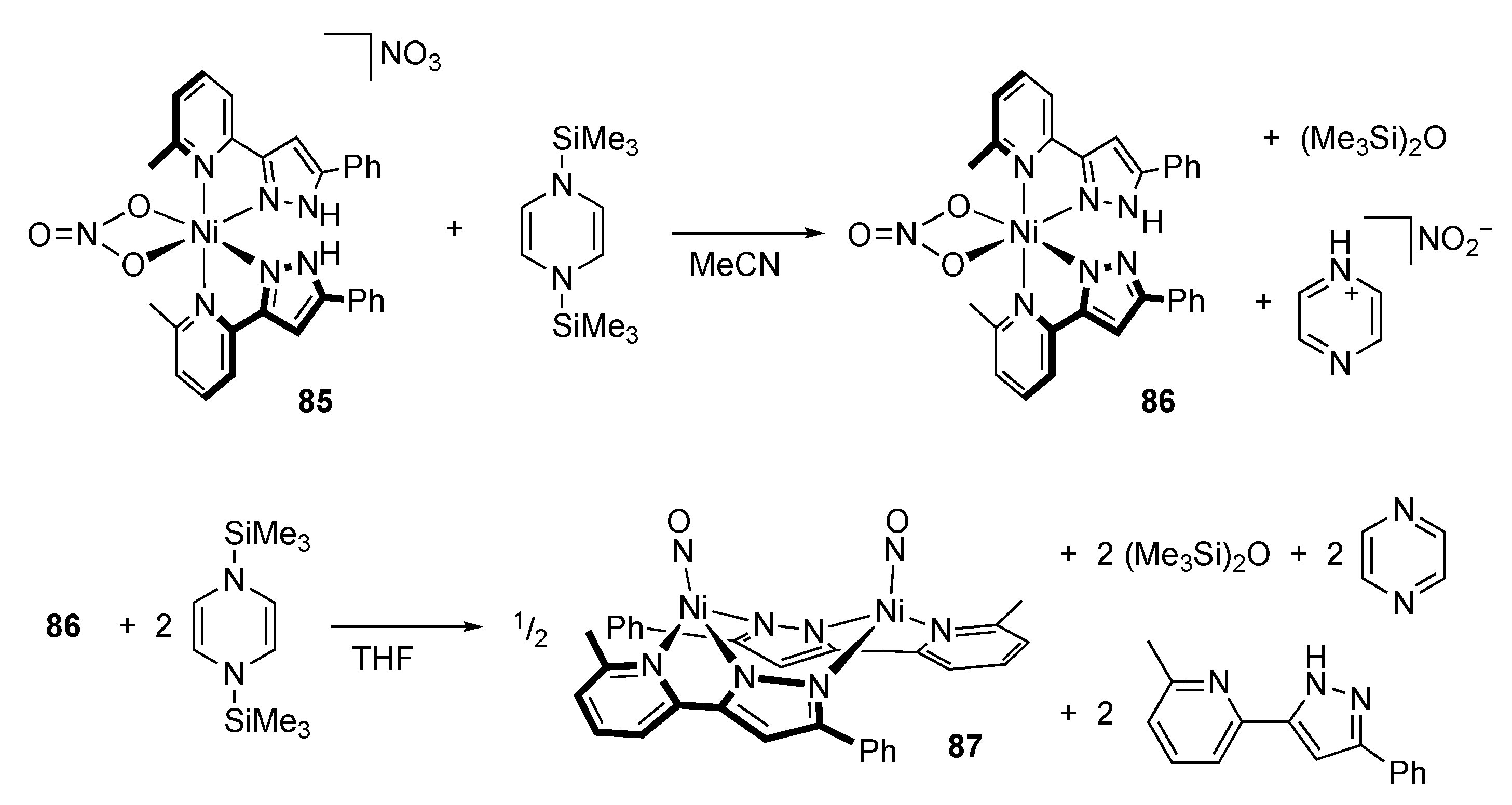
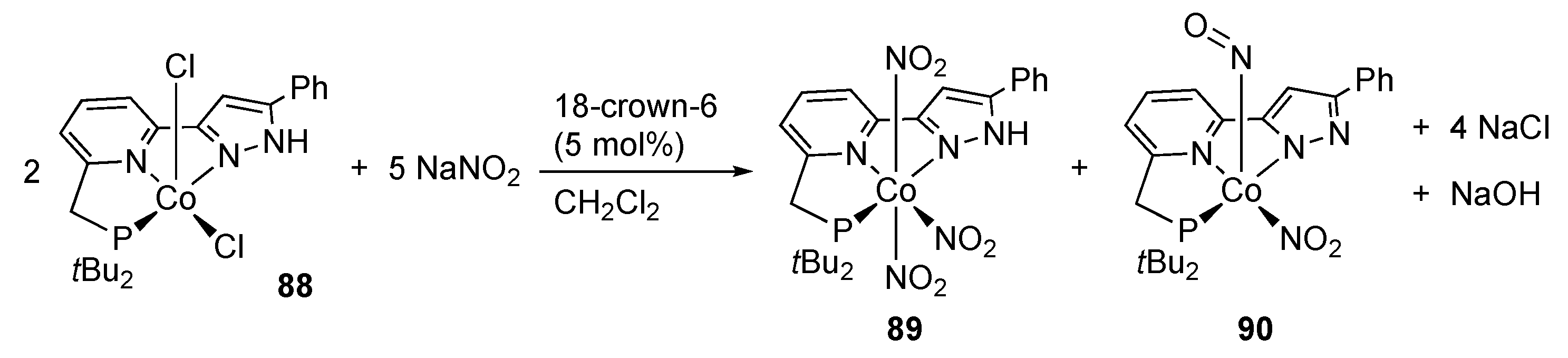
2.3. Nitrate Reduction
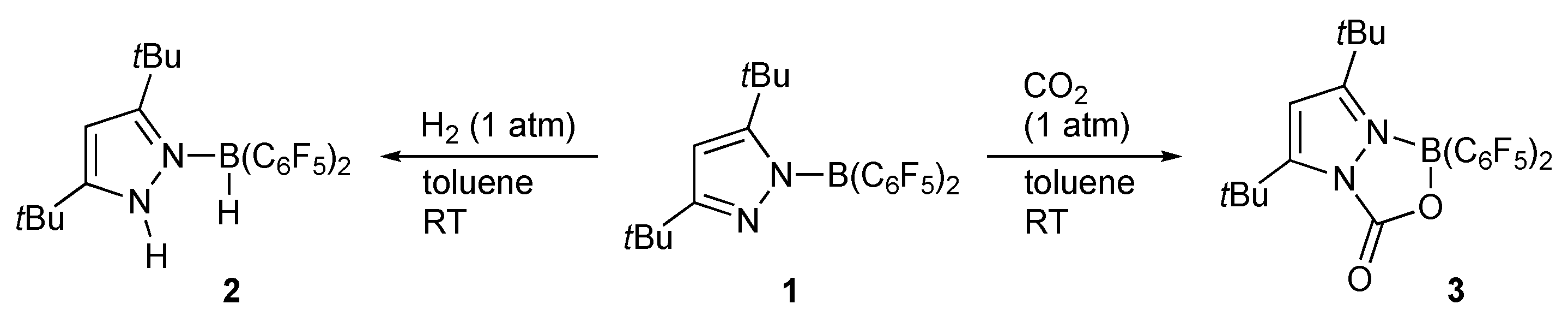
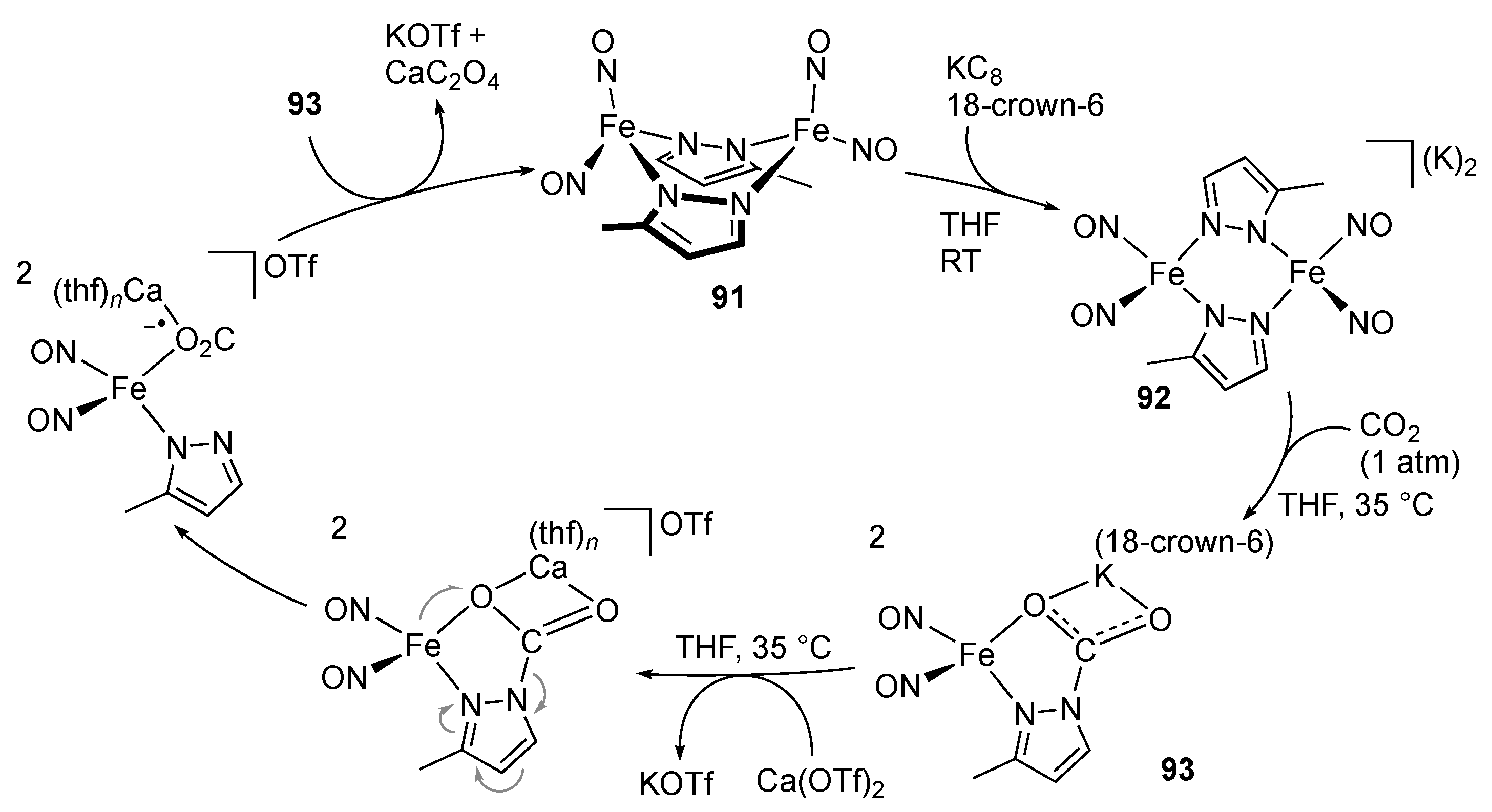
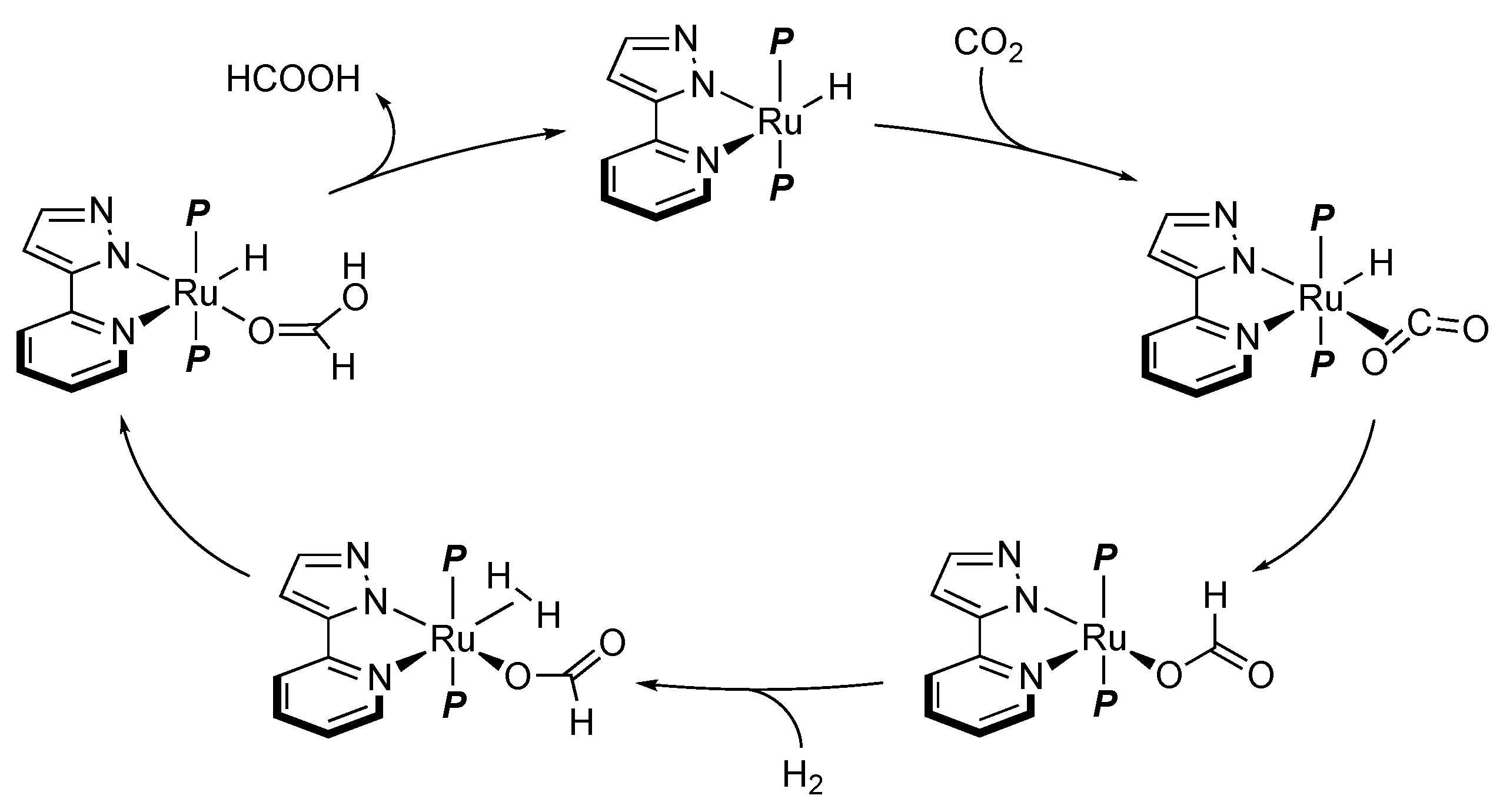
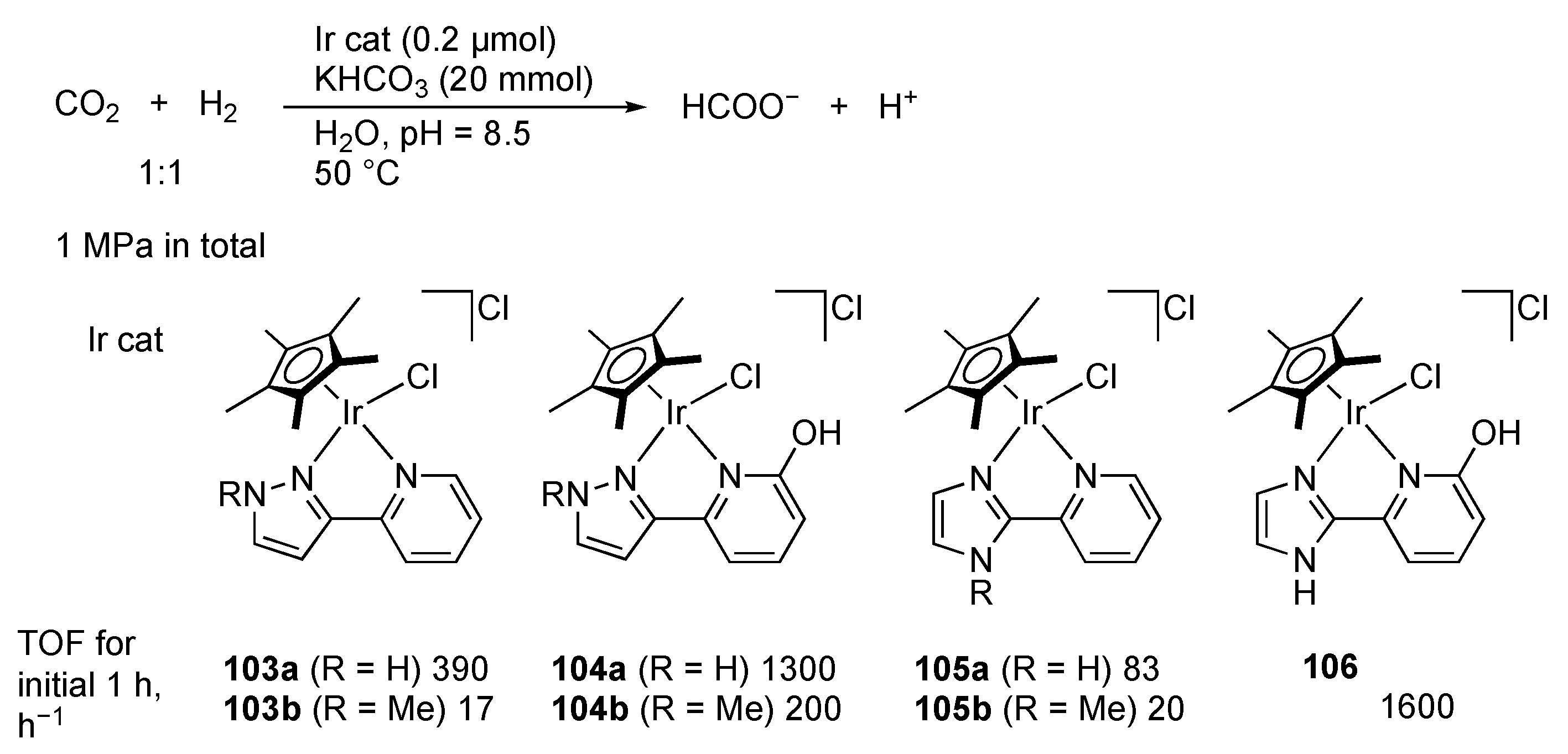
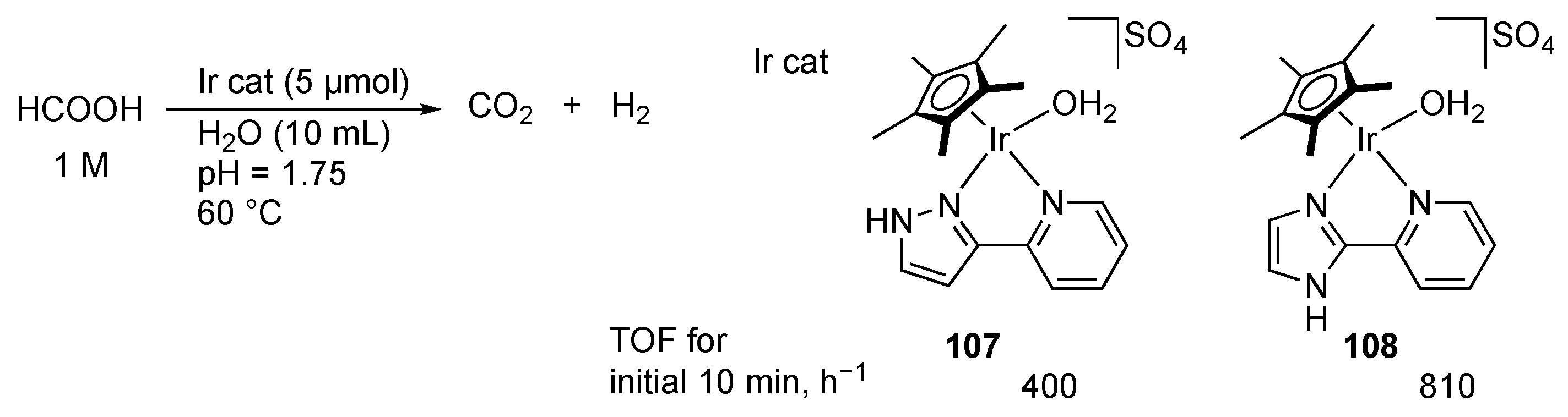
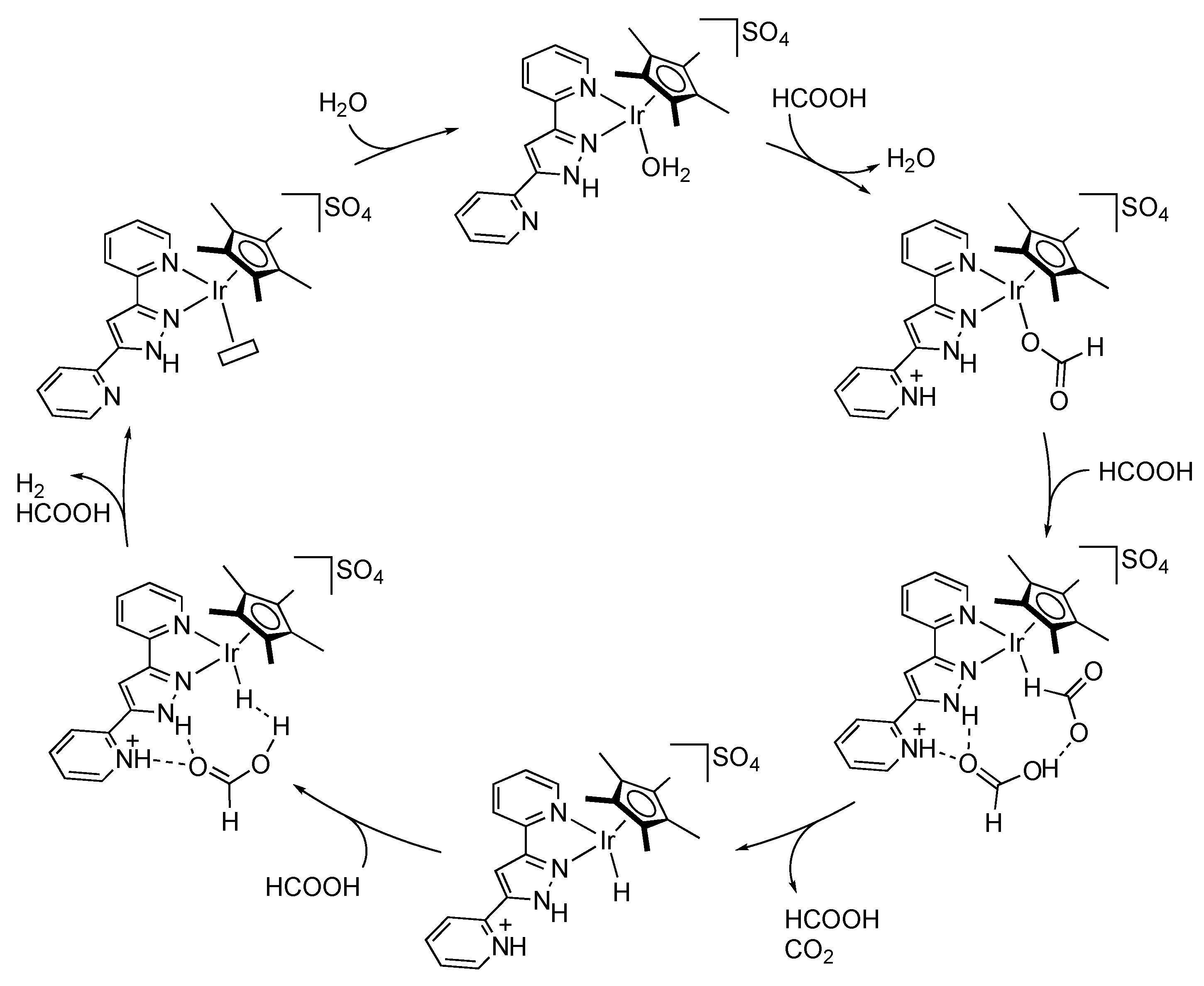
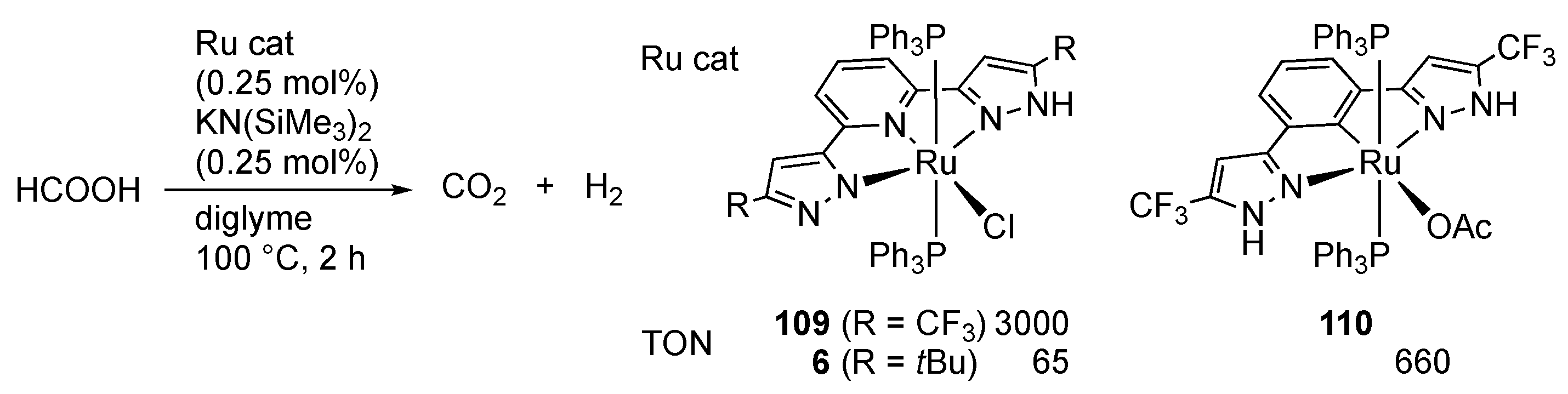
2.4. CO2 Reduction
3. Catalysis of Protic Pyrazole Complexes
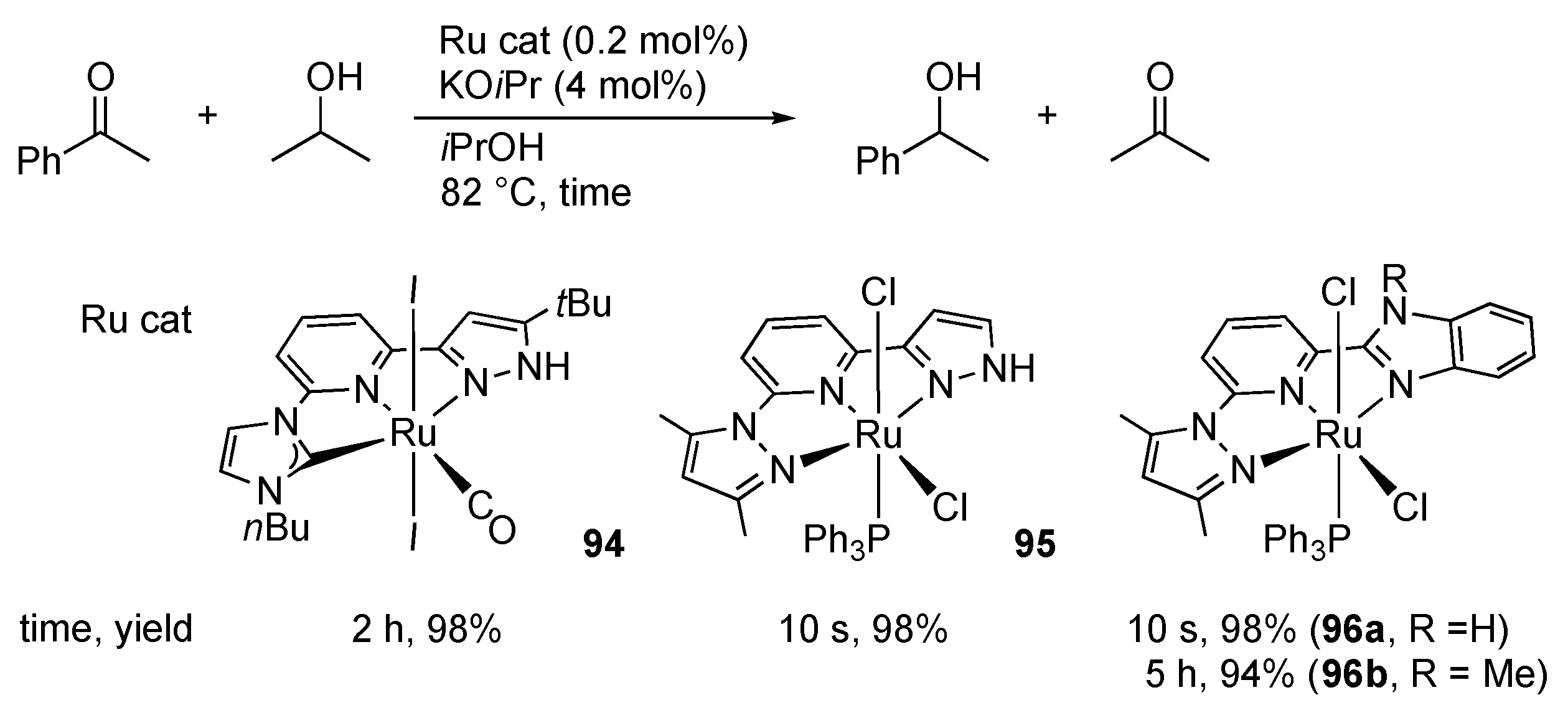
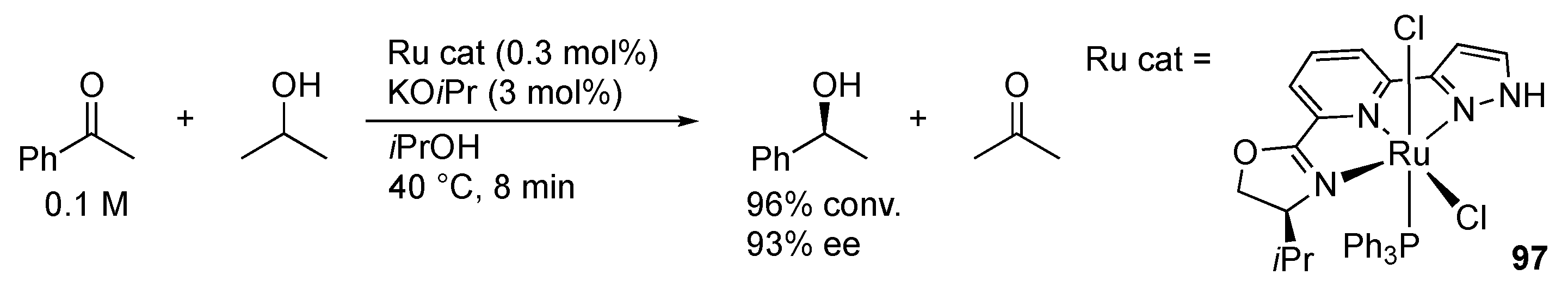
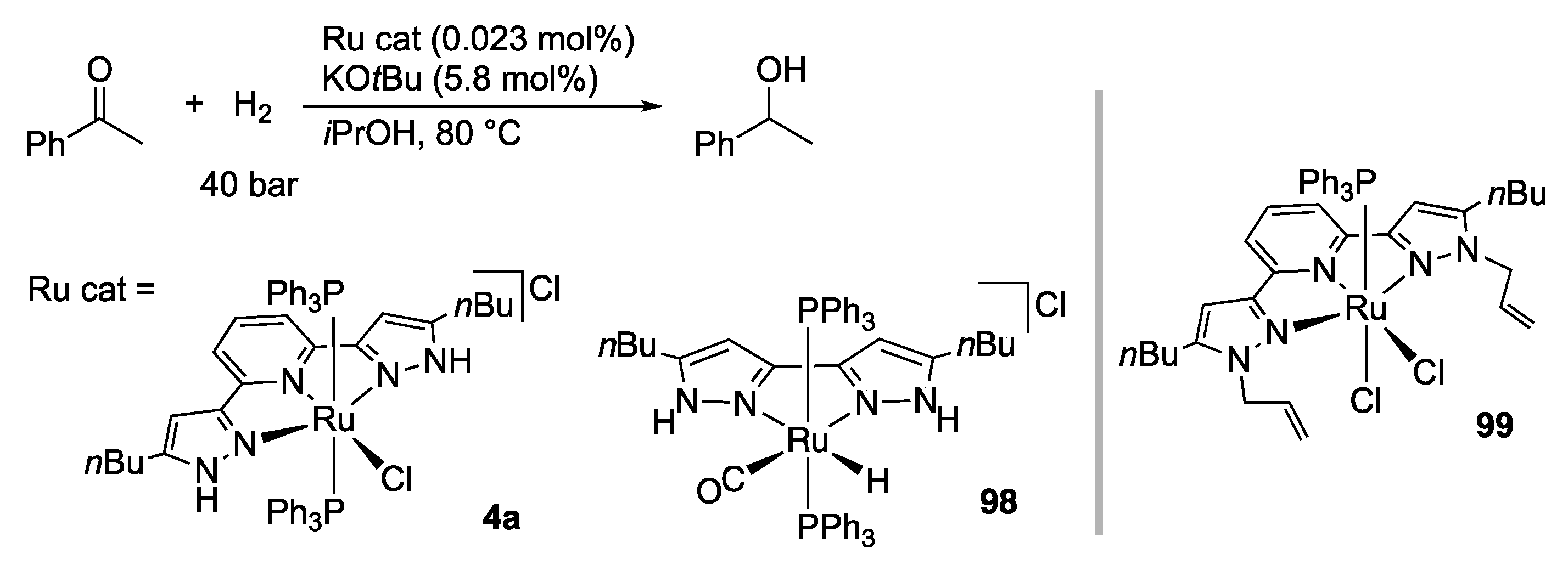
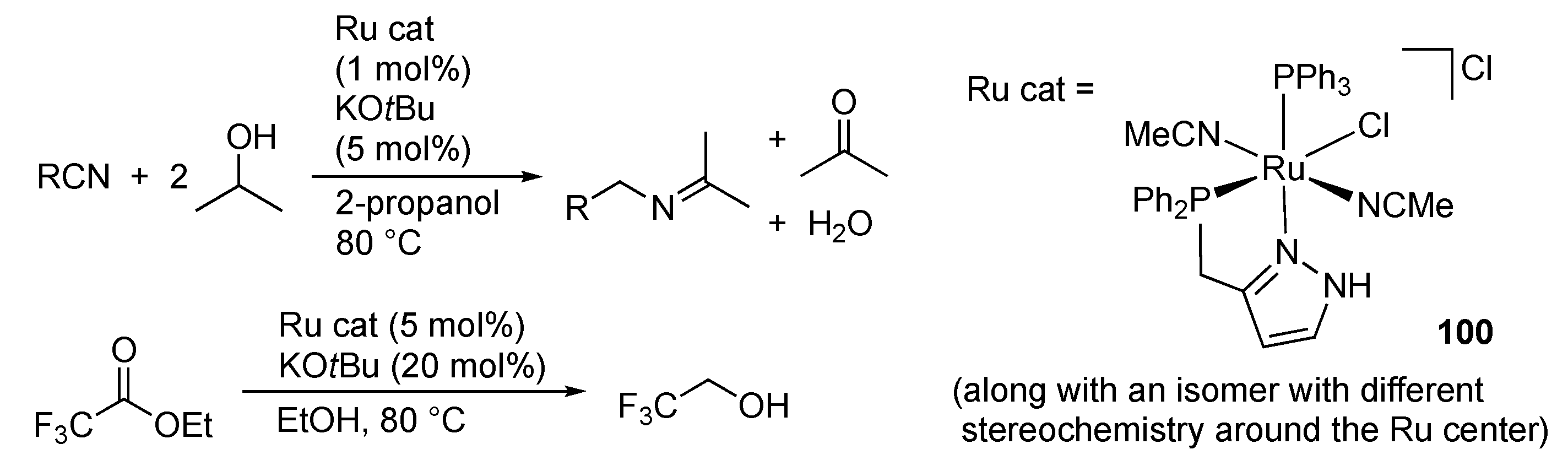
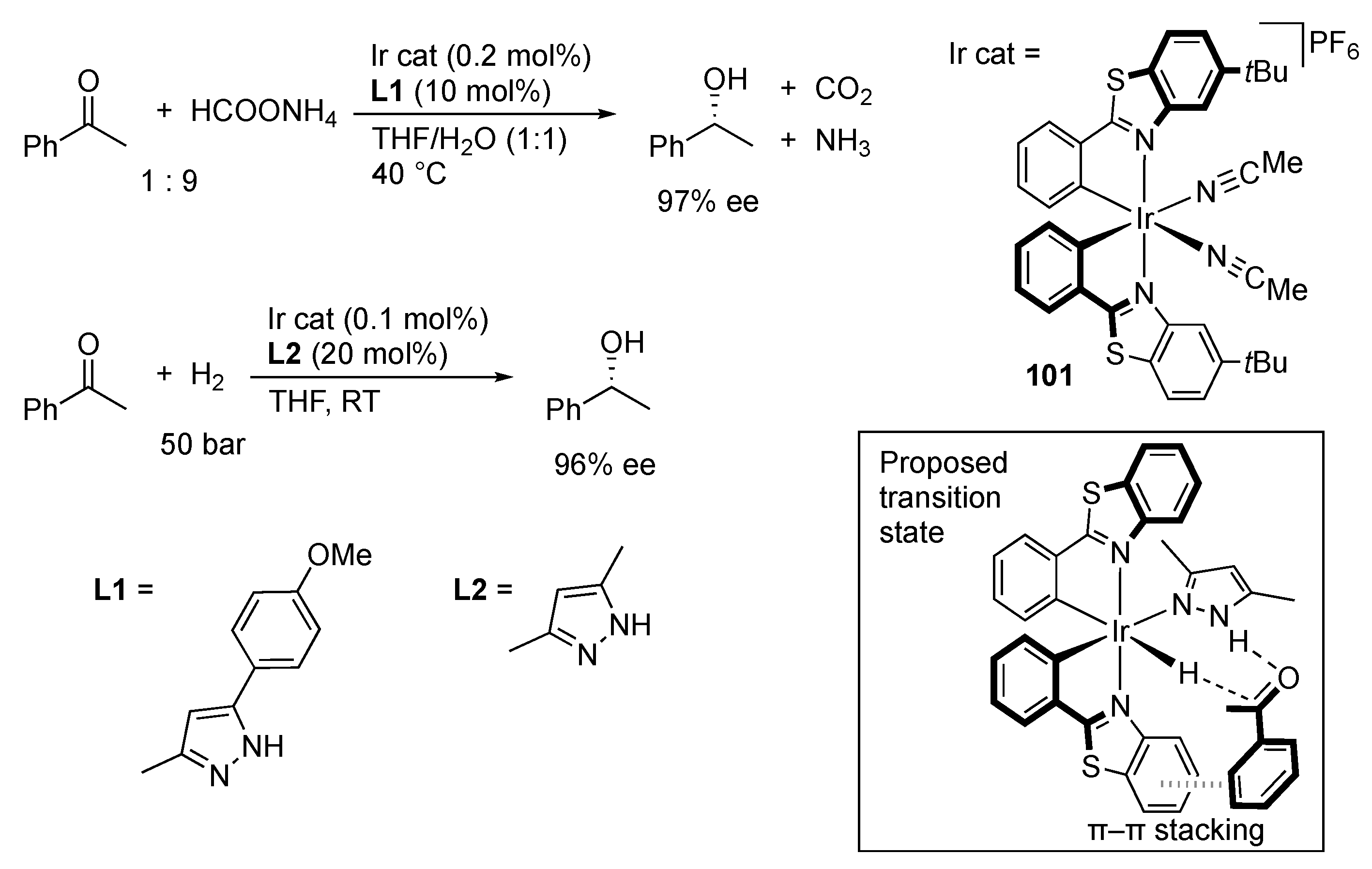
3.1. Hydrogenation and Transfer Hydrogenation
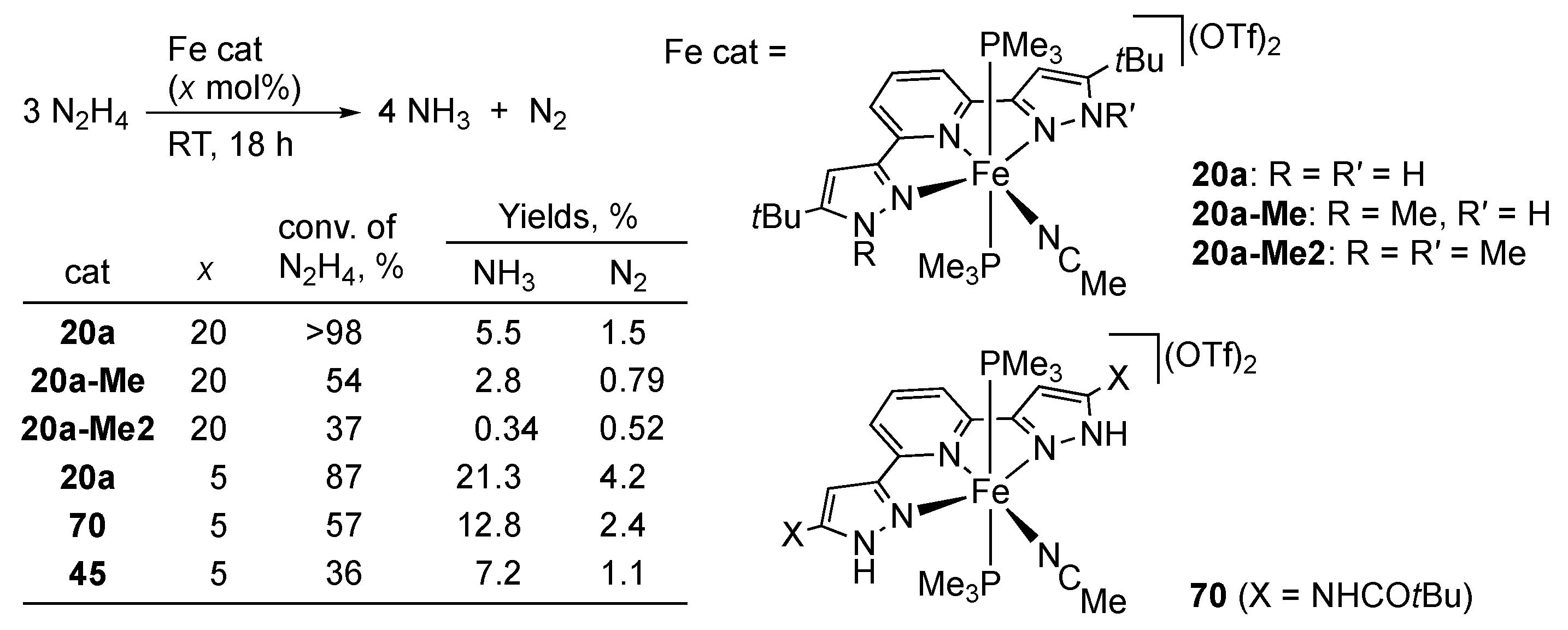
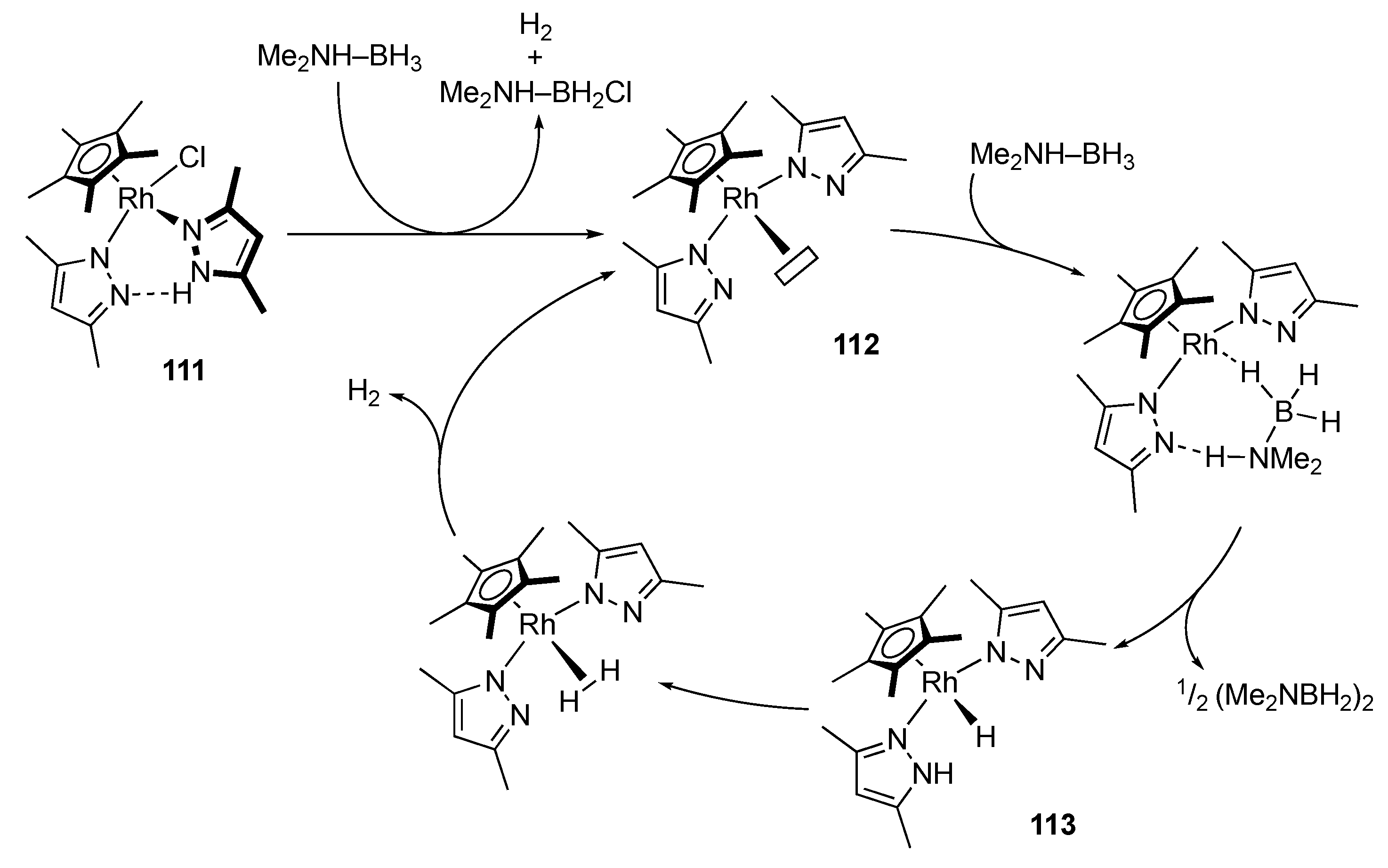
3.2. Hydrogen Evolution
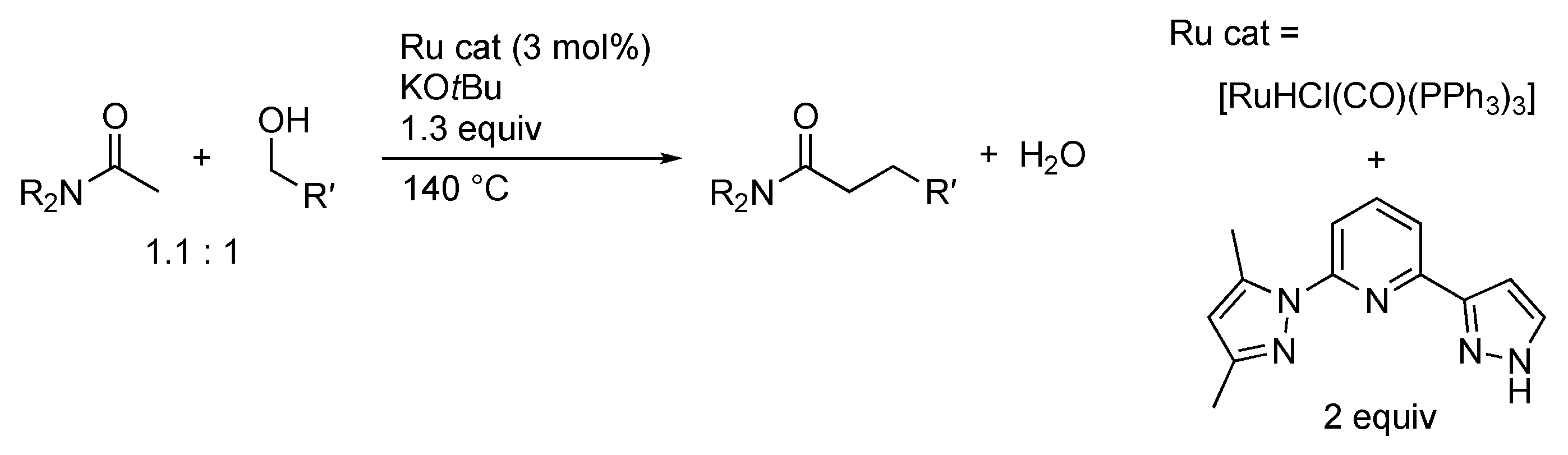
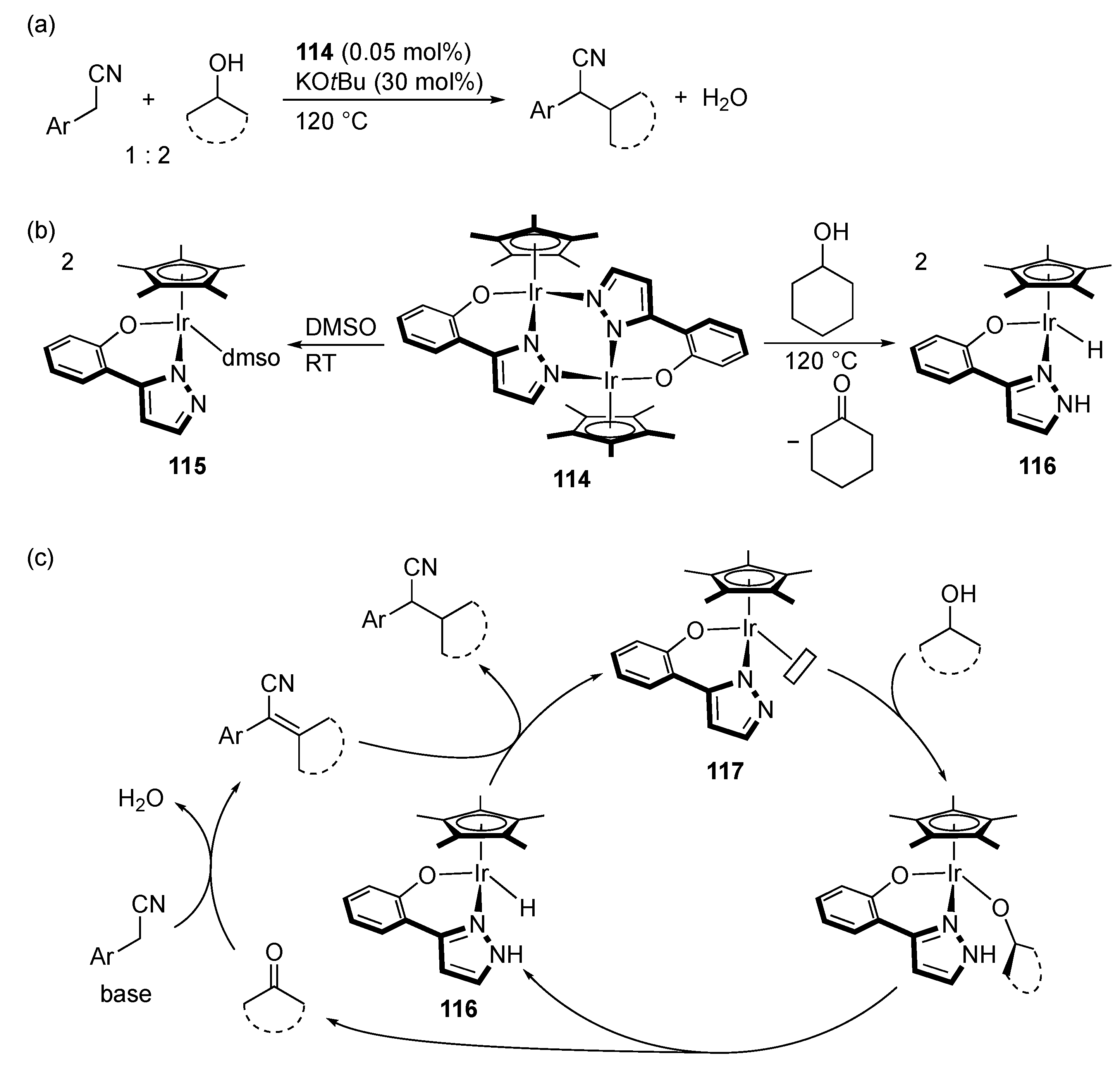
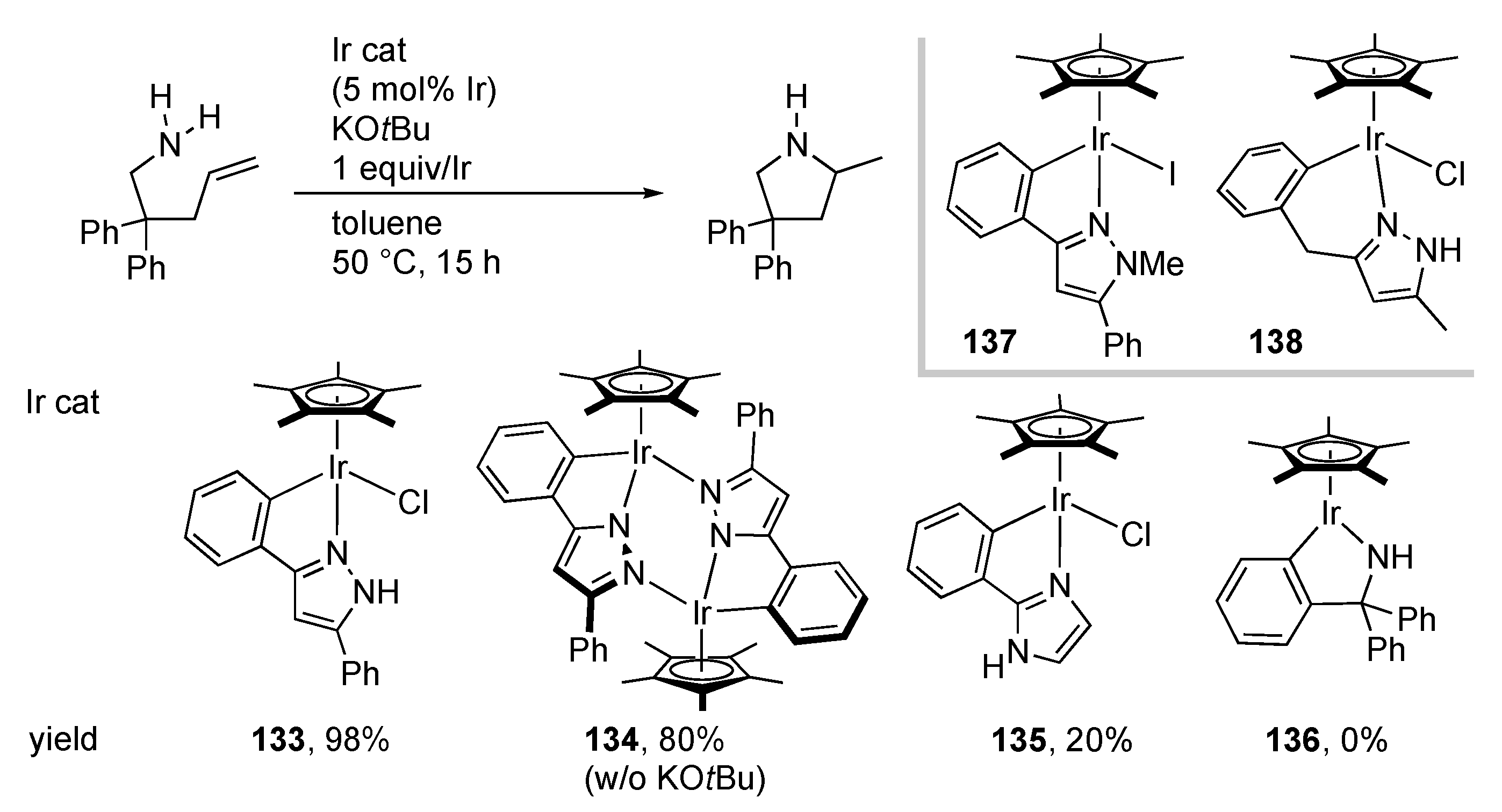
3.3. Borrowing Hydrogen Catalysis
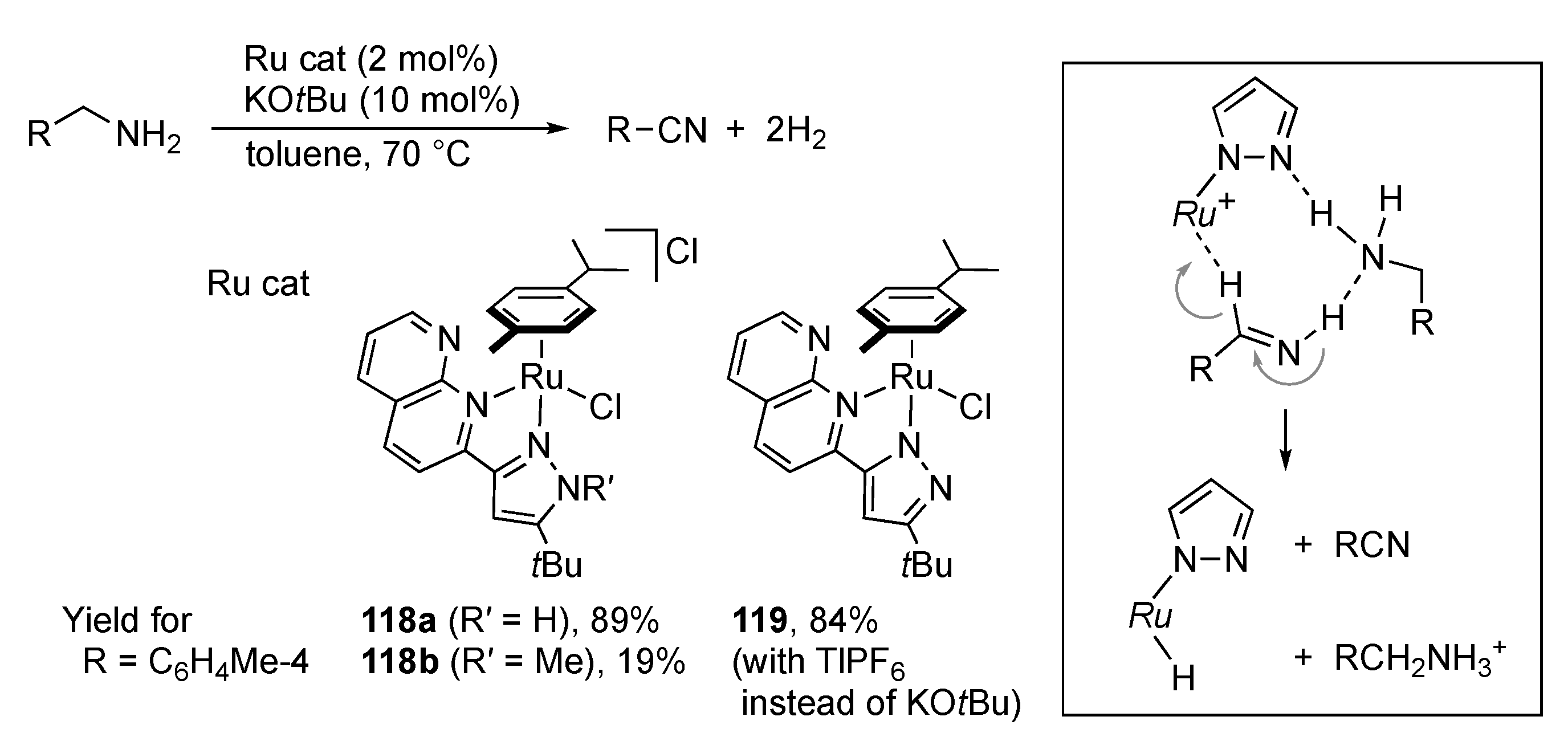
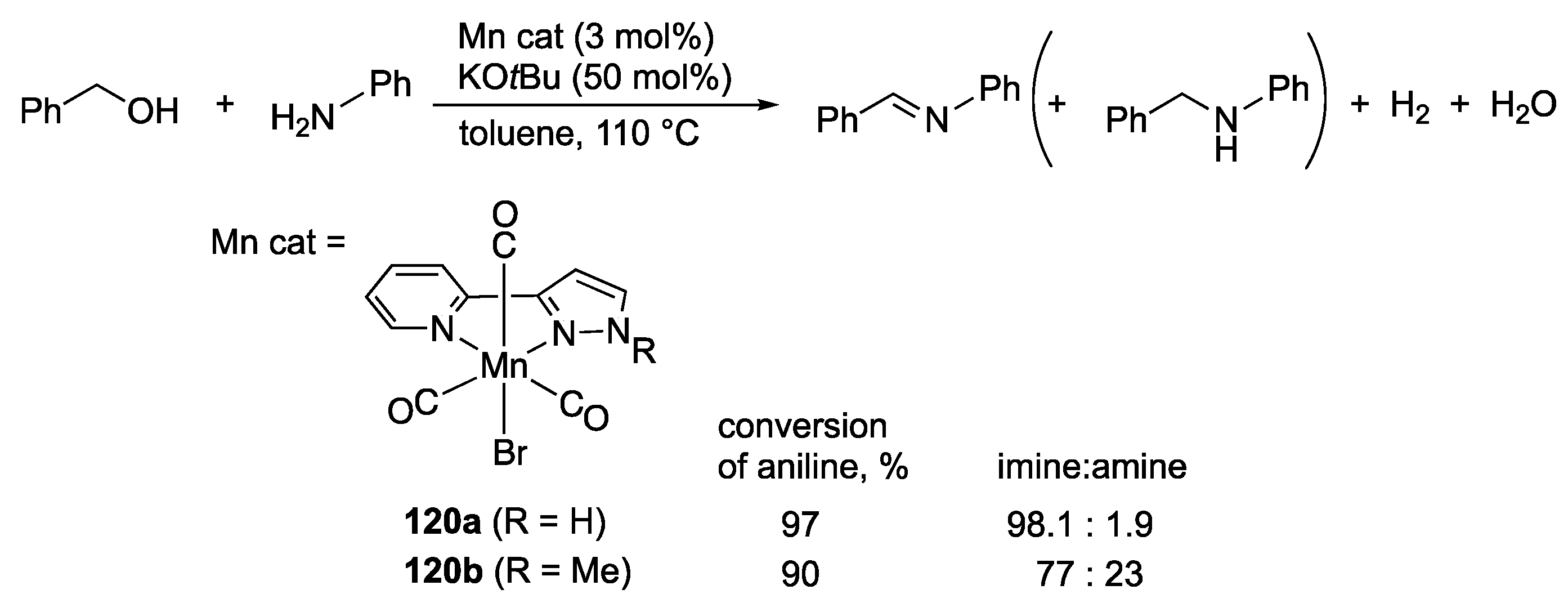
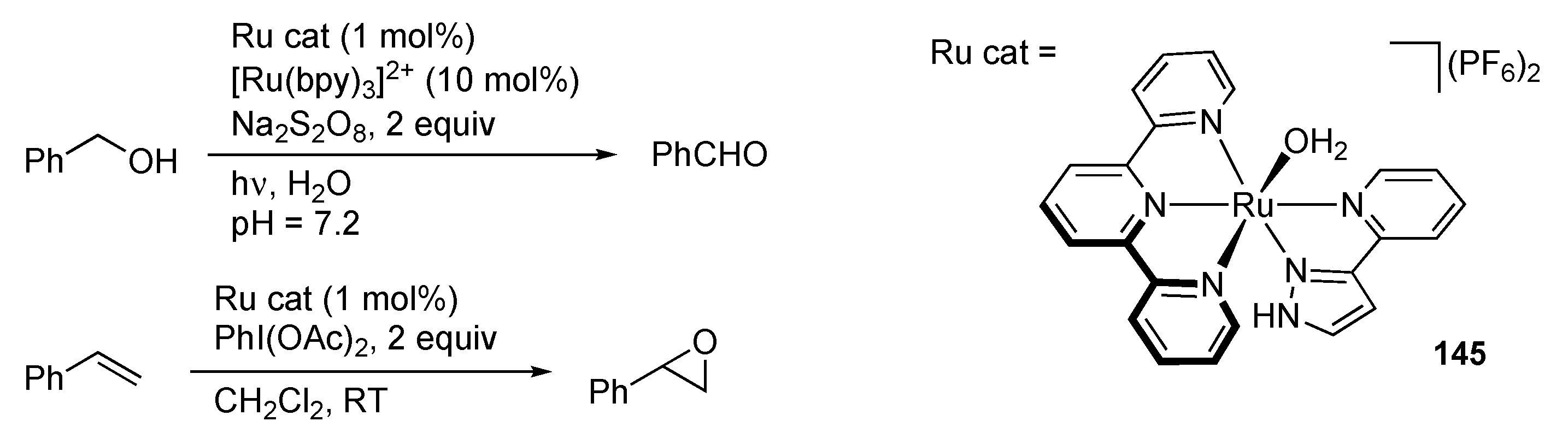
3.4. Dehydrogenative Oxidation
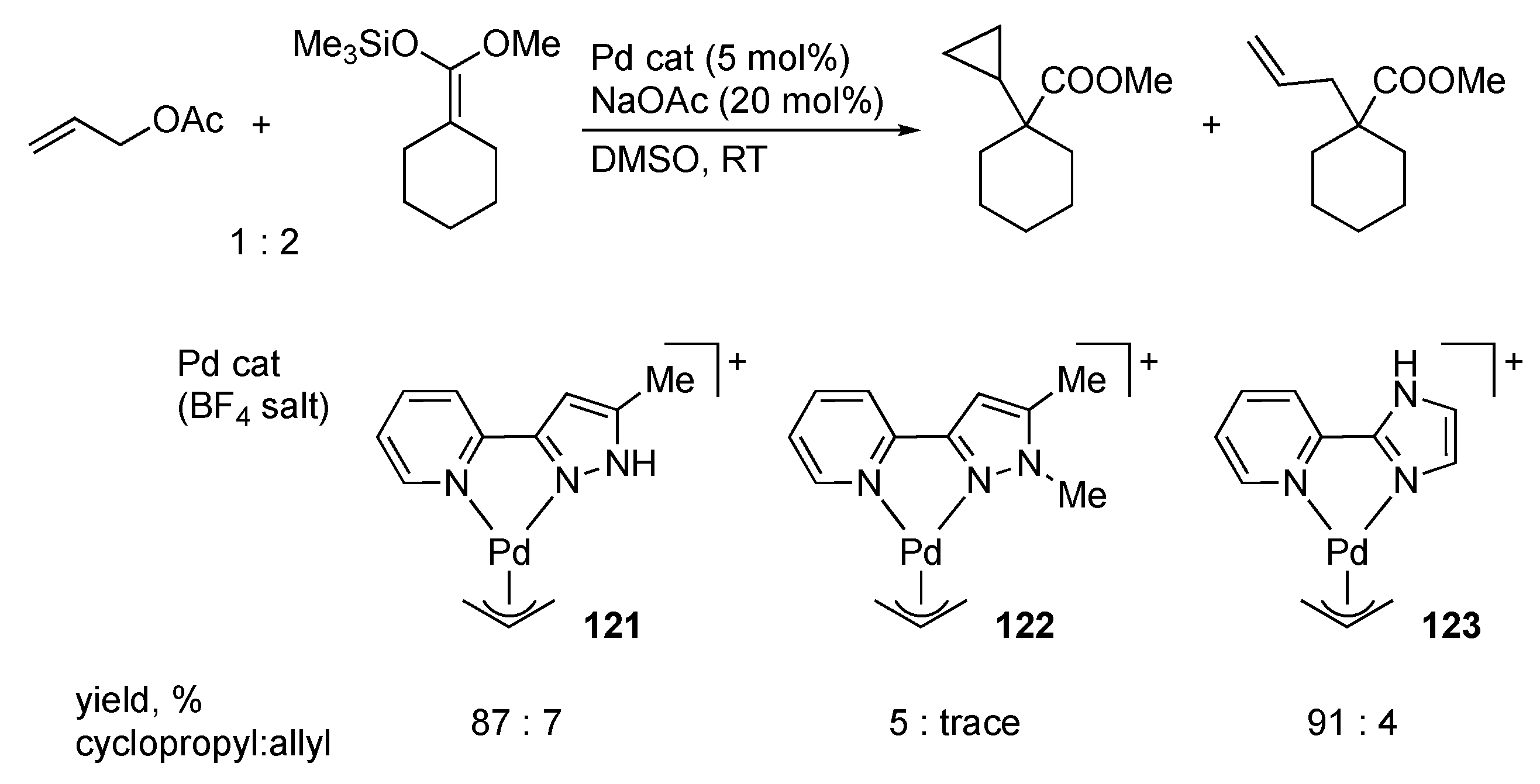
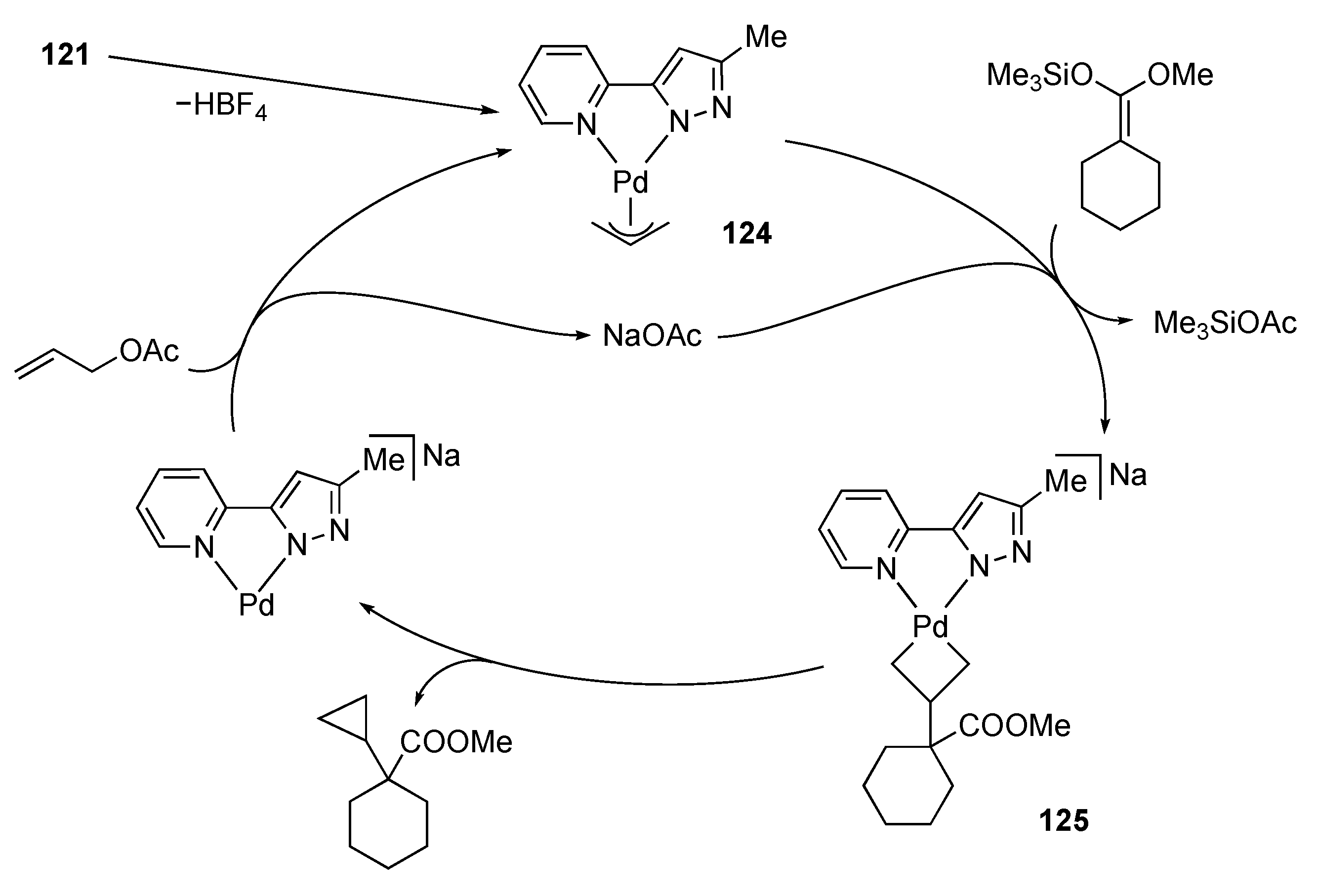
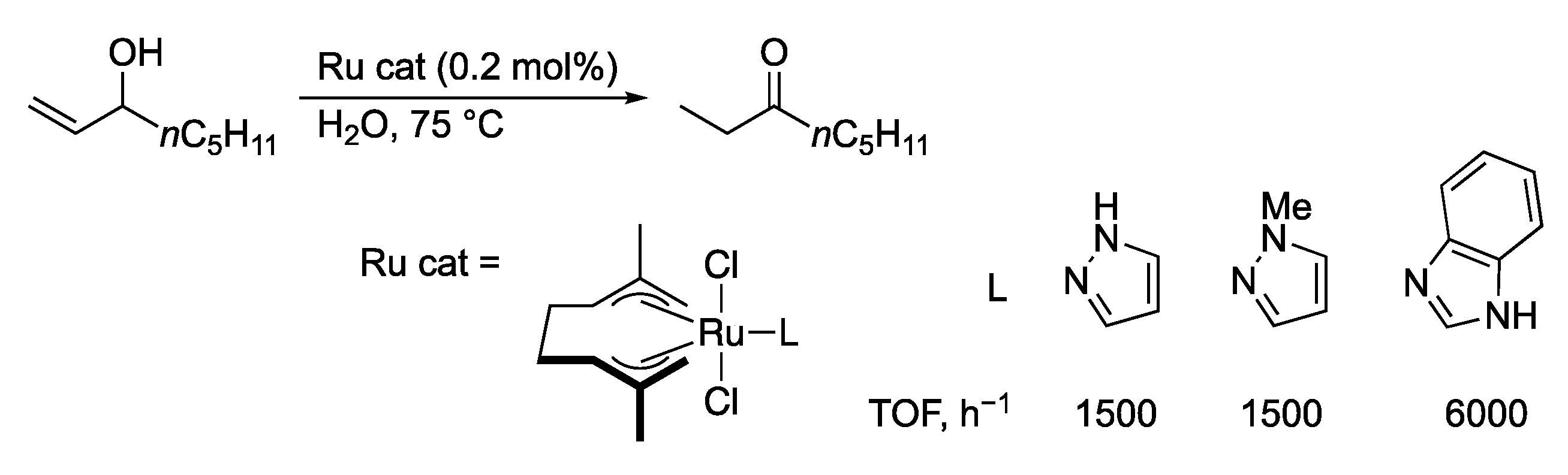
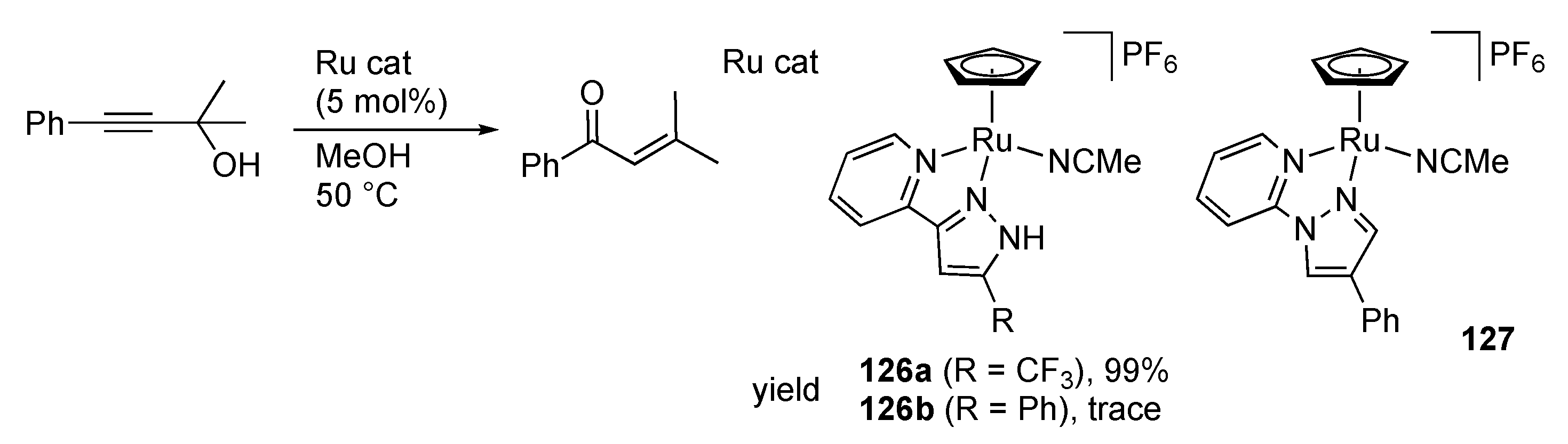
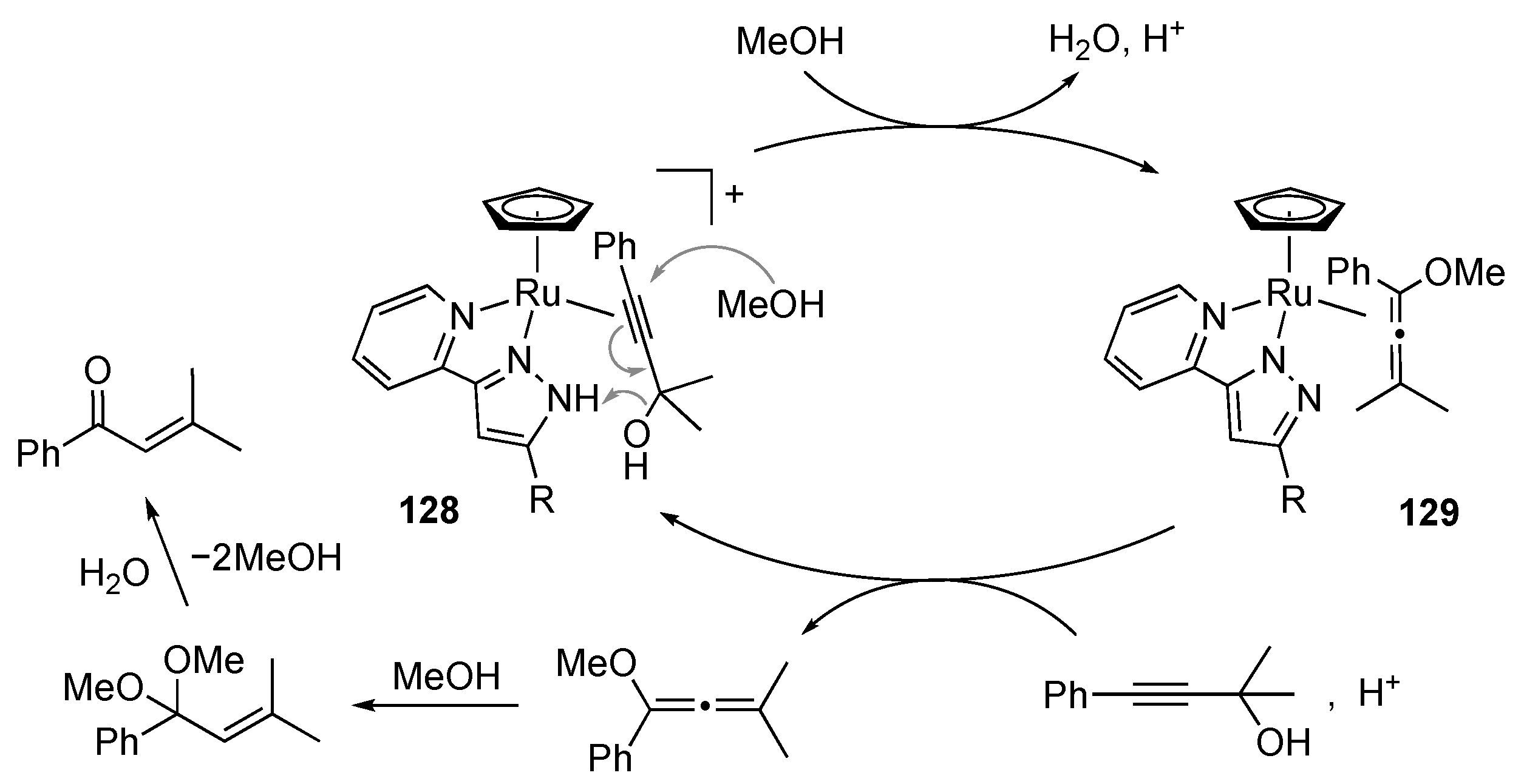
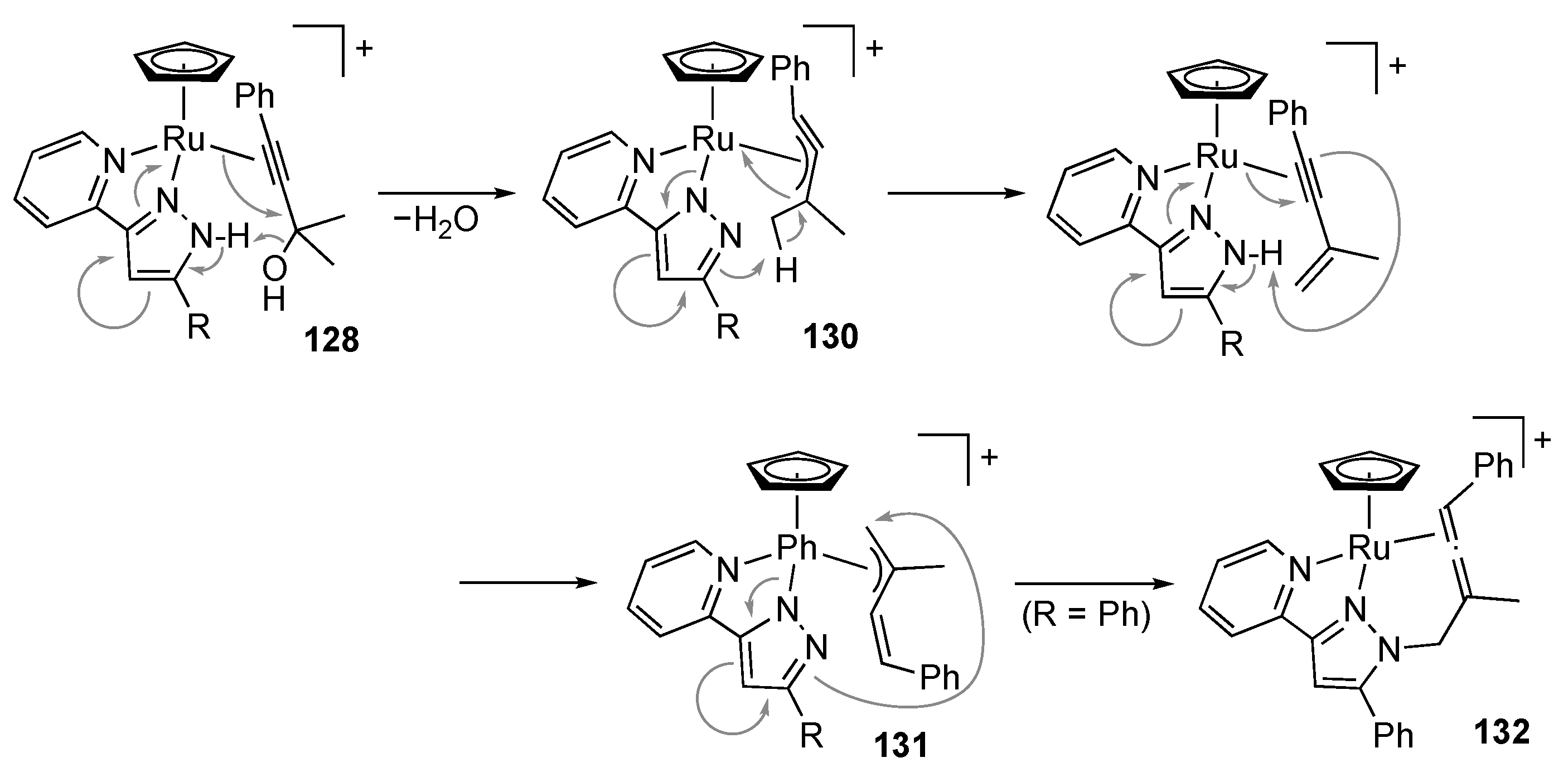
3.5. Transformation of Allylic and Propargylic Compounds
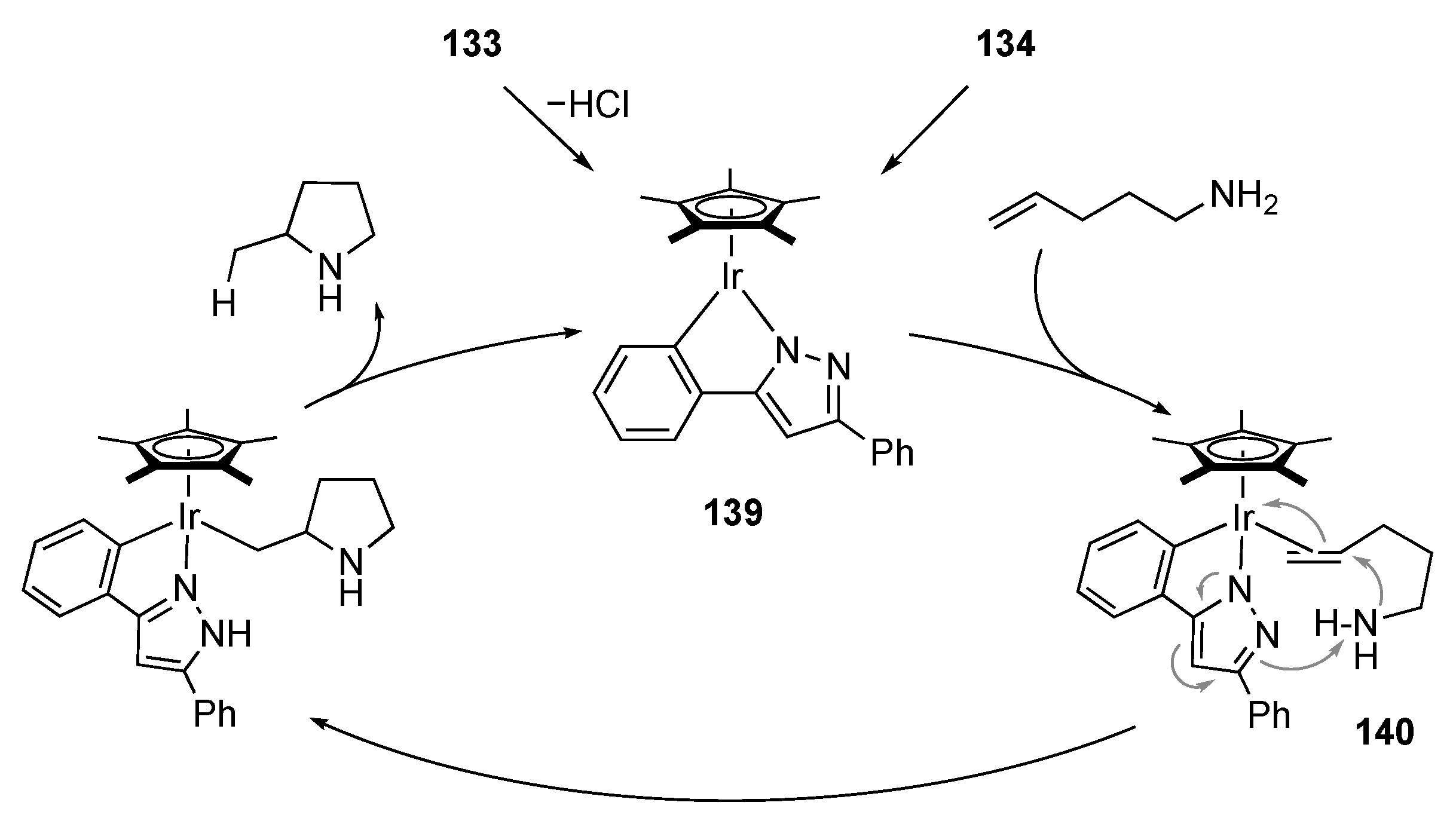
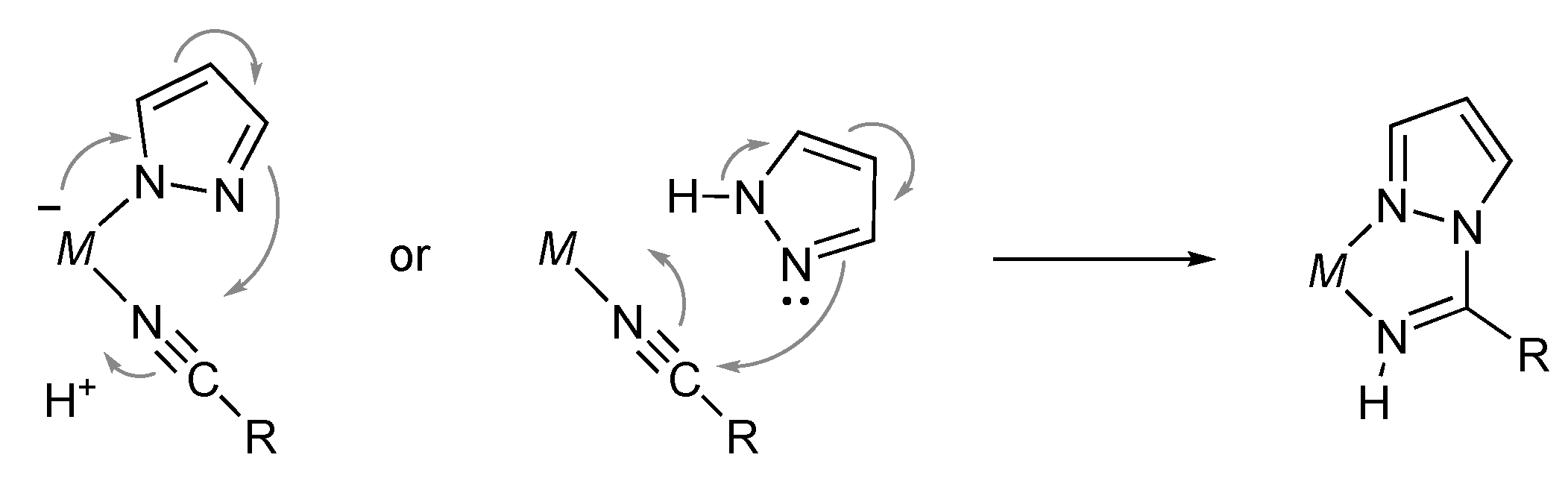
3.6. Hydroamination of Alkenes
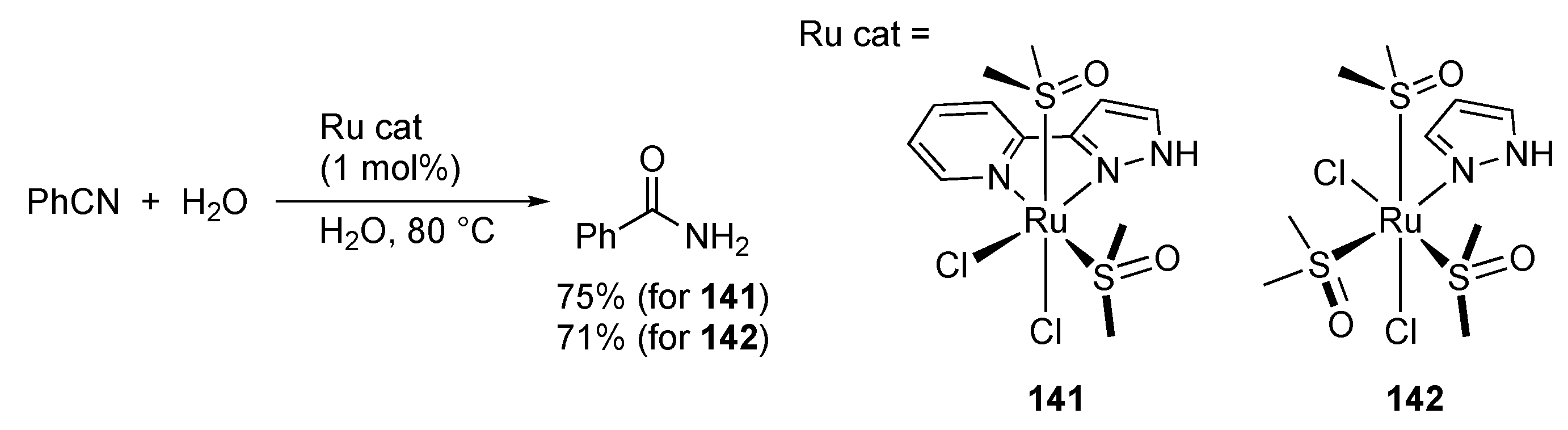
3.7. Hydration of Nitriles
3.8. Catalysis with Coordinatively Saturated Complexes
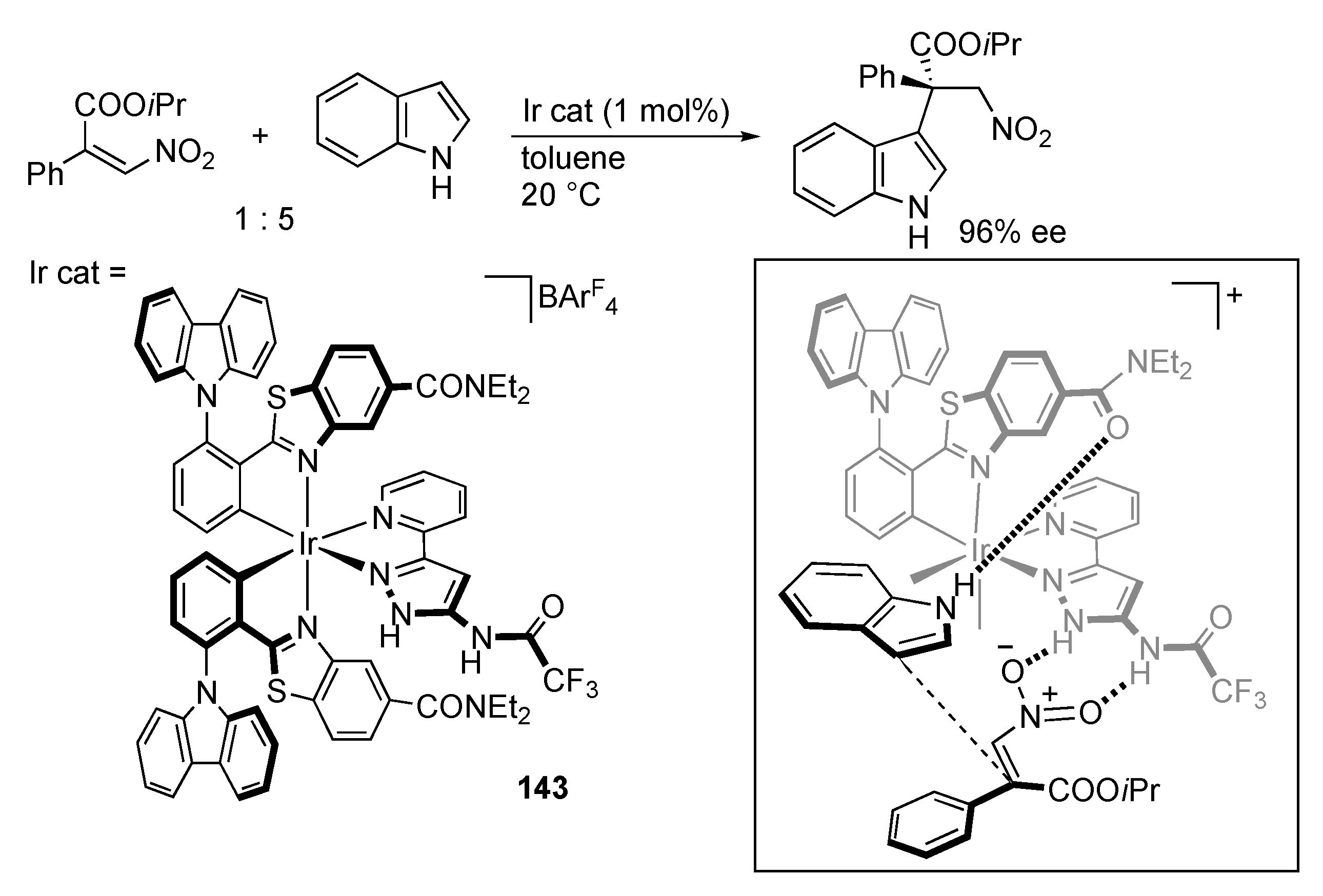
3.9. Miscellaneous
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Trofimenko, S. Recent Advances in Poly(pyrazolyl)borate (Scorpionate) Chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Halcrow, M.A. Pyrazoles and Pyrazolides—Flexible Synthons in Self-Assembly. Dalton Trans. 2009, 38, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.S.S.; Zeni, G. Alkynes and Nitrogen Compounds: Useful Substrates for the Synthesis of Pyrazoles. Chem. Eur. J. 2020, 26, 8175–8189. [Google Scholar] [CrossRef] [PubMed]
- Sloop, J.C.; Holder, C.; Henary, M. Selective Incorporation of Fluorine in Pyrazoles. Eur. J. Org. Chem. 2015, 2015, 3405–3422. [Google Scholar] [CrossRef]
- Bhaumik, P.K.; Ghosh, K.; Chattopadhyay, S. Synthetic Strategies, Crystal Structures and Biological Activities of Metal Complexes with the Members of Azole Family: A Review. Polyhedron 2021, 200, 115093. [Google Scholar] [CrossRef]
- Ionel, H. Inverse Coordination. Organic Nitrogen Heterocycles as Coordination Centers. A Survey of Molecular Topologies and Systematization. Part Five-Membered and Smaller Rings. J. Coord. Chem. 2019, 72, 2127–2159. [Google Scholar] [CrossRef]
- Sethi, S.; Jena, S.; Das, P.K.; Behera, N. Synthetic Approach and Structural Diversities of Pyridylpyrazole Derived Late Transition Metal Complexes. J. Mol. Struct. 2019, 1193, 495–521. [Google Scholar] [CrossRef]
- Doidge, E.D.; Roebuck, J.W.; Healy, M.R.; Tasker, P.A. Phenolic Pyrazoles: Versatile Polynucleating Ligands. Coord. Chem. Rev. 2015, 288, 98–117. [Google Scholar] [CrossRef]
- Lu, C.-W.; Wang, Y.; Chi, Y. Metal Complexes with Azolate-Functionalized Multidentate Ligands: Tactical Designs and Optoelectronic Applications. Chem. Eur. J. 2016, 22, 17892–17908. [Google Scholar] [CrossRef]
- Kuwata, S.; Ikariya, T. β-Protic Pyrazole and N-Heterocyclic Carbene Complexes: Synthesis, Properties, and Metal–Ligand Cooperative Bifunctional Catalysis. Chem. Eur. J. 2011, 17, 3542–3556. [Google Scholar] [CrossRef]
- Kuwata, S.; Ikariya, T. Metal–Ligand Bifunctional Reactivity and Catalysis of Protic N-Heterocyclic Carbene and Pyrazole Complexes Featuring β-NH Units. Chem. Commun. 2014, 50, 14290–14300. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, S. Design of Multiproton-Responsive Metal Complexes as Molecular Technology for Transformation of Small Molecules. In Molecular Technology: Energy Innovation; Yamamoto, H., Kato, T., Eds.; Wiley-VCH: Weinheim, Germany, 2018; pp. 81–103. ISBN 978-3-527-34163-4. [Google Scholar]
- Pérez, J.; Riera, L. Pyrazole Complexes and Supramolecular Chemistry. Eur. J. Inorg. Chem. 2009, 2009, 4913–4925. [Google Scholar] [CrossRef]
- Perez, J.; Riera, L. Organometallic Complexes as Anion Hosts. Chem. Commun. 2008, 44, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Riera, L. Stable Metal–Organic Complexes as Anion Hosts. Chem. Soc. Rev. 2008, 37, 2658–2667. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Shamsuzzaman Review: Biologically Active Pyrazole Derivatives. New J. Chem. 2016, 41, 16–41. [Google Scholar] [CrossRef]
- Khusnutdinova, J.R.; Milstein, D. Metal–Ligand Cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef]
- Kar, S.; Milstein, D. Sustainable Catalysis with Fluxional Acridine-Based PNP Pincer Complexes. Chem. Commun. 2022, 58, 3731–3746. [Google Scholar] [CrossRef]
- Elsby, M.R.; Baker, R.T. Strategies and Mechanisms of Metal–Ligand Cooperativity in First-Row Transition Metal Complex Catalysts. Chem. Soc. Rev. 2020, 49, 8933–8987. [Google Scholar] [CrossRef]
- Cotman, A.E. Escaping from Flatland: Stereoconvergent Synthesis of Three-Dimensional Scaffolds via Ruthenium(II)-Catalyzed Noyori–Ikariya Transfer Hydrogenation. Chem. Eur. J. 2021, 27, 39–53. [Google Scholar] [CrossRef]
- Akter, M.; Anbarasan, P. (Cyclopentadienone)iron Complexes: Synthesis, Mechanism and Applications in Organic Synthesis. Chem. Asian J. 2021, 16, 1703–1724. [Google Scholar] [CrossRef]
- Gonçalves, T.P.; Dutta, I.; Huang, K.-W. Aromaticity in Catalysis: Metal Ligand Cooperation via Ligand Dearomatization and Rearomatization. Chem. Commun. 2021, 57, 3070–3082. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K. Development and Application of New Iridium Catalysts for Efficient Dehydrogenative Reactions of Organic Molecules. Bull. Chem. Soc. Jpn. 2019, 92, 344–351. [Google Scholar] [CrossRef]
- Matsunami, A.; Kayaki, Y. Upgrading and Expanding the Scope of Homogeneous Transfer Hydrogenation. Tetrahedron Lett. 2018, 59, 504–513. [Google Scholar] [CrossRef]
- Hale, L.V.A.; Szymczak, N.K. Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere. ACS Catal. 2018, 8, 6446–6461. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The Role of the Metal-Bound N–H Functionality in Noyori-Type Molecular Catalysts. Nat. Rev. Chem. 2018, 2, 396–408. [Google Scholar] [CrossRef]
- Theuergarten, E.; Schlüns, D.; Grunenberg, J.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. Intramolecular Heterolytic Dihydrogen Cleavage by a Bifunctional Frustrated Pyrazolylborane Lewis Pair. Chem. Commun. 2010, 46, 8561–8563. [Google Scholar] [CrossRef]
- Theuergarten, E.; Schlösser, J.; Schlüns, D.; Freytag, M.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. Fixation of Carbon Dioxide and Related Small Molecules by a Bifunctional Frustrated Pyrazolylborane Lewis Pair. Dalton Trans. 2012, 41, 9101–9110. [Google Scholar] [CrossRef]
- Cook, L.J.K.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin State Behavior of Iron(II)/Dipyrazolylpyridine Complexes. New Insights from Crystallographic and Solution Measurements. Coord. Chem. Rev. 2015, 289–290, 2–12. [Google Scholar] [CrossRef]
- Halcrow, M.A. Recent Advances in the Synthesis and Applications of 2,6-Dipyrazolylpyridine Derivatives and Their Complexes. New J. Chem. 2014, 38, 1868–1882. [Google Scholar] [CrossRef]
- Craig, G.A.; Roubeau, O.; Aromí, G. Spin State Switching in 2,6-Bis(pyrazol-3-yl)pyridine (3-Bpp) Based Fe(II) Complexes. Coord. Chem. Rev. 2014, 269, 13–31. [Google Scholar] [CrossRef]
- Olguín, J.; Brooker, S. Spin Crossover Active Iron(II) Complexes of Selected Pyrazole-Pyridine/Pyrazine Ligands. Coord. Chem. Rev. 2011, 255, 203–240. [Google Scholar] [CrossRef]
- Halcrow, M.A. The Synthesis and Coordination Chemistry of 2,6-Bis(pyrazolyl)pyridines and Related Ligands—Versatile Terpyridine Analogues. Coord. Chem. Rev. 2005, 249, 2880–2908. [Google Scholar] [CrossRef]
- Li, T.-Y.; Wu, J.; Wu, Z.-G.; Zheng, Y.-X.; Zuo, J.-L.; Pan, Y. Rational Design of Phosphorescent Iridium(III) Complexes for Emission Color Tunability and Their Applications in OLEDs. Coord. Chem. Rev. 2018, 374, 55–92. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Van, S.; Combs, D.; Lev, D.A.; Schneider, C.; Incarvito, C.D.; Lam, K.-C.; Rossi, G.; Rheingold, A.L.; Rideout, M.; et al. Substituent Control of Hydrogen Bonding in Palladium(II)-Pyrazole Complexes. Inorg. Chem. 2003, 42, 3347–3355. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Van, S.; Combs, D.; Lev, D.A.; Schneider, C.; Rideout, M.; Meyer, C.; Hernandez, G.; Mejorado, L. New Flexible Synthesis of Pyrazoles with Different, Functionalized Substituents at C3 and C5. J. Org. Chem. 2002, 67, 9200–9209. [Google Scholar] [CrossRef]
- Grotjahn, D.B. Bifunctional Catalysts and Related Complexes: Structures and Properties. Dalton Trans. 2008, 37, 6497–6508. [Google Scholar] [CrossRef]
- Labrum, N.S.; Chen, C.-H.; Caulton, K.G. A New Face for Bis(pyrazol-3-yl)pyridine: Incompatible Geometric Preferences Dictates Unprecedented Pincer Ligand Connectivity. Inorg. Chim. Acta 2019, 485, 54–57. [Google Scholar] [CrossRef]
- Jozak, T.; Zabel, D.; Schubert, A.; Thiel, W.R. Ruthenium Complexes Bearing N–H Acidic Pyrazole Ligands. Eur. J. Inorg. Chem. 2010, 2010, 5135–5145. [Google Scholar] [CrossRef]
- Yoshinari, A.; Tazawa, A.; Kuwata, S.; Ikariya, T. Synthesis, Structures, and Reactivities of Pincer-Type Ruthenium Complexes Bearing Two Proton-Responsive Pyrazole Arms. Chem. Asian J. 2012, 7, 1417–1425. [Google Scholar] [CrossRef]
- Kashiwame, Y.; Watanabe, M.; Araki, K.; Kuwata, S.; Ikariya, T. Synthesis, Structure, and Proton-Transfer Reactions of Brønsted Acidic Pyridylpyrazole Complexes of Ruthenium. Bull. Chem. Soc. Jpn. 2011, 84, 251–258. [Google Scholar] [CrossRef]
- Liao, J.-L.; Chi, Y.; Su, Y.-D.; Huang, H.-X.; Chang, C.-H.; Liu, S.-H.; Lee, G.-H.; Chou, P.-T. Os(II) Metal Phosphors Bearing Tridentate 2,6-Di(pyrazol-3-yl)pyridine Chelate: Synthetic Design, Characterization and Application in OLED Fabrication. J. Mater. Chem. C 2014, 2, 6269–6282. [Google Scholar] [CrossRef]
- Petrović, A.Z.; Ćoćić, D.C.; Bockfeld, D.; Živanović, M.; Milivojević, N.; Virijević, K.; Janković, N.; Scheurer, A.; Vraneš, M.; Bogojeski, J.V. Biological Activity of Bis(pyrazolylpyridine) and Terpiridine Os(II) Complexes in the Presence of Biocompatible Ionic Liquids. Inorg. Chem. Front. 2021, 8, 2749–2770. [Google Scholar] [CrossRef]
- Kuo, J.L.; Goldberg, K.I. Metal/Ligand Proton Tautomerism Facilitates Dinuclear H2 Reductive Elimination. J. Am. Chem. Soc. 2020, 142, 21439–21449. [Google Scholar] [CrossRef] [PubMed]
- Milutinović, M.M.; Bogojeski, J.V.; Klisurić, O.; Scheurer, A.; Elmroth, S.K.C.; Bugarčić, Z.D. Synthesis and Structures of a Pincer-Type Rhodium(III) Complex: Reactivity toward Biomolecules. Dalton Trans. 2016, 45, 15481–15491. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Saitoh, K.; Yoshinari, A.; Ikariya, T.; Kuwata, S. Synthesis and Structures of NCN Pincer-Type Ruthenium and Iridium Complexes Bearing Protic Pyrazole Arms. Organometallics 2017, 36, 1188–1195. [Google Scholar] [CrossRef]
- Zahora, B.A.; Gau, M.R.; Goldberg, K.I. Synthesis and Reactivity of PtII Methyl Complexes Supported by Pyrazolate Pincer Ligands. Organometallics 2020, 39, 1230–1237. [Google Scholar] [CrossRef]
- Galstyan, A.; Naziruddin, A.R.; Cebrián, C.; Iordache, A.; Daniliuc, C.G.; Cola, L.D.; Strassert, C.A. Correlating the Structural and Photophysical Features of Pincer Luminophores and Monodentate Ancillary Ligands in PtII Phosphors. Eur. J. Inorg. Chem. 2015, 2015, 5822–5831. [Google Scholar] [CrossRef]
- Umehara, K.; Kuwata, S.; Ikariya, T. Synthesis, Structures, and Reactivities of Iron, Cobalt, and Manganese Complexes Bearing a Pincer Ligand with Two Protic Pyrazole Arms. Inorg. Chim. Acta 2014, 413, 136–142. [Google Scholar] [CrossRef]
- Umehara, K.; Kuwata, S.; Ikariya, T. N–N Bond Cleavage of Hydrazines with a Multiproton-Responsive Pincer-Type Iron Complex. J. Am. Chem. Soc. 2013, 135, 6754–6757. [Google Scholar] [CrossRef]
- Nikovsky, I.A.; Polezhaev, A.V.; Melnikova, E.K.; Nelyubina, Y.V. New Iron(III) Oxo Complex with Substituted 2,6-Bis(pyrazol-3-yl)pyridine. Russ. J. Inorg. Chem. 2020, 65, 864–869. [Google Scholar] [CrossRef]
- Cook, B.J.; Polezhaev, A.V.; Chen, C.-H.; Pink, M.; Caulton, K.G. Deprotonation, Chloride Abstraction, and Dehydrohalogenation as Synthetic Routes to Bis-Pyrazolate Pyridyl Iron(II) Complexes. Eur. J. Inorg. Chem. 2017, 2017, 3999–4012. [Google Scholar] [CrossRef]
- Cook, B.J.; Chen, C.-H.; Pink, M.; Caulton, K.G. Dehydrohalogenation of Proton Responsive Complexes: Versatile Aggregation Viapyrazolate Pincer Ligand Arms. Dalton Trans. 2018, 47, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.J.; Pink, M.; Chen, C.-H.; Caulton, K.G. Electrophile Recruitment as a Structural Element in Bis-Pyrazolate Pyridine Complex Aggregation. Eur. J. Inorg. Chem. 2018, 2018, 5160–5166. [Google Scholar] [CrossRef]
- Cook, B.J.; Chen, C.-H.; Caulton, K.G. A Multifunctional Pincer Ligand for Cobalt-Promoted Oxidation by N2O. Chem. Eur. J. 2018, 24, 5962–5966. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.J.; Pink, M.; Pal, K.; Caulton, K.G. Electron and Oxygen Atom Transfer Chemistry of Co(II) in a Proton Responsive, Redox Active Ligand Environment. Inorg. Chem. 2018, 57, 6176–6185. [Google Scholar] [CrossRef]
- Labrum, N.S.; Pink, M.; Chen, C.-H.; Caulton, K.G. Reactivity of an Unusual Divalent Chromium Aggregate Supported by a Multifunctional Bis(pyrazolate) Pincer Ligand. Eur. J. Inorg. Chem. 2019, 2019, 1932–1940. [Google Scholar] [CrossRef]
- Labrum, N.S.; Curtin, G.M.; Jakubikova, E.; Caulton, K.G. The Influence of Nucleophilic and Redox Pincer Character as Well as Alkali Metals on the Capture of Oxygen Substrates: The Case of Chromium(II). Chem. Eur. J. 2020, 26, 9547–9555. [Google Scholar] [CrossRef]
- Labrum, N.S.; Chen, C.-H.; Caulton, K.G. A Bis-Pyrazolate Pincer on Reduced Cr Deoxygenates CO2: Selective Capture of the Derived Oxide by CrII. Chem. Eur. J. 2019, 25, 7935–7940. [Google Scholar] [CrossRef]
- Toda, T.; Yoshinari, A.; Ikariya, T.; Kuwata, S. Protic N-Heterocyclic Carbene Versus Pyrazole: Rigorous Comparison of Proton- and Electron-Donating Abilities in a Pincer-Type Framework. Chem. Eur. J. 2016, 22, 16675–16683. [Google Scholar] [CrossRef]
- Toda, T.; Kuwata, S. Central N-Heterocyclic Carbene Moieties in Protic Pincer-Type Bis(pyrazole) Ligands: Perturbation on Steric and Electronic Properties of Ruthenium Center. J. Organomet. Chem. 2020, 917, 121270. [Google Scholar] [CrossRef]
- Toda, T.; Kuwata, S. Synthesis, Structures, and Reactivities of Iron Complexes Bearing an Isoindoline-Based, Polyprotic Pincer-Type Pyrazole Ligand. Z. Anorg. Allg. Chem. 2021, 647, 1471–1477. [Google Scholar] [CrossRef]
- Kuwata, S.; Hahn, F.E. Complexes Bearing Protic N-Heterocyclic Carbene Ligands. Chem. Rev. 2018, 118, 9642–9677. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Kuwata, S.; Ikariya, T. Unsymmetrical Pincer-Type Ruthenium Complex Containing β-Protic Pyrazole and N-Heterocyclic Carbene Arms: Comparison of Brønsted Acidity of NH Groups in Second Coordination Sphere. Chem. Eur. J. 2014, 20, 9539–9542. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Kuwata, S.; Ikariya, T. Synthesis and Structures of Ruthenium and Iron Complexes Bearing an Unsymmetrical Pincer-Type Ligand with Protic Pyrazole and Tertiary Aminoalkyl Arms. Z. Anorg. Allg. Chem. 2015, 641, 2135–2139. [Google Scholar] [CrossRef]
- Polezhaev, A.V.; Liss, C.J.; Telser, J.; Chen, C.-H.; Caulton, K.G. A PNNH Pincer Ligand Allows Access to Monovalent Iron. Chem. Eur. J. 2018, 24, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Polezhaev, A.V.; Chen, C.; Losovyj, Y.; Caulton, K.G. A Multifunctional Pincer Ligand Supports Unsaturated Cobalt: Five Functionalities in One Pincer. Chem. Eur. J. 2017, 23, 8039–8050. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, A.C.; Carta, V.; Chen, C.-H.; Pink, M.; Caulton, K.G. Pincers with Diverse Donors and Their Interconversion: Application to Ni(II). Z. Anorg. Allg. Chem. 2021, 647, 1524–1529. [Google Scholar] [CrossRef]
- Koo, C.-K.; Ho, Y.-M.; Chow, C.-F.; Lam, M.H.-W.; Lau, T.-C.; Wong, W.-Y. Synthesis and Spectroscopic Studies of Cyclometalated Pt(II) Complexes Containing a Functionalized Cyclometalating Ligand, 2-Phenyl-6-(1H-pyrazol-3-yl)-pyridine. Inorg. Chem. 2007, 46, 3603–3612. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.; Gómez-Iglesias, P.; Anton, N.; García-Rodríguez, R.; Alegria, E.C.B.A.; Pombeiro, A.J.L.; Miguel, D.; Villafañe, F. Homo- and Heteropolymetallic 3-(2-Pyridyl)pyrazolate Manganese and Rhenium Complexes. Dalton Trans. 2014, 43, 4009–4020. [Google Scholar] [CrossRef]
- Cabelof, A.C.; Carta, V.; Caulton, K.G. A Proton-Responsive Ligand Becomes a Dimetal Linker for Multisubstrate Assembly via Nitrate Deoxygenation. Chem. Commun. 2021, 57, 2780–2783. [Google Scholar] [CrossRef]
- Nakahara, Y.; Toda, T.; Kuwata, S. Iron and Ruthenium Complexes Having a Pincer-Type Ligand with Two Protic Amidepyrazole Arms: Structures and Catalytic Application. Polyhedron 2018, 143, 105–110. [Google Scholar] [CrossRef]
- Tanaka, H.; Hitaoka, S.; Umehara, K.; Yoshizawa, K.; Kuwata, S. Mechanistic Study on Catalytic Disproportionation of Hydrazine by a Protic Pincer-Type Iron Complex through Proton-Coupled Electron Transfer. Eur. J. Inorg. Chem. 2020, 2020, 1472–1482. [Google Scholar] [CrossRef]
- Yamagishi, H.; Konuma, H.; Kuwata, S. Stereoselective Synthesis of Chlorido–Phosphine Ruthenium Complexes Bearing a Pyrazole-Based Protic Tripodal Amine Ligand. Polyhedron 2017, 125, 173–178. [Google Scholar] [CrossRef]
- Yamagishi, H.; Nabeya, S.; Ikariya, T.; Kuwata, S. Protic Ruthenium Tris(pyrazol-3-ylmethyl)amine Complexes Featuring a Hydrogen-Bonding Network in the Second Coordination Sphere. Inorg. Chem. 2015, 54, 11584–11586. [Google Scholar] [CrossRef]
- Labrum, N.S.; Cabelof, A.C.; Caulton, K.G. A Dimeric Chromium(II) Pincer as an Electron Shuttle for N=N Bond Scission. Chem. Eur. J. 2020, 26, 13915–13926. [Google Scholar] [CrossRef]
- Labrum, N.S.; Caulton, K.G. [Cr(Pincer2−)]2 as an Electron Shuttle for Reductively Promoted Hydrazine Disproportionation. Dalton Trans. 2019, 48, 11642–11646. [Google Scholar] [CrossRef]
- Toda, T.; Suzuki, S.; Kuwata, S. Metallo-Supramolecular Assembly of Protic Pincer-Type Complexes: Encapsulation of Dinitrogen and Carbon Disulfide into a Multiproton-Responsive Diruthenium Cage. Chem. Commun. 2019, 55, 1028–1031. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, H.; Tanahashi, H.; Kawakita, K.; Tsurugi, H.; Mashima, K. 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes as Strong Salt-Free Reductants for Generating Low-Valent Early Transition Metals with Electron-Donating Ligands. J. Am. Chem. Soc. 2014, 136, 5161–5170. [Google Scholar] [CrossRef]
- Labrum, N.S.; Seo, J.; Chen, C.-H.; Pink, M.; Beagan, D.M.; Caulton, K.G. Di- and Trivalent Chromium Bis(pyrazol-3-yl)pyridine Pincer Complexes with Good Leaving Groups. Inorg. Chim. Acta 2019, 486, 483–491. [Google Scholar] [CrossRef]
- Seo, J.; Cabelof, A.C.; Chen, C.-H.; Caulton, K.G. Selective Deoxygenation of Nitrate to Nitrosyl Using Trivalent Chromium and the Mashima Reagent: Reductive Silylation. Chem. Sci. 2018, 10, 475–479. [Google Scholar] [CrossRef]
- Cabelof, A.C.; Erny, A.M.; Beagan, D.M.; Caulton, K.G. A Redox Cascade of NOx− Complexes: Structures and Nitrogen Deoxygenation Thermodynamics. Polyhedron 2021, 200, 115119. [Google Scholar] [CrossRef]
- Cabelof, A.C.; Carta, V.; Chen, C.-H.; Caulton, K.G. Nitrogen Oxyanion Reduction by Co(II) Augmented by a Proton Responsive Ligand: Recruiting Multiple Metals. Dalton Trans. 2020, 49, 7891–7896. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-T.; Ching, W.-M.; Liaw, W.-F.; Lu, T.-T. Dinitrosyl Iron Complex [K-18-crown-6-ether][(NO)2Fe(MePyrCO2)]: Intermediate for Capture and Reduction of Carbon Dioxide. Angew. Chem. Int. Ed. 2020, 59, 11819–11823. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Yu, Z. Ruthenium(II) Complexes Bearing a Pyridyl-Supported Pyrazolyl-N-Heterocyclic Carbene (NNC) Ligand and Their Catalytic Activity in the Transfer Hydrogenation of Ketones. Organometallics 2008, 27, 6025–6028. [Google Scholar] [CrossRef]
- Jin, W.; Wang, L.; Yu, Z. A Highly Active Ruthenium(II) Pyrazolyl–Pyridyl–Pyrazole Complex Catalyst for Transfer Hydrogenation of Ketones. Organometallics 2012, 31, 5664–5667. [Google Scholar] [CrossRef]
- Liu, T.; Wang, L.; Wu, K.; Wang, Q.; Yu, Z. Mono- and Multinuclear Pincer-Type Ru(II) Complex Catalysts and Their Catalytic Applications. Inorg. Chim. Acta 2023, 551, 121458. [Google Scholar] [CrossRef]
- Zeng, F.; Yu, Z. Exceptionally Efficient Unsymmetrical Ruthenium(II) NNN Complex Catalysts Bearing a Pyridyl-Based Pyrazolyl-Imidazolyl Ligand for Transfer Hydrogenation of Ketones. Organometallics 2008, 27, 2898–2901. [Google Scholar] [CrossRef]
- Zeng, F.; Yu, Z. Construction of Highly Active Ruthenium(II) NNN Complex Catalysts Bearing a Pyridyl-Supported Pyrazolyl-Imidazolyl Ligand for Transfer Hydrogenation of Ketones. Organometallics 2009, 28, 1855–1862. [Google Scholar] [CrossRef]
- Ye, W.; Zhao, M.; Yu, Z. Ruthenium(II) Pyrazolyl–Pyridyl–Oxazolinyl Complex Catalysts for the Asymmetric Transfer Hydrogenation of Ketones. Chem. Eur. J. 2012, 18, 10843–10846. [Google Scholar] [CrossRef]
- Roberts, T.D.; Halcrow, M.A. Supramolecular Assembly and Transfer Hydrogenation Catalysis with Ruthenium(II) Complexes of 2,6-Di(1H-pyrazol-3-yl)pyridine Derivatives. Polyhedron 2016, 103, 79–86. [Google Scholar] [CrossRef]
- Kashiwame, Y.; Kuwata, S.; Ikariya, T. Metal–Pyrazole Bifunction in Half-Sandwich C–N Chelate Iridium Complexes: Pyrazole–Pyrazolato Interconversion and Application to Catalytic Intramolecular Hydroamination of Aminoalkene. Chem. Eur. J. 2010, 16, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Kashiwame, Y.; Ikariya, T.; Kuwata, S. Synthesis, Structures, and Reactivities of Six-Membered C–N Chelate Protic Pyrazole Complexes of Iridium. Polyhedron 2021, 197, 115036. [Google Scholar] [CrossRef]
- Ghoochany, L.T.; Farsadpour, S.; Sun, Y.; Thiel, W.R. New N,N,N-Donors Resulting in Highly Active Ruthenium Catalysts for Transfer Hydrogenation at Room Temperature. Eur. J. Inorg. Chem. 2011, 2011, 3431–3437. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, J.; Fu, H.; Yuan, M.; Zheng, X.; Chen, H.; Li, R. Construction of Pincer-Type Symmetrical Ruthenium(II) Complexes Bearing Pyridyl-2,6-Pyrazolyl Arms: Catalytic Behavior in Transfer Hydrogenation of Ketones. RSC Adv. 2014, 4, 52734–52739. [Google Scholar] [CrossRef]
- Dayan, O.; Dayan, S.; Kani, İ.; Cetinkaya, B. Ruthenium(II) Complexes Bearing Pyridine-Based Tridentate and Bidentate Ligands: Catalytic Activity for Transfer Hydrogenation of Aryl Ketones. Adv. Organomet. Chem. 2012, 26, 663–670. [Google Scholar] [CrossRef]
- Gunnaz, S.; Ozdemir, N.; Dayan, S.; Dayan, O.; Cetinkaya, B. Synthesis of Ruthenium(II) Complexes Containing Tridentate Triamine (‘NNN’) and Bidentate Diamine Ligands (NN′): As Catalysts for Transfer Hydrogenation of Ketones. Organometallics 2011, 30, 4165–4173. [Google Scholar] [CrossRef]
- Alshakova, I.D.; Korobkov, I.; Kuzmina, L.G.; Nikonov, G.I. Ruthenium Complexes with a Pyrazole-Phosphine Ligand. J. Organomet. Chem. 2017, 853, 68–73. [Google Scholar] [CrossRef]
- Alshakova, I.D.; Gabidullin, B.; Nikonov, G.I. Ru-Catalyzed Transfer Hydrogenation of Nitriles, Aromatics, Olefins, Alkynes and Esters. ChemCatChem 2018, 10, 4860–4869. [Google Scholar] [CrossRef]
- Dubey, A.; Khaskin, E. Catalytic Ester Metathesis Reaction and Its Application to Transfer Hydrogenation of Esters. ACS Catal. 2016, 6, 3998–4002. [Google Scholar] [CrossRef]
- Tian, C.; Gong, L.; Meggers, E. Chiral-at-Metal Iridium Complex for Efficient Enantioselective Transfer Hydrogenation of Ketones. Chem. Commun. 2016, 52, 4207–4210. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, J.; Huang, X.; Meggers, E. Sequential Asymmetric Hydrogenation and Photoredox Chemistry with a Single Catalyst. Org. Chem. Front. 2018, 5, 166–170. [Google Scholar] [CrossRef]
- Muller, K.; Sun, Y.; Heimermann, A.; Menges, F.; Niedner-Schatteburg, G.; van Wüllen, C.; Thiel, W.R. Structure–Reactivity Relationships in the Hydrogenation of Carbon Dioxide with Ruthenium Complexes Bearing Pyridinylazolato Ligands. Chem. Eur. J. 2013, 19, 7825–7834. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, B.G.; Young, K.J.H.; Yousufuddin, M.; Goddard, W.A.; Periana, R.A. Acceleration of Nucleophilic CH Activation by Strongly Basic Solvents. J. Am. Chem. Soc. 2010, 132, 12542–12545. [Google Scholar] [CrossRef] [PubMed]
- Suna, Y.; Himeda, Y.; Fujita, E.; Muckerman, J.T.; Ertem, M.Z. Iridium Complexes with Proton-Responsive Azole-Type Ligands as Effective Catalysts for CO2 Hydrogenation. ChemSusChem 2017, 10, 4535–4543. [Google Scholar] [CrossRef] [PubMed]
- Onishi, N.; Xu, S.; Manaka, Y.; Suna, Y.; Wang, W.-H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. CO2 Hydrogenation Catalyzed by Iridium Complexes with a Proton-Responsive Ligand. Inorg. Chem. 2015, 54, 5114–5123. [Google Scholar] [CrossRef]
- Manaka, Y.; Wang, W.-H.; Suna, Y.; Kambayashi, H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Efficient H2 Generation from Formic Acid Using Azole Complexes in Water. Catal. Sci. Technol. 2014, 4, 34–37. [Google Scholar] [CrossRef]
- Wang, W.-H.; Wang, H.; Yang, Y.; Lai, X.; Li, Y.; Wang, J.; Himeda, Y.; Bao, M. Synergistic Effect of Pendant N Moieties for Proton Shuttling in the Dehydrogenation of Formic Acid Catalyzed by Biomimetic IrIII Complexes. ChemSusChem 2020, 13, 5015–5022. [Google Scholar] [CrossRef]
- Nakahara, Y.; Toda, T.; Matsunami, A.; Kayaki, Y.; Kuwata, S. Protic NNN and NCN Pincer-Type Ruthenium Complexes Featuring (Trifluoromethyl)pyrazole Arms: Synthesis and Application to Catalytic Hydrogen Evolution from Formic Acid. Chem. Asian J. 2018, 13, 73–80. [Google Scholar] [CrossRef]
- Pal, S.; Iwasaki, T.; Nozaki, K. Metal–Ligand Cooperative κ1-N-Pyrazolate Cp*RhIII-Catalysts for Dehydrogenation of Dimethylamine-Borane at Room Temperature. Dalton Trans. 2021, 50, 7938–7943. [Google Scholar] [CrossRef]
- Reed-Berendt, B.G.; Latham, D.E.; Dambatta, M.B.; Morrill, L.C. Borrowing Hydrogen for Organic Synthesis. ACS Cent. Sci. 2021, 7, 570–585. [Google Scholar] [CrossRef]
- Kuwahara, T.; Fukuyama, T.; Ryu, I. Ruthenium Hydride/Nitrogen Tridentate Ligand-Catalyzed α-Alkylation of Acetamides with Primary Alcohols. RSC Adv. 2013, 3, 13702–13704. [Google Scholar] [CrossRef]
- Panda, S.; Saha, R.; Sethi, S.; Ghosh, R.; Bagh, B. Efficient α-Alkylation of Arylacetonitriles with Secondary Alcohols Catalyzed by a Phosphine-Free Air-Stable Iridium(III) Complex. J. Org. Chem. 2020, 85, 15610–15621. [Google Scholar] [CrossRef] [PubMed]
- Dutta, I.; Yadav, S.; Sarbajna, A.; De, S.; Hölscher, M.; Leitner, W.; Bera, J.K. Double Dehydrogenation of Primary Amines to Nitriles by a Ruthenium Complex Featuring Pyrazole Functionality. J. Am. Chem. Soc. 2018, 140, 8662–8666. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Yu, K.; Liu, B.; Tan, W.; Zhang, G. A Highly Selective Manganese-Catalyzed Synthesis of Imines under Phosphine-Free Conditions. Organometallics 2020, 39, 217–226. [Google Scholar] [CrossRef]
- Satake, A.; Nakata, T. Novel η3-Allylpalladium–Pyridinylpyrazole Complex: Synthesis, Reactivity, and Catalytic Activity for Cyclopropanation of Ketene Silyl Acetal with Allylic Acetates. J. Am. Chem. Soc. 1998, 120, 10391–10396. [Google Scholar] [CrossRef]
- Satake, A.; Koshino, H.; Nakata, T. Cyclopropanation of Ketene Silyl Acetals with Allylic Acetates Using η3-Allylpalladium-Pyridinylimidazole Catalysts. Chem. Lett. 1999, 28, 49–50. [Google Scholar] [CrossRef]
- Aranyos, A.; Szabó, K.J.; Castaño, A.M.; Bäckvall, J.-E. Central versus Terminal Attack in Nucleophilic Addition to (π-Allyl)palladium Complexes. Ligand Effects and Mechanism. Organometallics 1997, 16, 1058–1064. [Google Scholar] [CrossRef]
- Satake, A.; Kadohama, H.; Koshino, H.; Nakata, T. Asymmetric Cyclopropanation of Ketene Silyl Acetal with Allylic Acetate Catalyzed by a Palladium Complex. Tetrahedron Lett. 1999, 40, 3597–3600. [Google Scholar] [CrossRef]
- Diez, J.; Gimeno, J.; Lledós, A.; Suárez, F.J.; Vicent, C. Imidazole Based Ruthenium(IV) Complexes as Highly Efficient Bifunctional Catalysts for the Redox Isomerization of Allylic Alcohols in Aqueous Medium: Water as Cooperating Ligand. ACS Catal. 2012, 2, 2087–2099. [Google Scholar] [CrossRef]
- Bellarosa, L.; Diez, J.; Gimeno, J.; Lledós, A.; Suárez, F.J.; Ujaque, G.; Vicent, C. Highly Efficient Redox Isomerisation of Allylic Alcohols Catalysed by Pyrazole-Based Ruthenium(IV) Complexes in Water: Mechanisms of Bifunctional Catalysis in Water. Chem. Eur. J. 2012, 18, 7749–7765. [Google Scholar] [CrossRef]
- Tashima, N.; Ohta, S.; Kuwata, S. Metal–Ligand Cooperative C–O Bond Cleavage of Propargylic Alcohol with Protic Pyrazole Complexes of Ruthenium. Faraday Discuss. 2019, 220, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Justaud, F.; Hachem, A.; Grée, R. Recent Developments in the Meyer-Schuster Rearrangement. Eur. J. Org. Chem. 2021, 2021, 514–542. [Google Scholar] [CrossRef]
- Cadierno, V.; Crochet, P.; García-Garrido, S.E.; Gimeno, J. Metal-Catalyzed Transformations of Propargylic Alcohols into α,β-Unsaturated Carbonyl Compounds: From the Meyer–Schuster and Rupe Rearrangements to Redox Isomerizations. Dalton Trans. 2010, 39, 4015–4031. [Google Scholar] [CrossRef] [PubMed]
- Wojcicki, A. Allenyls and Propargyls: Versatile Ligands in Transition-Metal Chemistry. Inorg. Chem. Commun. 2002, 5, 82–97. [Google Scholar] [CrossRef]
- Ferrer, Í.; Rich, J.; Fontrodona, X.; Rodríguez, M.; Romero, I. Ru(II) Complexes Containing DMSO and Pyrazolyl Ligands as Catalysts for Nitrile Hydration in Environmentally Friendly Media. Dalton Trans. 2013, 42, 13461–13469. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, Í.; Fontrodona, X.; Rodríguez, M.; Romero, I. Ru(II)-DMSO Complexes Containing Azole-Based Ligands: Synthesis, Linkage Isomerism and Catalytic Behaviour. Dalton Trans. 2016, 45, 3163–3174. [Google Scholar] [CrossRef]
- Zhang, L.; Meggers, E. Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res. 2017, 50, 320–330. [Google Scholar] [CrossRef]
- Zhang, L.; Meggers, E. Stereogenic-Only-at-Metal Asymmetric Catalysts. Chem. Asian J. 2017, 12, 2335–2342. [Google Scholar] [CrossRef]
- Chen, L.-A.; Tang, X.; Xi, J.; Xu, W.; Gong, L.; Meggers, E. Chiral-at-Metal Octahedral Iridium Catalyst for the Asymmetric Construction of an All-Carbon Quaternary Stereocenter. Angew. Chem. Int. Ed. 2013, 52, 14021–14025. [Google Scholar] [CrossRef]
- Vera, S.; García-Urricelqui, A.; Mielgo, A.; Oiarbide, M. Progress in (Thio)urea- and Squaramide-Based Brønsted Base Catalysts with Multiple H-Bond Donors. Eur. J. Org. Chem. 2023, 26, e202201254. [Google Scholar] [CrossRef]
- Agarwal, P.; Thirupathi, N.; Nethaji, M. Syntheses, Structural Aspects, Solution Behavior, and Catalytic Utility of Cyclopalladated N,N′,N″-Triarylguanidines [κ2(C,N)Pd(Pyrazole)2X] (X = Br, OC(O)CF3, and PF6) in Suzuki–Miyaura Coupling Reactions of Aryl Bromides. Organometallics 2016, 35, 3112–3123. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, L.; Lei, L.; Zheng, X.-L.; Fu, H.; Yuan, M.; Chen, H.; Li, R.-X. PdCl2-2,6-Bis(1,5-diphenyl-1H-pyrazol-3-yl)pyridine Catalyzed Suzuki–Miyaura Cross-Coupling. Catal. Commun. 2012, 29, 194–197. [Google Scholar] [CrossRef]
- Nacianceno, V.S.; Azpeitia, S.; Ibarlucea, L.; Mendicute-Fierro, C.; Rodriguez-Dieguez, A.; Seco, J.M.; Sebastian, E.S.; Garralda, M.A. Stereoselective Formation and Catalytic Activity of Hydrido(acylphosphane)(chlorido)(pyrazole)rhodium(III) Complexes. Experimental and DFT Studies. Dalton Trans. 2015, 44, 13141–13155. [Google Scholar] [CrossRef] [PubMed]
- Manrique, E.; Fontrodona, X.; Rodríguez, M.; Romero, I. A Ruthenium(II) Aqua Complex as Efficient Chemical and Photochemical Catalyst for Alkene and Alcohol Oxidation. Eur. J. Inorg. Chem. 2019, 2019, 2124–2133. [Google Scholar] [CrossRef]


























































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.-S.; Kuwata, S. Recent Developments in Reactions and Catalysis of Protic Pyrazole Complexes. Molecules 2023, 28, 3529. https://doi.org/10.3390/molecules28083529
Lin W-S, Kuwata S. Recent Developments in Reactions and Catalysis of Protic Pyrazole Complexes. Molecules. 2023; 28(8):3529. https://doi.org/10.3390/molecules28083529
Chicago/Turabian StyleLin, Wei-Syuan, and Shigeki Kuwata. 2023. "Recent Developments in Reactions and Catalysis of Protic Pyrazole Complexes" Molecules 28, no. 8: 3529. https://doi.org/10.3390/molecules28083529
APA StyleLin, W.-S., & Kuwata, S. (2023). Recent Developments in Reactions and Catalysis of Protic Pyrazole Complexes. Molecules, 28(8), 3529. https://doi.org/10.3390/molecules28083529