Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive memory loss and cognitive impairment due in part to a severe loss of cholinergic neurons in specific brain areas. AD is the most common type of dementia in the aging population. Although several acetylcholinesterase (AChE) inhibitors are currently available, their performance sometimes yields unexpected results. Thus, research is ongoing to find potentially therapeutic AChE inhibitory agents, both from natural and synthetic sources. Here, we synthesized 13 new lupinine triazole derivatives and evaluated them, along with 50 commercial lupinine-based esters of different carboxylic acids, for AChE inhibitory activity. The triazole derivative 15 [1S,9aR)-1-((4-(4-(benzyloxy)-3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)methyl)octahydro-2H-quinolizine)] exhibited the most potent AChE inhibitory activity among all 63 lupinine derivatives, and kinetic analysis demonstrated that compound 15 was a mixed-type AChE inhibitor. Molecular docking studies were performed to visualize interaction between this triazole derivative and AChE. In addition, a structure-activity relationship (SAR) model developed using linear discriminant analysis (LDA) of 11 SwissADME descriptors from the 50 lupinine esters revealed 5 key physicochemical features that allowed us to distinguish active versus non-active compounds. Thus, this SAR model could be applied for design of more potent lupinine ester-based AChE inhibitors.
1. Introduction
Alzheimer’s disease (AD) and other neurodegenerative disorders are predicted to become the second leading cause of death worldwide because of the increasing elderly population in most countries. AD produces gradual cognitive dysfunction, including difficulty in making decisions, language problems, mood swings, learning, orientation, and other behavioral issues [1]. The loss of cognitive function due to AD is associated with the rapid hydrolysis of acetylcholine by cholinesterases, including acetylcholinesterase (AChE). Consequently, inhibition of AChE has been proposed to be neuroprotective [2]. Indeed, AChE inhibitors represent the first line of symptomatic drug treatment for mild-to-moderate AD. AChE inhibitors were initially utilized in the treatment of myasthenia gravis, a neuromuscular condition associated with loss of ACh receptors at the neuromuscular junction, followed by skeletal muscle weakening [3,4]. AChE also represents a therapeutic target for controlling glaucoma, Parkinson’s disease, senile dementia, myasthenia gravis, and ataxia [3].
Natural products are often used as starting points for drug discovery and have been considered as the most important resource for the identification of lead compounds due to their diverse molecular architectures and a wide range of bioactivities [5,6]. Thus, natural products represent a valuable source from which novel AChE inhibitors may be discovered [4,7], and plant alkaloids, flavonoids, chalcones, xanthones and their derivatives have been screened for AChE inhibitory activity (e.g., see [8,9,10,11,12,13]). Considering the paucity of new AChE inhibitors, we explored the possibility of developing inhibitors based on transformation of the plant alkaloid lupinine. Lupinine ([(1R,9aR)-octahydro-2H-quinolizin-1-yl]methane) is found mainly in Lupinus and Anabasis plants [14,15,16] and is of interest as a pharmacophore. For example, lupinine has been reported to inhibit the fungal metalloprotease Mpr1 [17]. Likewise, compounds with octahydro quinolizine nuclei have been reported as ligands of serotonin receptors 5-HT3 and 5-HT4 [18]. Similar compounds have also been shown to exhibit antimalarial [19,20], antitubercular [21], and anticholinesterase activities [22,23,24,25]. The quinolizidine nucleus of lupinine is simultaneously bulky and highly lipophilic and can be used for replacement with heterocyclic groups, or the ring could be connected with bi- and tricyclic groups to develop AChE inhibitors [25,26]. On the other hand, it should be noted that simple esters of lupinine have also been reported to exhibit some anti-AChE activity [23].
We synthesized 13 lupinine-based triazole derivatives and evaluated them, together with 50 additional commercial lupinine-based esters of different carboxylic acids, for AChE inhibitory activity. This screening resulted in the identification of some novel AChE inhibitors, with the most potent being compound 15. Molecular docking allowed us to characterize 15 for its potential interaction with the AChE binding site. We also developed a structure-activity relationship (SAR) model to predict AChE inhibitory activity of lupinine esters.
2. Results and Discussion
2.1. Chemistry
We synthesized three novel compounds by reacting lupinine azide 1 with terminal alkynes 3-(prop-2-yn-1-yl-thio)-1H-1,2,4-triazole-5-amine (2), (2R,2S)-3-methylpent-4-yne-2,3-diol (3), and 3-ethoxy-4-(prop-2-ynyloxy)benzaldehyde (4) under the conditions of Cu-catalyzed 1,3-dipolar cycloaddition. As a result, (1S,9aR)-1-[(1,2,3-triazole-1-yl)methyl]octahydro-1H-quinolizines 5–7, which contained various substituents at position C-4 of the 1,2,3-triazole ring were obtained (Scheme 1). Structures of the synthesized compounds 5–7 were confirmed by 1H and 13C NMR spectroscopy, mass spectrometry, and two-dimensional COSY (1H-1H), HMQC (1H-13C) and HMBC (1H-13C) NMR spectroscopy (see Supplementary Figures S1–S6), which established the homo- and heteronuclear spin-spin couplings. In describing the spectra, we used the numbering of core atoms shown in Scheme 1 structure 5.
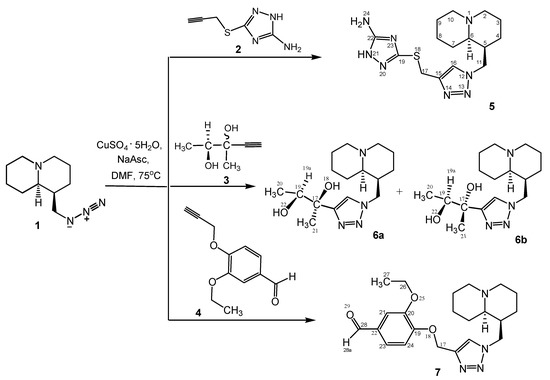
Scheme 1.
Synthesis of quinolizine-triazoles 5–7.
Reaction of lupinine azide 1 with a diastereomeric mixture of alkyne 3 resulted in a mixture of unseparated diastereomers 6a,b (ratio 4:1, as in initial alkyne 3). Relative stereochemistry and the ratio of diastereomers 6a,b was determined by analysis of 1H NMR data. The proton on the substituent at C-15 of the triazole ring (H-19) appeared at 3.88 and 4.05 ppm (multiplet signals). The other triazole derivatives 8–17 were synthesized as single compounds, as previously described [27,28,29]; however, this is the first report of their effects on AChE activity.
2.2. Biological Results
We screened our library of lupinine derivatives for their effects on AChE activity in comparison with galantamine, a known AChE inhibitor used in the treatment of mild Alzheimer’s disease. The library was assembled from two sets of compounds: the first set contained 13 lupinine-based triazole derivatives 5–17 (Table 1), and the second set contained 50 lupinine-based esters 18–67 of different carboxylic acids containing aliphatic, aromatic, or heterocyclic moieties. Structures of the lupinine-based esters are shown in Supplementary Table S1. AChE inhibitory activity of all triazole derivatives, and structures of the active lupinine derivatives are shown in Table 1 and Table 2, respectively.

Table 1.
AChE inhibitory activity of lupinine-based triazole derivatives.
Table 2.
AChE inhibitory activity of lupinine-based esters of different carboxylic acids.
Due to the small number of compounds, it is difficult to draw definite conclusions on structure-activity relationships. However, it can be noted that replacing the methyl group in compound 12 with a methoxy substituent resulted in AChE inhibitory activity (compound 13), whereas increasing the number of methoxy groups in the benzene ring to three resulted in a loss of activity (compound 14). On the other hand, the presence of a benzyloxy group in the para position of the benzene ring resulted in the maximum activity among all derivatives (compound 15; IC50 = 7.2 µM). Compound 15 has a 4-benzyloxyphenyl moiety, which was also present on several other previously reported AChE inhibitors, including compounds A–C with IC50 values in the micromolar/submicromolar range [30,31,32] (see chemical structures and AChE inhibitory activity in Table 3). AChE inhibitory activity of compound 15 was comparable to that of galantamine (IC50 = 8.2 ± 1.3 μM).
A visual inspection of the ester lupinine derivatives showed that a compound should contain a sufficiently bulky R group (e.g., compounds 25, 44, and 64), or the linker between the ester oxygen and the terminal hydrophobic moiety should consist of four chemical bonds (e.g., compounds 22, 43, and 49) for it to exhibit AChE inhibitory activity. Compound 25 (IC50 = 24.4 µM) bearing a 6,6-dimethyl-6,7-dihydrobenzofuran-4(5H)-one group was the most active of the lupinine-based esters. Noticeably, the 3,3-dimethylcyclohexanone substructure in this fragment was also present in previously reported AChE inhibitors [33].
The mechanism of AChE inhibition was determined for the most active compound 15. The Lineweaver–Burk reciprocal plot (Figure 1) revealed a series of lines converging on the same point near the x-axis, indicating that 15 caused a mixed type of inhibition, as expected for dual binding site inhibitors of AChE [34,35].
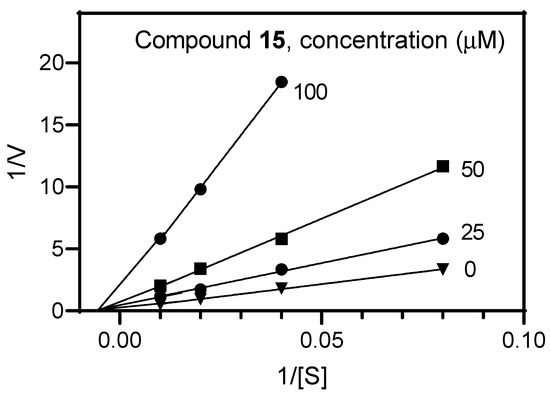
Figure 1.
Representative double-reciprocal Lineweaver–Burk plot illustrating the mixed-type mechanism of AChE inhibition by compound 15.
Table 3.
Chemical structures and AChE inhibitory activities of lupinine derivative 15 and previously reported AChE inhibitors with a 4-benzyloxyphenyl moiety [36,37,38].
Table 3.
Chemical structures and AChE inhibitory activities of lupinine derivative 15 and previously reported AChE inhibitors with a 4-benzyloxyphenyl moiety [36,37,38].
| Name | Chemical Structure | IC50 (μM) |
|---|---|---|
| Compound 15 |  | 7.2 |
| Compound A |  | 11.8 |
| Compound B |  | 1.2 |
| Compound C |  | 0.6 |
Chemical names: Compound 15, (1S,9aR)-1-((4-(4-(benzyloxy)-3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)methyl)octahydro-2H-quinolizine; Compound A, (E)-2-((2-(4-(benzyloxy)phenyl)hydrazineylidene)methyl)-1-methylbenzimidazole; Compound B, (E)-5-((E)-4-(benzyloxy)benzylidene)-2-((5-ethyl-1,3,4-thiadiazol-2-yl)imino)thiazolidin-4-one; Compound C, (E)-3-(4-(benzyloxy)-3-methoxyphenyl)-N-(2-((2-methylquinolin-4-yl)amino)ethyl)acrylamide.
Lupinine and all synthesized derivatives (5–17) were evaluated for their cytotoxicity in vitro using human THP-1 monocytic cells. These compounds had no cytotoxicity when tested at concentrations up to 50 μM. Thus, the lupinine derivatives reported here could be used for further biological evaluation in cell culture and in vivo models.
2.3. Molecular Docking
We performed molecular docking of compound 15 into the AChE binding site (PDB code 4EY7) using the Rosetta ligand docking protocol implemented in the ROSIE server, which accounts for full flexibility of the main chain and side-chain residues in the vicinity of the docking area [36]. According to our modeling results, the best docking pose of compound 15 had a calculated interface energy of −24.05 kcal/mol. In this pose, the ligand forms H-bonds with residues Tyr337 (with the participation of both benzyloxy and methoxy oxygen atoms), Tyr124 (with methoxy oxygen), and Phe295 (with two nitrogen atoms of the triazole heterocycle) (Figure 2). These general features of the ligand-binding site interactions can be responsible for the AChE inhibitory activity of compound 15. For reference, the 2D diagram of ligand-receptor interactions obtained on docking of compound 15 into AChE is shown in Supplementary Figure S7.
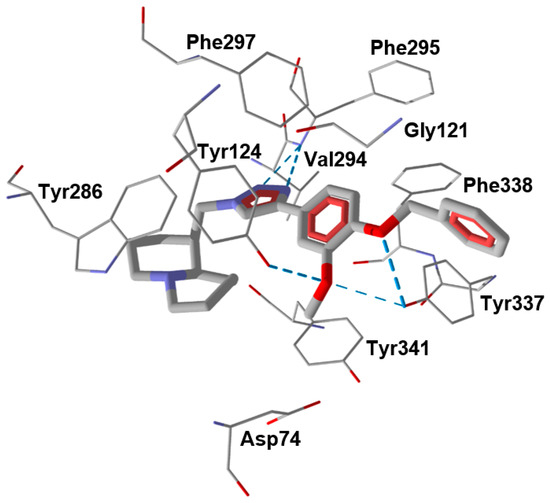
Figure 2.
Docking poses of compound 15 in AChE (PDB code 4EY7). H-bonds are shown in blue dashed lines. Residues within 2.5 Å of each pose are visible.
For comparative purposes, we modeled three other previously reported AchE inhibitors (A–C) with molecular topology analogous to compound 15 [36,37,38] (Table 3). The docking computations for these ligands led to docking poses positioned within the AChE binding site similarly to 15 (Figure 3), and with interface energies indicating strong binding: −23.34 (compound A), −24.70 (compound B), and −23.65 kcal/mol (compound C).
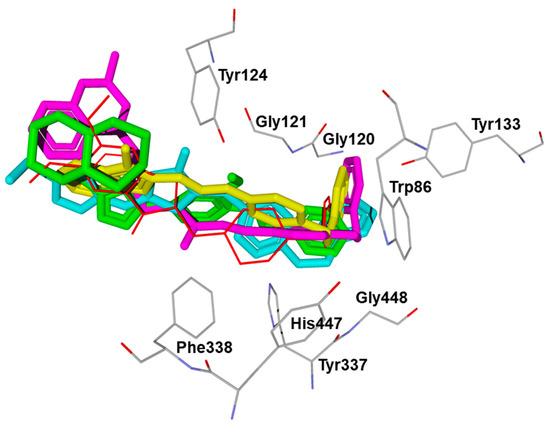
Figure 3.
Superimposed docking poses of compounds 15 (green), A (yellow), B (light blue), and C (magenta) in AChE (PDB code 4EY7). The co-crystallized ligand donepezil is shown in thin red sticks. The residues embracing the hydrophobic pocket of AChE are visible. The positions of the co-crystallized donepezil and AChE residues correspond to their “native” location in the 4EY7 structure.
It is noteworthy that the 4-benzyloxyphenyl moieties of all docked inhibitors and the N-benzylpiperidine fragment of the co-crystallized ligand donepezil occupy the same area of space in the hydrophobic pocket surrounded by residues Trp86, Gly120, Gly121, Tyr124, Tyr133, Tyr337, Phe338, His447, and Gly448 (Figure 2 and Figure 3), although the H-bonding patterns of A–C differed from those of compound 15. These molecules formed H-bonds with Tyr124 (compounds A and C), Tyr337 (compound A), His447, Ser293, and Arg296 (compound B). In addition, the terminal cyclic moieties of these inhibitors, including the quinolizidine heterocycle of compound 15 and the indanone fragment of donepezil, match well with each other in the AChE binding site. It should be noted that for the investigated compounds, most of the above-mentioned residues surrounding the pocket are among the top ten residues tightly interacting with the ligands, according to the partial MolDock scores as evaluated by the “Energy Inspector” tool of Molegro 6.0 software, which is due to significant van der Waals interactions of the molecules with these residues. In terms of the reported AChE functional domains [37], the subpocket residues identified belong to important functional domains, including the catalytic triad (His447), the anionic domain (Trp86, Tyr337, Phe338), and the oxyanion domain (Gly121). Additionally, we obtained high partial MolDock scores for Tyr124 and Trp286, which are located in the peripheral anionic site at the entrance of the binding gorge [37].
2.4. Classification SAR Model
Lupinine derivatives containing an ester moiety are, in general, more synthetically accessible. Thus, we used the lupinine ester derivatives to build an SAR model using linear discriminant analysis (LDA) to determine if it would be helpful for further drug design within this subgroup of substituted lupinines. LDA is a statistical technique used to categorize data points into two or more classes using a linear formalism [38]. The compounds containing an ester or carbamate moiety were separated into two classes (“Active” and “NA”) according to the data shown in Table 2, which includes only active AChE inhibitors found within the entire set of the lupinine esters (see Supplementary Table S1). Selected physicochemical and ADME parameters calculated using the SwissADME online tool were considered as independent variables (predictors) for LDA analysis along with two manually defined structural descriptors Nam and Q. Based on the 11 selected predictors, the LDA models in the form of classification functions (1) and (2) were built by STATISTICA 6.0 software with the “Best subset” option switched on.
We found that the best subset included 5 of the 11 descriptors (D1–D5, Table 2), which were sufficient for good LDA classification of the compounds, with 41 of the 50 lupinine derivatives classified correctly as AChE inhibitors (the class “Active”) or inactive compounds (the class “NA”). The values of SwissADME descriptors appearing in the classification functions are shown in Table S2 (see Supplementary Materials).
The best subset of predictors included molecular weight (MW), number of rotatable bonds (Nrot), molar refractivity (MR), water solubility measure SILICOS-IT Log Sw (sLogS) [39], and the indicator Q of the quaternary carbon atom. This relatively simple LDA model can be expressed by the following two classification functions:
where D1–D5 are the values of descriptors from the best subset; a0, b0 are the intercepts from Table 4; a1–a5, b1–b5 are coefficients of the linear classification functions from the corresponding columns of Table 4.
F(Active) = a0 + a1·D1 + a2·D2 + … + a5·D5
F(NA) = b0 + b1·D1 + b2·D2 + … + b5·D5
Table 4.
Physicochemical descriptors from the best subset and the corresponding coefficients of classification functions.
According to the LDA model, a compound is classified as active if F (Active) > F (NA), and vice versa. Hence, the influence of each predictor can be evaluated based on the corresponding pair coefficients in the two classification functions. For example, a higher molecular weight favors activity because the coefficient for MW is larger in F (Active). The same refers to molar refractivity and water solubility. Conversely, higher molecular flexibility and the presence of a quaternary carbon atom disfavor activity in view of lower (more negative) coefficients for Nrot and Q predictors in F (Active).
The classification matrix for the investigated compounds is shown in Table 5. According to this matrix, the LDA model correctly classifies 6 of 7 (85.7%) active AChE inhibitors and 35 of 43 (81.4%) inactive compounds. In spite of the noticeable number of false positives among the “NA” class, a total of 82.0% of the compounds were recognized properly by the model. The per-compound classifications are presented in Table S3 (see Supplementary Materials).
Table 5.
Classification matrices for the LDA model built based on 50 lupinine ester derivatives.
The single compound which was erroneously classified as inactive (19) contains a chorine atom at the terminal position of the ester tail. This is a significant structural difference from other active lupinine derivatives, which contain cyclic substructures at the terminal position of each molecule.
Leave-one-out (LOO) validation of the model (i.e., predicting the activity of a discarded compound by a model built on the basis of the remaining 49 molecules) showed that 32 out of 43 inactive compounds (74.1%) and 5 out of 7 active compounds (71.4%) were correctly predicted (74 % in the total set).
The SAR model based on physicochemical descriptors of the lupinine-based esters revealed key features distinguishing AChE inhibitors versus non-active compounds. One of the weak points of the model is the imbalanced character of the data set, which contained many more inactive compounds than active ones. However, the reasonable quality and predictive ability of the model, as well as the simplicity and rapidity of the calculations associated with the LDA algorithm, suggest promise in using this model for large database mining and virtual screening of lupinine-based AChE inhibitors.
3. Experimental Section
3.1. Chemistry
1H and 13C NMR spectra were recorded on a JNN-ECA Jeol 400 spectrometer (frequency 399.78 and 100.53 MHz, respectively) with deuterated dimethyl sulfoxide (DMSO-d6) as the solvent. The chemical shifts were measured with reference to signals of the residual protons or carbon atoms of DMSO-d6. The multiplicity of signals in the 13C NMR spectra was determined from spectra recorded in the J-modulation mode (JMOD). The assignment of signals in the 1H and 13C NMR spectra were confirmed by two-dimensional homonuclear (1H-1H COSY) and heteronuclear 1H-13C (HMBC, HSQC) spectroscopy and literature data for quinolizine. High-resolution mass spectra were recorded on a ThermoScientific DFS spectrometer (evaporator temperature of 200–250 °C, electron ionization 70 eV). Melting points were determined on a Mettler Toledo FP900 system. The process of chemical reactions was monitored by thin-layer chromatography (TLC) on Sorbfil UV-254 plates using CH3Cl and CH3Cl–EtOH (10:1) as eluents. The plates were visualized with iodine vapor and ultraviolet (UV) light (254 nm). The reaction products were isolated by recrystallization or column chromatography using Acros silicagel (0.035–0.240 mm) and CHCl3 and CHCl3–EtOH (100:1→10:1) as eluents.
Alkynes of 3-(prop-2-yn-1-yl-thio)-1H-1,2,4-triazole-5-amine (2), (2R,2S)-3-methylpent-4-yne-2,3-diol (4:1, diastereomeric mixture) (3) and 3-ethoxy-4-(prop-2-ynyloxy)benzaldehyde (4) were purchased from Alfa Aesar.
Compounds 1 and 8–17 were synthesized as described previously [27,28,29]. The synthesized structures were confirmed by analytical and spectral data. Sample purity was >99%.
(–)-Lupinine (m.p. 69–71 °C (EtOH), [α]D25–30.5° (c 0.41, MeOH); (literature data: m.p. 68–69 °C (EtOH), [α]D25–23.5°) [40] was isolated from the Anabasis aphyla L., as described previously [41].
Lupinine azide 1 was obtained from lupinine in two stages, as described previously [28]. Briefly, the reaction of (–)-lupinine with methanesulfonyl chloride in the presence of Et3N in CH2Cl2 resulted in (1R,9aR)-(octahydro-2H-quinolizine-1-yl)methyl methanesulfonate, which was treated with NaN3 in dimethylformamide (DMF), resulting in the organic quinolizine azide (1) [29].
3.1.1. General Procedure for Compounds (5–7)
A mixture of lupinine azide (1) (0.29 g, 1.5 mmol), substituted acetylene [3-(prop-2-yn-1-yl-thio)-1H-1,2,4-triazole-5-amine (2), (2S)-3-methylpent-4-yne-2,3-diol (3), and 3-ethoxy-4-(prop-2-ynyloxy)benzaldehyde (4) (1.35 mmol), CuSO4 × 5H2O (0.017 g, 0.0675 mmol) and sodium ascorbate (0.013 g, 0.0675 mmol) in DMF (6 mL) was stirred at 75 °C for 6–8 h using TLC monitoring. After cooling, the residue was filtered, washed with hexane, and dried. Triazoles 5–7 were isolated from the residue by chromatography on silicagel (eluent: CH3Cl, CH3Cl–EtOH, 100:1 → 10:1).
3.1.2. 3-((1-(((1S,9aR)-Octahydro-1H-quinolizine-1-yl)methyl)-1H-1,2,3-triazole-4-yl)methylthio)-1H-1,2,4-triazole-5-amine (5)
Yield 0.22 g (75.86%). Dark-brown powder, m.p. 177–179 °C (decomp.). 1H NMR spectrum (DMSO-d6), δ, ppm: 1.15–1.66 m (10H, H-3ax,3eq, 4ax,4eq,7ax,7eq,8ax,8eq,9ax,9eq), 1.89 s (2H, H-2ax,10ax), 2.05 s (2H, H-5,6), 2.75 s (2H, H-2eq,10eq), 4.19 s (2H, H-17,17), 4.41 s (2H, H-11,11), 5.97 s (2H, H-24,24), 7.90 s (1H, H-16), 11.95 br. s (1H, H-21). 13C NMR spectrum (DMSO-d6), δ, ppm: 20.67 (C-3), 24.70 (C-8,9), 26.73 (C-17), 28.85 (C-4,7), 39.27 (C-5), 48.76 (C-11), 57.38 (C-2,10), 64.17 (C-6), 124.09 (C-16), 144.44 (C-15) and 156.20 (C-19,22). Mass spectrum, m/z (Irel., %) (2): 348.2 (7.23), 232.2 (17.40), 151.1 (100.0), 96.0 (21.73), 55.0 (16.38). Found m/z: 348.1839 [M]+. C15H24N8S. Calculated m/z: 348.1838.
3.1.3. (2R,S)-2-(1-(((1S,9aR)-Octahydro-1H-quinolizine-1-yl)methyl)-1H-1,2,3-triazole-4-yl)butane-2,3-diol (6)
Yield 0.25 g (86.20%). Cream-colored, m.p. 158–161 °C. 1H NMR (DMSO-d6), δ, ppm: 1.03 d (3H, H-22,22,22), 1.04–1.95 m (10H, H-3ax,3eq, 4ax,4eq,7ax,7eq,8ax,8eq,9ax,9eq), 1.52 s (3H, H-21,21,21), 1.93–1.98 m (2H, H-2ax,10ax), 1.97–2.06 m (1H, H-6), 2.15–2.21 m (1H, H-5), 2.81 s (2H, H-2eq,10eq), 3.41 br. s (2H, H-18,22), 4.46–4.57 m (2H, H-11,11), 7.47 s (1H, H-16). 13C NMR (DMSO-d6), δ, ppm: 17.86 (C-22), 22.98 (C-21), 24.32 (C-3), 24.88 (C-8), 25.38 (C-9), 26.20 (C-4), 29.73 (C-7), 39.26 (C-5), 57.23 (C-2,10), 64.27 (C-6), 73.07 (C-17), 74.54 (C-19), 121.89 (C-16), 151.81 (C-15). Mass spectrum, m/z (Irel., %): 308.3 (12.45), 219.2 (1.66), 151.1 (100.0), 98.0 (9.77), 43.2 (6.68). Found m/z: 308.2207 [M]+. C16H28N4O2. Calculated m/z: 308.2211.
3.1.4. 3-Ethoxy-4-((1-(((1S,9aR)-octahydro-1H-quinolizine-1-yl)methyl)-1H-1,2,3-triazole-4-yl)methoxy)benzaldehyde (7)
Yield 0.23 g (76.66%). White powder, m.p. 166–168 °C. 1H NMR (DMSO-d6), δ, ppm: 1.12–1.23 m (3H, H-4ax, H-7ax, H-3ax), 1.27 t (3H, H-27,27,27, 3J 7.6 Hz), 1.34–1.41 m (2H, H-4eq, H-7eq), 1.46–1.49 m (2H, H-8ax, H-8eq), 1.47–1.51 m (2H, H-11,11), 1.63–1.74 m (3H, H-2ax, H-10ax, H-3eq), 1.92–1.95 m (1H, H-6), 2.70–2.72 m (2H, H-2eq, H-10eq), 2.09 br. s (1H, H-5), 4.03 q (2H, H-26,26, 3J 7.6 Hz), 5.23 s (2H, H-17,17), 7.32 d (1H, H-24, 3J 9.2 Hz), 7.48 d (1H, H-23, 3J 9.2 Hz), 8.24 s (1H, H-16), 9.79 s (1H, H-28). 13C NMR (DMSO-d6), δ, ppm: 15.13 (C-27), 21.82 (C-3), 25.03 (C-8), 25.77 (C-9), 25.98 (C-4), 26.36 (C-7), 39.07 (C-5), 57.09 (C-2,10), 48.34 (C-11), 62.60 (C-17), 64.67 (C-6), 64.86 (C-26), 111.57 (C-21), 113.4 (C-24), 126.26 (C-23,16), 130.49 (C-22), 149.09 (C-19), 153.50 (C-20), 191.96 (C-28). Mass spectrum, m/z (Irel., %): 398.3 (28.87), 256.2 (4.49), 151.1 (100.0), 84.9 (24.34), 55.0 (23.62). Found m/z: 398.2312 [M]+. C22H30N4O3. Calculated m/z: 398.2314.
3.2. Commercial Compounds
Fifty lupinine-based esters of different carboxylic acids (compounds 18–67) were purchased from the Vitas-M laboratory (Champaign, IL, USA). All compounds were dissolved in DMSO at a stock concentration of 10 mM and stored at −20 °C.
3.3. AChE Inhibition Assay
The inhibitory effect of test compounds and galantamine (Tocris Bioscience, San Francisco, CA, USA) on AChE activity was determined using an AChE inhibitor screening kit from the Sigma-Aldrich Chemical Co., (St. Louis, MO, USA). The kit is based on an improved Ellman method, whereby thiocholine produced from AChE activity forms a yellow color with 5,5′-dithiobis(2-nitrobenzoic acid), and the intensity the color (412 nm) is proportional to the enzyme activity. The concentration of compound required to cause 50% inhibition (IC50) was determined by graphing the % inhibition of enzyme activity versus the logarithm of concentration of the test compound using 5–7 tested concentrations.
3.4. Cytotoxicity Assay
The cytotoxicity of the synthesized compounds was analyzed using a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Human THP-1 monocytic cells obtained from ATCC (Manassas, VA, USA) were cultured in RPMI 1640 medium (Mediatech Inc., Herndon, VA, USA) supplemented with 10% (v/v) FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin. For the cytotoxicity assay, the cells were cultured at a density of 104 cells/well with different concentrations of the test compounds added (3, 6.125, 12.5, 25, 50 μM; final concentration of DMSO was 1%) for 24 h at 37 °C and 5% CO2. Following treatment, substrate was added to the cells, and the samples were analyzed with a Fluoroscan Ascent FL microplate reader.
3.5. Molecular Docking
Docking of compounds into the acetyl cholinesterase binding site (structure 4EY7 from the Protein Data Bank) was performed using the ROSIE server [42]. The docking area was chosen around the geometric center of co-crystallized donepezil (1-benzyl-4-[(5,6-dimethoxy-1-indanon-2-yl)methyl]piperidine) occupying the binding site of the receptor in the 4EY7 structure. For each of the investigated compounds, up to 2000 ligand conformers were generated with the BCL algorithm [43] switched on. The number of intermediately generated docking poses was set to 2000. Other options were used as defaults within the ROSIE ligand docking protocol, which accounted for full flexibility of the main chain and side-chains for residues in the vicinity of the docking area [36]. After completion of the computations, PDB files containing the best poses of compounds docked into AChE were downloaded from the server and imported into Molegro Virtual Docker 6.0 (MVD) for visualization and analysis using the built-in “Pose Organizer” tool of MVD.
3.6. Linear Discriminant Analysis (LDA)
Structures of the 50 lupinine-based esters were built using ChemOffice 2016, represented as SMILES strings, and imported into the SwissADME online tool [39]. The calculated physicochemical and ADME parameters were subjected to correlation analysis to select descriptors with low mutual pairwise correlations. The following descriptors were selected: molecular weight (MW), fraction of sp3 carbon atoms (Csp3), number of rotatable bonds (Nrot), number of hydrogen bond donors and acceptors (NHBD and NHBA, respectively), molar refractivity (MR), topological polar surface area (tPSA), consensus LogP (cLogP), and water solubility SILICOS-IT Log Sw (sLogS). Two structural indicators were added, which indicated absence or presence (value of 0 or 1, respectively) of an amide unit -C(O)NH- or a quaternary sp3 carbon atom (descriptors Nam and Q, respectively). Although useful computational methods have been developed for finding molecular subunits (e.g., [44]), the Nam and Q indicators were assigned manually because of their simplicity. The data sheet containing columns with the values of the independent predictors enumerated above was supplemented with a column indicating compound activity (values “Active” or “NA”) as a categorical dependent variable. The resulting data sheet was imported in STATISTICA 6.0 program (StatSoft, Inc., Tulsa, OK, USA), and the LDA procedure was performed with the “Best subset” option switched on using equal prior probabilities for the compound classes. All 50 lupinine-based esters were used as a training set. To validate the model, the leave-one-out (LOO) procedure was performed by sequentially discarding one of the compounds and predicting its activity class (i.e., the dependent categorical variable) by an LDA model obtained on the basis of the remaining 49 compounds.
4. Conclusions
We identified compound 15 as a novel AChE inhibitor and showed that it exhibited mixed-type inhibitory activity. Molecular docking modeling indicated that compound 15 meets structural requirements necessary to reproduce important intermolecular interactions described in the literature as fundamental for AChE inhibition. Thus, this compound could be a promising candidate for evaluation in AD models. Our results also indicate that the 4-benzyloxyphenyl moiety attached to different molecular scaffolds can play an important role in ligand binding to AChE due to the interaction with the receptor subpocket. This finding, as well as the derived classification SAR model, may be useful in the design of other novel AChE inhibitors.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28083357/s1, Supplementary Figures S1–S3. Schemes of correlations in the COSY and HMQC spectra of 5–7. Supplementary Figure S4.1. 1H NMR spectrum of 5. Supplementary Figure S4.2. 13C NMR spectrum of 5. Supplementary Figure S4.3. The mass spectrum of 5. Supplementary Figure S5.1. 1H NMR spectrum of 6a,b. Supplementary Figure S5.2. 13C NMR spectrum of 6a,b. Supplementary Figure S5.3. The mass spectrum of 6a,b. Supplementary Figure S6.1. 1H NMR spectrum of 7. Supplementary Figure S6.2. 13C NMR spectrum of 7. Supplementary Figure S6.3. The mass spectrum of 7. Supplementary Table S1. Chemical structures of lupinine-based esters of different carboxylic acids under investigation. Supplementary Figure S7. 2D diagram of ligand-receptor interactions obtained on docking of compound 15 in AChE. Blue dashed lines—hydrogen bonding interactions. Red dashed line—steric interaction. Supplementary Table S2. Chemical formulas, selected ADME parameters of the lupinine-based esters calculated with SwissADME web tool, and manually added indicator variable for the quaternary sp3 carbon atom (Q). Supplementary Table S3. Experimentally determined, calculated, and LOO-predicted classes for AChE inhibitory activity of the lupinine-based esters of different carboxylic acids.
Author Contributions
I.A.S., Z.S.N. and M.T.Q. conceived and designed the project; Z.S.N., S.D.F., O.A.N., T.M.S., A.S.K. and E.E.S. synthesized and characterized compounds; I.A.S. performed the enzymatic assay; A.I.K. conducted the molecular modeling study; I.A.S., Z.S.N., S.D.F., O.A.N., A.I.K., T.M.S., A.S.K. and E.E.S. analyzed and interpreted the data; I.A.S., Z.S.N., A.I.K. and M.T.Q. drafted and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by National Institutes of Health IDeA Program grant GM103474; USDA National Institute of Food and Agriculture Hatch project 1009546; the Montana State University Agricultural Experiment Station; project No. AP08855567 under the grant funding from the Committee of Science of the Ministry of Education and Science of the Republic of Kazakhstan, and the Tomsk Polytechnic University Development Program (Project Priority-2030-NIP/IZ-009-0000-2023).
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of synthesized compounds are available from the authors.
References
- Robichaud, A.J. Approaches to palliative therapies for Alzheimer’s disease. Curr. Top. Med. Chem. 2006, 6, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Umar, T.; Meena, R.; Mustehasan; Kumar, P.; Khan, A.A. Recent updates in the development of small molecules as potential clinical candidates for Alzheimer’s disease: A review. Chem. Biol. Drug Des. 2022, 100, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, S.; Saeedi, M.; Larijani, B.; Mahdavi, M. Recent developments in the design and synthesis of benzylpyridinium salts: Mimicking donepezil hydrochloride in the treatment of Alzheimer’s disease. Front. Chem. 2022, 10, 936240. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Satheeshkumar, N.; Venkatesh, P.; Venkatesh, M. Lead finding for acetyl cholinesterase inhibitors from natural origin: Structure activity relationship and scope. Mini Rev. Med. Chem. 2011, 11, 247–262. [Google Scholar] [CrossRef]
- Ahn, K. The worldwide trend of using botanical drugs and strategies for developing global drugs. BMB Rep. 2017, 50, 111–116. [Google Scholar] [CrossRef]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef]
- Ayaz, M.; Nawaz, A.; Naz, F.; Ullah, F.; Sadiq, A.; Islam, Z.U. Phytochemicals-based Therapeutics against Alzheimer’s Disease: An Update. Curr. Top. Med. Chem. 2022, 22, 1811–1820. [Google Scholar] [CrossRef]
- Suciati; Poerwantoro, D.; Widyawaruyanti, A.; Ingkaninan, K. Acetylcholinesterase inhibitory activity of extract and fractions from the root of Rauvolfia serpentine (L.) Bth.ex Kurz. J. Basic Clin. Physiol. Pharmacol. 2021, 32, 313–317. [Google Scholar] [CrossRef]
- Tomassini, L.; Ventrone, A.; Frezza, C.; Fabbri, A.M.; Fortuna, S.; Volpe, M.T.; Cometa, M.F. Phytochemical analysis of Viburnum davidii Franch. and cholinesterase inhibitory activity of its dihydrochalcones. Nat. Prod. Res. 2021, 35, 5794–5800. [Google Scholar] [CrossRef]
- Saenkham, A.; Jaratrungtawee, A.; Siriwattanasathien, Y.; Boonsri, P.; Chainok, K.; Suksamrarn, A.; Namsa-Aid, M.; Pattanaprateeb, P.; Suksamrarn, S. Highly potent cholinesterase inhibition of geranylated xanthones from Garcinia fusca and molecular docking studies. Fitoterapia 2020, 146, 104637. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, M.; Sun, C.; Peng, C.; Zhang, Y.; Li, X. Tunicyclin L, a cyclic peptide from Psammosilene tunicoides: Isolation, characterization, conformational studies and biological activity. Fitoterapia 2020, 145, 104628. [Google Scholar] [CrossRef]
- Lou, H.; Yi, P.; Hu, Z.; Li, Y.; Zeng, Y.; Gu, W.; Huang, L.; Yuan, C.; Hao, X. Polycyclic polyprenylated acylphloroglucinols with acetylcholinesterase inhibitory activities from Hypericum perforatum. Fitoterapia 2020, 143, 104550. [Google Scholar] [CrossRef]
- Tuzimski, T.; Petruczynik, A. Determination of Anti-Alzheimer’s Disease Activity of Selected Plant Ingredients. Molecules 2022, 27, 3222. [Google Scholar] [CrossRef]
- Omodeo-Salè, F.; Cortelezzi, L.; Basilico, N.; Casagrande, M.; Sparatore, A.; Taramelli, D. Novel antimalarial aminoquinolines: Heme binding and effects on normal or Plasmodium falciparum-parasitized human erythrocytes. Antimicrob. Agents Chemother. 2009, 53, 4339–4344. [Google Scholar] [CrossRef]
- Van Eijk, J.L.; Radema, M.H. Virgiboidine, A New Alkaloid from Virgilia oroboides and Virgilia divaricata. Planta Med. 1982, 44, 224–226. [Google Scholar] [CrossRef]
- Suzuki, H.; Murakoshi, I.; Saito, K. A novel O-tigloyltransferase for alkaloid biosynthesis in plants. Purification, characterization, and distribution in Lupinus plants. J. Biol. Chem. 1994, 269, 15853–15860. [Google Scholar] [CrossRef]
- Aaron, P.A.; Vu, K.; Gelli, A. An Antivirulence Approach for Preventing Cryptococcus neoformans from Crossing the Blood-Brain Barrier via Novel Natural Product Inhibitors of a Fungal Metalloprotease. mBio 2020, 11, e01249-20. [Google Scholar] [CrossRef]
- Sparatore, A.; Cagnotto, A.; Sparatore, F. Quinolizidinyl derivatives of 2,3-dihydro-2-oxo-1H-benzimidazole-1-carboxylic acid and 1-homolupinanoyl benzimidazolones as ligands for 5-HT3 and 5-HT4 receptors. Farmaco 1999, 54, 248–254. [Google Scholar] [CrossRef]
- Basilico, N.; Parapini, S.; Sparatore, A.; Romeo, S.; Misiano, P.; Vivas, L.; Yardley, V.; Croft, S.L.; Habluetzel, A.; Lucantoni, L.; et al. In vivo and in vitro activities and ADME-tox profile of a quinolizidine-modified 4-aminoquinoline: A potent anti-P. falciparum and anti-P. vivax blood-stage antimalarial. Molecules 2017, 22, 2102. [Google Scholar] [CrossRef]
- Rusconi, C.; Vaiana, N.; Casagrande, M.; Basilico, N.; Parapini, S.; Taramelli, D.; Romeo, S.; Sparatore, A. Synthesis and comparison of antiplasmodial activity of (+), (-) and racemic 7-chloro-4-(N-lupinyl)aminoquinoline. Bioorg. Med. Chem. 2012, 20, 5980–5985. [Google Scholar] [CrossRef]
- Vazzana, I.; Novelli, F.; Sparatore, F.; Sparatore, A.; Fadda, G.; Manca, C. Quinolizidine derivatives with antitubercular activity. Farmaco 1994, 49, 105–110. [Google Scholar] [PubMed]
- Basova, N.E.; Kormilitsyn, B.N.; Perchenok, A.; Rozengart, E.V.; Saakov, V.S.; Suvorov, A.A. Reversible lupininin inhibitors of cholinesterases of mammalian blood and of optical ganglia of individuals of the commander squid Berryteuthis magister from different zones of species areal. Zh. Evol. Biokhim. Fiziol. 2012, 48, 213–218. [Google Scholar] [PubMed]
- Rozengart, E.V. Bisalkaloid derivatives of dicarboxylic acids on the basis of lupinine, anabasine, and cytisine as reversible cholinesterase inhibitors. Dokl. Biochem. Biophys. 2003, 388, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A.; et al. Quinolizidinyl derivatives of bi- and tricyclic systems as potent inhibitors of acetyl- and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Catto, M.; Tasso, B.; Novelli, F.; Canu, C.; Iusco, G.; Pisani, L.; Stradis, A.D.; Denora, N.; Sparatore, A.; et al. Multitarget Therapeutic Leads for Alzheimer’s Disease: Quinolizidinyl Derivatives of Bi- and Tricyclic Systems as Dual Inhibitors of Cholinesterases and β-Amyloid (Aβ) Aggregation. ChemMedChem 2015, 10, 1040–1053. [Google Scholar] [CrossRef]
- Obaid, R.J.; Naeem, N.; Mughal, E.U.; Al-Rooqi, M.M.; Sadiq, A.; Jassas, R.S.; Moussa, Z.; Ahmed, S.A. Inhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase. RSC Adv. 2022, 12, 19764–19855. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Nurkenov, O.A.; Fazylov, S.D.; Mukusheva, G.K.; Gazaliev, A.M.; Muldakhmetov, Z.M. Synthesis of 1,2,3-triazolo-quinolizidines based on the quinolizidine alkaloid lupinine. Chem. J. Kazakhstan 2021, 3, 108–118. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Savelyev, V.A.; Gatilov, Y.V.; Nurkenov, O.A.; Seidakhmetova, R.B.; Shulgau, Z.T.; Mukusheva, G.K.; Fazylov, S.D.; Shults, E.E. Synthesis and analgesic activity of 1-[(1,2,3-triazol-1-yl)methyl]quinolizines based on the alkaloid lupinine. Chem. Heterocycl. Compd. 2021, 57, 911–919. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Fazylov, S.D.; Turdybekov, K.M.; Nurkenov, O.A.; Turdybekov, D.M.; Mukusheva, G.K.; Minayeva, Y.V.; Khabdolda, G. Synthesis and structure of 4-substituted (1S,9aR)-1-[(1,2,3-triazol-1-yl)-methyl]octahydro-1H-quinolysines of lupinine. Bull. Univ. Karaganda Chem. 2022, 2, 12–22. [Google Scholar] [CrossRef]
- Khodja, I.A.; Boulebd, H.; Bensouici, C.; Belfaitah, A. Design, synthesis, biological evaluation, molecular docking, DFT calculations and in silico ADME analysis of (benz)imidazole-hydrazone derivatives as promising antioxidant, antifungal, and anti- acetylcholinesterase agents. J. Mol. Struct. 2020, 1218, 128527. [Google Scholar] [CrossRef]
- Aggarwal, N.; Jain, S.; Chopra, N. Hybrids of Thiazolidin-4-Ones and 1,3,4-Thiadiazole: Synthesis and Biological Screening of A Potential New Class of Acetylcholinesterae Inhibitors. Biointerface Res. Appl. Chem. 2022, 12, 2800–2812. [Google Scholar]
- Mo, J.; Yang, H.; Chen, T.; Li, Q.; Lin, H.; Feng, F.; Liu, W.; Qu, W.; Guo, Q.; Chi, H.; et al. Design, synthesis, biological evaluation, and molecular modeling studies of quinoline-ferulic acid hybrids as cholinesterase inhibitors. Bioorg. Chem. 2019, 93, 103310. [Google Scholar] [CrossRef]
- Lotfi, S.; Rahmani, T.; Hatami, M.; Pouramiri, B.; Kermani, E.T.; Rezvannejad, E.; Mortazavi, M.; Fathi Hafshejani, S.; Askari, N.; Pourjamali, N.; et al. Design, synthesis and biological assessment of acridine derivatives containing 1,3,4-thiadiazole moiety as novel selective acetylcholinesterase inhibitors. Bioorg. Chem. 2020, 105, 104457. [Google Scholar] [CrossRef]
- Catto, M.; Pisani, L.; Leonetti, F.; Nicolotti, O.; Pesce, P.; Stefanachi, A.; Cellamare, S.; Carotti, A. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg. Med. Chem. 2013, 21, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Luz, R.; Almeida, R.B.M.; Albuquerque, M.M.S.; Cerqueira, A.P.M.; Tavares, J.F.; Silva, M.S.D.; Filho, R.B.; Dos Santos Junior, M.C.; Branco, A.; Botura, M.B. Two new dilactonized glycerol glycosides of the dual anticholinesterase active extract from Ocotea daphnifolia using bioguided fractionation and molecular docking studies. Chem. Biol. Drug Des. 2022, 101, 855–864. [Google Scholar] [CrossRef]
- DeLuca, S.; Khar, K.; Meiler, J. Fully Flexible Docking of Medium Sized Ligand Libraries with RosettaLigand. PLoS ONE 2015, 10, e0132508. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorg. Med. Chem. 2015, 23, 5382–5389. [Google Scholar] [CrossRef]
- Mitteroecker, P.; Bookstein, F. Linear Discrimination, Ordination, and the Visualization of Selection Gradients in Modern Morphometrics. Evol. Biol. 2011, 38, 100–114. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Winterfeld, K.; Holschneider, F.W. Über die Konstitution des Lupinins (I. Mitteil). Chem. Ber. Recl. 1931, 64, 137–150. [Google Scholar] [CrossRef]
- Nurkenov, O.A.; Nurmaganbetov, Z.S.; Fazylov, S.D.; Satpaeva, Z.B.; Turdybekov, K.M.; Seilkhanov, T.M.; Talipov, S. Synthesis, Structure, and Properties of New Lupinine O-Acyl Derivatives. Chem. Nat. Compd. 2019, 55, 506–508. [Google Scholar] [CrossRef]
- Lyskov, S.; Chou, F.C.; Conchúir, S.; Der, B.S.; Drew, K.; Kuroda, D.; Xu, J.; Weitzner, B.D.; Renfrew, P.D.; Sripakdeevong, P.; et al. Serverification of molecular modeling applications: The Rosetta Online Server that Includes Everyone (ROSIE). PLoS ONE 2013, 8, e63906. [Google Scholar] [CrossRef]
- Kothiwale, S.; Mendenhall, J.L.; Meiler, J. BCL::Conf: Small molecule conformational sampling using a knowledge based rotamer library. J. Cheminform. 2015, 7, 47. [Google Scholar] [CrossRef]
- Mukherjee, G.; Braka, A.; Wu, S. Quantifying functional-group-like structural fragments in molecules and its applications in drug design. J. Chem. Inf. Model. 2023, 63, 2073–2083. [Google Scholar] [CrossRef]






















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).