
One-Pot Phosphonylation of Heteroaromatic Lithium Reagents: The Scope and Limitations of Its Use for the Synthesis of Heteroaromatic Phosphonates



Abstract
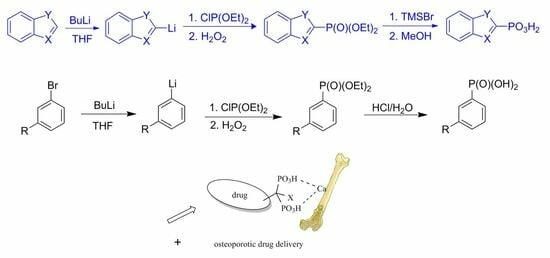
1. Introduction
2. Results
2.1. Chemistry
2.2. In Vitro Antiproliferative Evaluation
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Crystallography
4.3. General Procedure for the Synthesis
4.3.1. Preparation of Diethyl 1H-indol-2-ylphosphonate (1)
4.3.2. Preparation of 1H-indolin-2-ylphosphonic Acid (4)
4.3.3. General Procedure for the Preparation of Benzo[b]tiophen-2-ylphosphonic Acid (8), Tri-2-benzofuryl Phosphine Oxide (13), Benzofuran-2-ylphosphonic Acid (14), Tri-2-furyl oxide (15), 5-Methyltiophen-2-ylphosphonic Acid (17), Phenylphosphonic Acid (24), 3-Methoxyphenylphosphonic Acid (25), and Aphtha-1-ylphosphonic Acid (26)
4.3.4. Mixture of Diethyl Benzofuran-2-ylphosphonate (10) and Diethyl Benzofuran-2-ylphosphinate (11)
4.3.5. Preparation of Benzo[b]thiazol-2-ylphosphonic Acid (7)
4.3.6. Preparation of Ethyl Bis(N-methylpyrrol-2-yl)phosphinate (19)
4.4. Antiproliferative Activity
4.4.1. Cell Culture
4.4.2. SRB Antiproliferative Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Bałczewski, P.; Skalik, J. Quinquevalent phosphorus acids. In Organophosphorus Chemistry; Allen, D.W., Loakes, D., Tebby, J.C., Eds.; Royal Society of Chemistry: Cambridge, UK, 2013; Volume 42, pp. 81–196. [Google Scholar] [CrossRef]
- Van der Jeught, S.; Stevens, C. Direct phosphonylation of aromatic azaheterocycles. Chem. Rev. 2009, 109, 2672–2702. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Mandal, S.; Banerjee, D.; Kumar Yadav, A. Transition metal free α−C−H functionalization of six membered heteroaromatic-N-oxides. ChemSelect 2021, 6, 2832–2854. [Google Scholar] [CrossRef]
- Mehwish, H.; Chen, X.-L.; Yu, B.; Qu, L.-B.; Zhao, Y.-F. Applications of H-phosphonates for C element bond formation. Pure Appl. Chem. 2019, 91, 33–41. [Google Scholar] [CrossRef]
- Popovics-Tóth, N.; Bálint, E. Multicomponent synthesis of sotentially siologically active heterocycles containing a chosphonate or a chosphine oxide moiety. Acta Chim. Slov. 2022, 69, 735–755. [Google Scholar] [CrossRef] [PubMed]
- Demmer, C.S.; Krogsgaard-Larsen, N.; Bunch, L. Review on modern advances of chemical methods for the introduction of a phosphonic acid group. Chem. Rev. 2011, 111, 7981–8001. [Google Scholar] [CrossRef]
- Bock, T.; Mőhwald, H.; Műlhaupt, R. Arylphosphonic acid-functionalized polyelectrolytes as fuel cell membrane material. Macromol. Chem. Phys. 2007, 208, 1324–1340. [Google Scholar] [CrossRef]
- Dang, Q.; Kasibhatla, S.R.; Jiang, T.; Fan, K.; Liu, Y.; Taplin, F.; Schulz, W.; Cashion, D.K.; Reddy, K.R.; van Poelje, P.D.; et al. Discovery of phosphonic diamide prodrugs and their use for the oral delivery of a series of fructose 1,6-bisphosphatase inhibitors. J. Med. Chem. 2008, 51, 4331–4339. [Google Scholar] [CrossRef]
- Dang, Q.; Liu, Y.; Cashion, D.K.; Kasibhatla, S.R.; Jing, T.; Taplin, F.; Jacintho, J.D.; Li, H.; Sun, Z.; Fan, Y.; et al. Discovery of a series of phosphonic acid-containing thiazoles and orally bioavailable diamide prodrugs that lower glucose in diabetic animals through inhibition of fructose-1,6-bisphosphatase. J. Med. Chem. 2011, 54, 153–156. [Google Scholar] [CrossRef]
- Teixeira, F.; Rangel, C.M.; Teixeira, A.P.S. New azaheterocyclic aromatic diphosphonates for hybrid materials for fuel cell applications. New J. Chem. 2013, 37, 3084–3091. [Google Scholar] [CrossRef]
- Dziuganowska, Z.A.; Ślepokura, A.; Volle, J.-N.; Virieux, D.; Pirat, J.-L.; Kafarski, P. Structural analogues of Selfotel. J. Org. Chem. 2016, 81, 4947–4954. [Google Scholar] [CrossRef]
- Iwanejko, J.; Brol, A.; Szyja, B.; Daszkiewicz, M.; Wojaczyńska, E.; Olszewski, T.K. Hydrophosphonylation of chiral hexahydroquinoxalin-2(1H)-one derivatives as an effective route to new bicyclic compounds: Aminophosphonates, enamines and imines. Tetrahedron 2019, 75, 1431–1439. [Google Scholar] [CrossRef]
- Sabat, N.; Poštova Slavětinská, L.; Klepetářová, B.; Hocek, M.C.-H. Phosphonylation of pyrrolopyrimidines: Synthesis of substituted 7- and 9-dezapurine-8-phosphonate derivatives. J. Org. Chem. 2016, 81, 9507–9514. [Google Scholar] [CrossRef]
- Chmielewska, E.; Miszczyk, P.; Kozłowska, J.; Prokopowicz, M.; Młynarz, P.; Kafarski, P. Reaction of benzolactams with triethyl phosphite prompted by phosphoryl chloride affords benzoannulated monophosphonates instead of expected bisphosphonates. J. Organomet. Chem. 2015, 785, 84–91. [Google Scholar] [CrossRef]
- Cervera-Villanueva, J.M., Jr.; Viveros-Ceballos, J.L.; Ordóñez, M. First practical synthesis of novel 1-phosphonlated pyrrolo[1,2-α]pyrazine derivatives. Heteroatom Chem. 2017, 28, e21398. [Google Scholar] [CrossRef]
- Shety, M.; Huang, H.; Kang, J.Y. Regioselective synthesis of α- and γ-amino quinolilyl phosphonamides using N-heterocyclic phosphines (NHPs). Org. Lett. 2018, 20, 700–703. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, Y.; Yang, S.-D. Visible-light-induced tandem phosphorylation cyclization of vinyl azides under mild conditions. Org. Chem. Front. 2018, 5, 822–826. [Google Scholar] [CrossRef]
- Niu, L.; Wang, S.; Liu, J.; Yi, H.; Liang, X.-A.; Liu, T.; Lei, A. Visible-light-mdiated oxidative C(sp3)-H phosphonylaion for α–aminophosphonates under oxidnt-free conditions. Chem. Commun. 2018, 54, 1659–1662. [Google Scholar] [CrossRef]
- Su, X.; Yang, F.; Wu, Y.; Wu, Y. Direct C4-H phosphonylation of 8-hydroxyquinoline derivatives employing photoredox catalysis and silver catalysis. Org. Biomol. Chem. 2018, 16, 2753–2756. [Google Scholar] [CrossRef]
- Wahab, A.; Gao, Z.; Gou, J.; Yu, B. The construction of phosphonate triazolyl by copper(ii)-catalyzed furan dearomatized [3 + 2] cycloaddition. Org. Biomol. Chem. 2022, 20, 6319–6323. [Google Scholar] [CrossRef]
- Ali, T.E.; Assiri, M.A.; Hassanin, N.M. One-pot synthesis, antimicrobial activities, and drug-likeness analysis of some novel 1,2-benzoxaphosphinines, phospholobenzofuran, and chromonyl/coumarinyl/indenonyl phosphonate. Synth. Commun. 2022, 52, 1967–1980. [Google Scholar] [CrossRef]
- Polat-Cakir, S. 1,3-Dipolar cycloaddition reactions of acyl phosphonates with nitrile oxides: Synthesis of phosphonate-containing dioxazole derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2020, 196, 461–467. [Google Scholar] [CrossRef]
- Sreelakshmi, P.; Krishna, B.S.; Sandhisudha, S.; Murali, S.; Reddy, G.R.; Venkataramaiah, C.; Rao, P.V.; Reddy, A.V.K.; Swetha, V.; Zyryanov, G.V.; et al. Synthesis and biological evaluation of novel dialkyl (4-amino-5H-chromeno[2,3-d]pyrimidin-5-yl)phosphonates. Bioorg. Chem. 2022, 129, 106121. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Popovics-Tóth, N.; Tajti, A.; Rávai, B.; Szabó, K.E.; Perdih, F. Microwave-assisted multicomponentmsyntheses of heterocyclic phosphonates. Chem. Proc. 2021, 3, 108. [Google Scholar] [CrossRef]
- Philippov, I.; Gatilov, Y.; Sonina, A.; Vorob’ev, A. Oxidative [3+2]cycloaddition of alkynylphosphonates with heterocyclic N-imines: Synthesis of pyrazolo[1,5-a]pyridine-3-phosphonates. Molecules 2022, 27, 7913. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J. Organolithiums: Selectivity for Synthesis. In Tetrahedron Organic Chemistry; Series 23; Pergamon Press: Oxford, UK, 2002. [Google Scholar]
- Nájera, C.; Sansano, J.M.; Yus, M. Recent synthetic uses of functionalised aromatic and heteroaromatic organolithium reagents prepared by non-deprotonating methods. Tetrahedron 2003, 59, 9255–9303. [Google Scholar] [CrossRef]
- Ila, H.; Markiewicz, J.T.; Malakhov, V.; Knochel, P. Metalated indoles, indazoles, benzimidazoles, and azaindoles and their synthetic applications. Synthesis 2013, 45, 2343–2371. [Google Scholar] [CrossRef]
- Power, M.; Alcock, E.; McGlaken, P. Organolithium bases in flow chemistry: A review. Org. Process Res. Dev. 2020, 24, 1814–1838. [Google Scholar] [CrossRef]
- Bisseret, P.; Thielges, S.; Bourg, S.; Miethke, M.; Marahiel, M.A.; Eustache, J. Synthesis of a 2-indolylphosphonamide derivative with inhibitory activity against yersiniabactin biosynthesis. Tetrahedron Lett. 2007, 48, 6080–6083. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Akutagawa, K. Carbon dioxide: A reagent for the protection of nucleophilic centres and the simultaneous activation of alternative locations to electrophilic attack: Part I. A new synthetic method for the 2-substitution of 1-unsubstituted indoles. Tetrahedron Lett. 1985, 26, 5935–5938. [Google Scholar] [CrossRef]
- Du, Y.; Wiemer, D.F. Preparation of α-phosphonolactams using electrophilic phosphorus reagents: Application in the synthesis of lactam-based farnesyl transferase inhibitors. J. Org. Chem. 2002, 67, 5709–5717. [Google Scholar] [CrossRef]
- Haelters, J.P.; Corbel, B.; Sturtz, G. Synthese D’indole phosphonates par cyclisation selon fischer d’arylhydrazones phosphonates. Phosphorus Sulfur Silicon Relat. Elem. 1988, 37, 41–64. [Google Scholar] [CrossRef]
- Iddon, B. Synthesis and reactions of lithiated monocyclic azoles containing two or more hetero-toms. Part II: Oxazoles. Hetrocycles 1994, 37, 1321–1346. [Google Scholar] [CrossRef]
- Jenkins, D.; Sykora, R.E.; Assefa, Z. Tri-2-furyl-phosphine oxide: An oxidation product of the weak Lewis base tri-2-furylphosphine. Acta Cryst. 2007, E63, 3510–3511. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Cryst. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Gjøs, N.; Gronowitz, S. Thienylpyrroles I. Synthesis via Copper intermediates. Acta Chem. Scand. 1971, 25, 2596–2608. [Google Scholar] [CrossRef]
- Merly, I.; Smith, S.L. Murine RAW 264.7 cell line as an immune target: Are we missing something? Immunopharmacol. Immunotoxicol. 2017, 39, 55–58. [Google Scholar] [CrossRef]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef]
- Raja Singh, P.; Sugantha Priya, E.; Balakrishnan, S.; Arunkumar, R.; Sharmila, G.; Rajalakshmi, M.; Arunakaran, J. Inhibition of cell survival and proliferation by nimbolide in human androgen-independent prostate cancer (PC-3) cells: Involvement of the PI3K/Akt pathway. Mol. Cell Biochem. 2017, 427, 69–79. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- CrysAlis PRO Software System. Version 1 171.37.57. Oxford Diffraction Ltd.: Abingdon, UK, 2005.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sek C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Chen, C.; Jie, D.; Liying, L.; Yujie, H.; Bolin, Z. Palladium-catalyzed domino cyclization/phosphorylation of gem-dibromoolefins with P(O)H compounds: Synthesis of phosphorylated heteroaromatics. Adv. Synt. Catal. 2022, 364, 200–205. [Google Scholar] [CrossRef]
- Chunya, L.; Yuta, S.; Shun-Ya, O.; Shu, K.; Kazuhiko, S.; Norihisa, F.; Li-Biao, H. Wet and dry processes for the selective transformation of phosphonates to phosphonic acids catalyzed by brønsted acids. J. Org. Chem. 2020, 85, 14411–14419. [Google Scholar] [CrossRef]
- Koohgard, M.; Hosseini-Sarvar, M. Visible-light-mediated phosphonylation reaction: Formation of phosphonates from alkyl/arylhydrazines and trialkylphosphites using zinc phthalocyanine. Org. Biomol. Chem. 2021, 19, 5905–5911. [Google Scholar] [CrossRef]
- Grabiak, R.C.; Miles, J.A.; Schwenzer, G.M. Synthesis of phosphonic dichlorides and correlation of their p-31 chemical shifts. Phosphorus Sulfur Silicon Relat. Elem. 1980, 9, 197–202. [Google Scholar] [CrossRef]
- Kuimov, V.A.; Malysheva, S.F.; Gusarova, N.K.; Vakul’skaya, T.I.; Khutsishvili, S.S.; Trofimov, B.A. The reaction of red phosphorus with 1-bromonaphthalene in the KOH–DMSO system: Synthesis of tri(1-naphthyl)phosphane. Heteroat. Chem. 2011, 22, 198–203. [Google Scholar] [CrossRef]
- Soumyadip, H.; Abhijeet, S.; Singh, R.P. Cu-Catalyzed D\direct C–P B\bond formation through dehydrogenative cross-coupling reactions between azoles and dialkyl phosphites. J. Org. Chem. 2019, 84, 6868–6878. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Nevozhay, D. Cheburator software for automatically calculating drug inhibitory concentrations from in vitro screening assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13 | 15 | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C144H92O22P6 | C12H9O4P |
| Mr | 2359.99 | 248.16 |
| Crystal system, space group | Trigonal, R | Orthorhombic, P212121 |
| a, b, c (Å) | 30.3245 (9), 30.3245 (9), 10.5270 (3) | 7.9719 (2), 10.8701 (3), 13.3094 (4) |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 |
| V (Å3) | 8383.4 (6) | 1153.33 (6) |
| Z | 3 | 4 |
| µ (mm−1) | 0.18 | 0.24 |
| Crystal size (mm) | 0.3 × 0.25 × 0.2 | 0.4 × 0.3 × 0.2 |
| Data collection | ||
| No. of measured, independent and observed [I > 2σ(I)] reflections | 19,039, 3651, 2079 | 5500, 2274, 2103 |
| Rint | 0.072 | 0.022 |
| (sin θ/λ)max (Å−1) | 0.617 | 0.617 |
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.039, 0.063, 0.83 | 0.027, 0.066, 1.02 |
| No. of reflections | 3651 | 2274 |
| No. of parameters | 271 | 154 |
| Δρmax, Δρmin (e Å−3) | 0.30, −0.21 | 0.31, −0.25 |
| Compound | Structure | IC50 a [μM] | ||
|---|---|---|---|---|
| RAW 264.7 | PC-3 | MCF-7 | ||
| 4 |  | 6.49 ± 0.139 | 4.78 ± 3.69 | 1.25 ± 0.47 |
| 7 |  | 16.95 ± 2.11 | 4.942± 4.08 | 5.83 ± 2.26 |
| 8 |  | 28.07 ± 17.8 | 16.03 ± 3.40 | 5.40 ± 1.55 |
| 14 |  | 18.21 ± 2.11 | 3.95 ± 4.18 | 15.78 ±7.55 |
| 17 |  | 15.08 ± 2.47 | 10.00 ± 2.83 | 4.00 ± 2.83 |
| 24 |  | 38.91 ± 0.132 | 6.18 ± 1.02 | 8.55 ±1.48 |
| 26 |  | 14.05 ± 3.37 | 3.96 ± 1.63 | 6.09 ± 1.73 |
| Zoledronic acid |  | 42.2 ± 8.40 | 146.0 ± 67.4 | 115.3 ± 87.6 |
| Cisplatin | (H2N)2PtCl2 | 0.93 ± 0.40 | 9.83 ± 1.70 | 6.37 ± 0.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmielewska, E.; Miodowska, N.; Dziuk, B.; Psurski, M.; Kafarski, P. One-Pot Phosphonylation of Heteroaromatic Lithium Reagents: The Scope and Limitations of Its Use for the Synthesis of Heteroaromatic Phosphonates. Molecules 2023, 28, 3135. https://doi.org/10.3390/molecules28073135
Chmielewska E, Miodowska N, Dziuk B, Psurski M, Kafarski P. One-Pot Phosphonylation of Heteroaromatic Lithium Reagents: The Scope and Limitations of Its Use for the Synthesis of Heteroaromatic Phosphonates. Molecules. 2023; 28(7):3135. https://doi.org/10.3390/molecules28073135
Chicago/Turabian StyleChmielewska, Ewa, Natalia Miodowska, Błażej Dziuk, Mateusz Psurski, and Paweł Kafarski. 2023. "One-Pot Phosphonylation of Heteroaromatic Lithium Reagents: The Scope and Limitations of Its Use for the Synthesis of Heteroaromatic Phosphonates" Molecules 28, no. 7: 3135. https://doi.org/10.3390/molecules28073135
APA StyleChmielewska, E., Miodowska, N., Dziuk, B., Psurski, M., & Kafarski, P. (2023). One-Pot Phosphonylation of Heteroaromatic Lithium Reagents: The Scope and Limitations of Its Use for the Synthesis of Heteroaromatic Phosphonates. Molecules, 28(7), 3135. https://doi.org/10.3390/molecules28073135