


Potential Therapeutic Value of the STING Inhibitors

Abstract
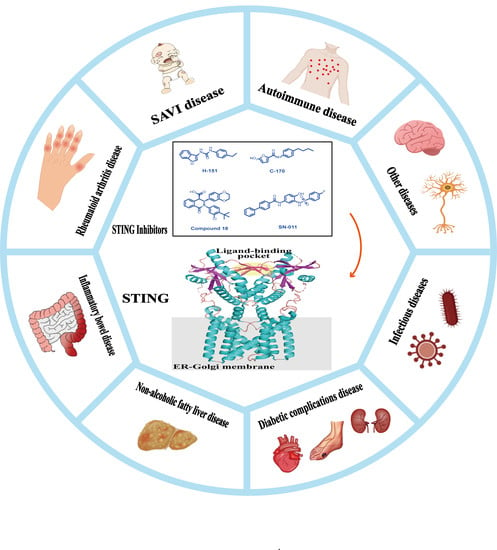
1. Introduction
2. The Structure and Location of STING
3. The Function of STING
4. Genotype
5. DNA Sensors Upstream of STING
6. The Regulation of the STING Pathway
7. STING Inhibitors
7.1. STING Inhibitors Targeting the Palmitoylation Site
7.2. STING Inhibitors Targeting CDN Binding Site
7.3. STING Inhibitor with an Unknown Site of Action
7.4. STING Protein Degraders
8. STING-Related Diseases
8.1. STING and Psoriasis
8.2. STING and Systemic Lupus Erythematosus
8.3. STING and Infectious Diseases
8.4. STING and SAVI
8.5. STING and CNS Diseases
8.6. STING and Inflammatory Bowel Disease
8.7. STING and NAFLD
8.8. STING and Diabetic Complications
8.9. STING and Other Diseases
9. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhao, J.; Xiao, R.; Zeng, R.; He, E.; Zhang, A. Small molecules targeting cGAS-STING pathway for autoimmune disease. Eur. J. Med. Chem. 2022, 238, 114480. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct Target. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Saferding, V.; Blüml, S. Innate Immunity as the trigger of systemic autoimmune diseases. J. Autoimmun. 2020, 110, 102382. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Szodoray, P.; Zeher, M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2016, 50, 1–17. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Wicherska-Pawłowska, K.; Wróbel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e11. [Google Scholar] [CrossRef]
- Khan, S.; Godfrey, V.; Zaki, M.H. Cytosolic Nucleic Acid Sensors in Inflammatory and Autoimmune Disorders. Int. Rev. Cell Mol. Biol. 2019, 344, 215–253. [Google Scholar] [CrossRef]
- Zhang, H.; You, Q.D.; Xu, X.L. Targeting Stimulator of Interferon Genes (STING): A Medicinal Chemistry Perspective. J. Med. Chem. 2020, 63, 3785–3816. [Google Scholar] [CrossRef]
- Cerboni, S.; Jeremiah, N.; Gentili, M.; Gehrmann, U.; Conrad, C.; Stolzenberg, M.C.; Picard, C.; Neven, B.; Fischer, A.; Amigorena, S.; et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 2017, 214, 1769–1785. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Joshi, J.C.; Mehta, D. Regulation of cGAS Activity and Downstream Signaling. Cells 2022, 11, 2812. [Google Scholar] [CrossRef]
- Benmerzoug, S.; Ryffel, B.; Togbe, D.; Quesniaux, V.F.J. Self-DNA Sensing in Lung Inflammatory Diseases. Trends Immunol. 2019, 40, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, B.; Liu, S.Y.; Iyer, S.S.; Yu, Y.; Wu, A.; Cheng, G. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 2015, 194, 1545–1554. [Google Scholar] [CrossRef]
- Ma, F.; Li, B.; Yu, Y.; Iyer, S.S.; Sun, M.; Cheng, G. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. 2015, 16, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Panchanathan, R.; Liu, H.; Xin, D.; Choubey, D. Identification of a negative feedback loop between cyclic di-GMP-induced levels of IFI16 and p202 cytosolic DNA sensors and STING. Innate Immun. 2014, 20, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, R.; Guo, W.; Xie, L.; Qiao, Z.; Chen, S.; Zhu, J.; Huang, C.; Huang, J.; Chen, B.; et al. STING-Mediated IFI16 Degradation Negatively Controls Type I Interferon Production. Cell Rep. 2019, 29, 1249–1260.e4. [Google Scholar] [CrossRef]
- Hong, Z.; Mei, J.; Guo, H.; Zhu, J.; Wang, C. Intervention of cGAS-STING signaling in sterile inflammatory diseases. J. Mol. Cell Biol. 2022, 14, mjac005. [Google Scholar] [CrossRef]
- Gota, C.; Calabrese, L. Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity 2003, 36, 511–518. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Gyongyosi, B.; Satishchandran, A.; Lowe, P.; Kodys, K.; Catalano, D.; Calenda, C.D.; Kurt-Jones, E.A.; Fitzgerald, K.A.; et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 2016, 291, 26794–26805. [Google Scholar] [CrossRef]
- Li, S.; Hong, Z.; Wang, Z.; Li, F.; Mei, J.; Huang, L.; Lou, X.; Zhao, S.; Song, L.; Chen, W.; et al. The Cyclopeptide Astin C Specifically Inhibits the Innate Immune CDN Sensor STING. Cell Rep. 2018, 25, 3405–3421.e3407. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Li, G.; Tao, J.; Wu, N.N.; Kandadi, M.R.; Bi, Y.; Wang, S.; Pei, Z.; Ren, J. Double knockout of Akt2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS-STING-mediated mechanism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165855. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Geng, K.; Law, B.Y.; Wang, P.; Pu, Y.L.; Chen, Q.; Xu, H.W.; Tan, X.Z.; Jiang, Z.Z.; Xu, Y. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol. Toxicol. 2023, 39, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhao, R.; He, Q.; Cui, C.; Song, J.; Guo, X.; Zang, N.; Yang, M.; Zou, Y.; Yang, J.; et al. cGAS-STING mediates cytoplasmic mitochondrial-DNA-induced inflammatory signal transduction during accelerated senescence of pancreatic β-cells induced by metabolic stress. FASEB J. 2022, 36, e22266. [Google Scholar] [CrossRef]
- Pan, Y.; You, Y.; Sun, L.; Sui, Q.; Liu, L.; Yuan, H.; Chen, C.; Liu, J.; Wen, X.; Dai, L.; et al. The STING antagonist H-151 ameliorates psoriasis via suppression of STING/NF-κB-mediated inflammation. Br. J. Pharmacol. 2021, 178, 4907–4922. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Gao, Y.; Gao, R.; Wang, Y.; Qu, Y.; Yang, J.; Wei, X.; Zhang, F.; Ge, J. The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int. Immunopharmacol. 2022, 107, 108658. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Lu, L.; Zhou, Y.; Liu, J.; Ma, H.; Fu, L.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. The novel STING antagonist H151 ameliorates cisplatin-induced acute kidney injury and mitochondrial dysfunction. Am. J. Physiol Ren. Physiol. 2021, 320, F608–F616. [Google Scholar] [CrossRef]
- Wu, B.; Xu, M.M.; Fan, C.; Feng, C.L.; Lu, Q.K.; Lu, H.M.; Xiang, C.G.; Bai, F.; Wang, H.Y.; Wu, Y.W.; et al. STING inhibitor ameliorates LPS-induced ALI by preventing vascular endothelial cells-mediated immune cells chemotaxis and adhesion. Acta Pharm. Sin. 2022, 43, 2055–2066. [Google Scholar] [CrossRef]
- Wu, J.J.; Zhao, L.; Hu, H.G.; Li, W.H.; Li, Y.M. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rev. 2020, 40, 1117–1141. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates Innate Immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Skopelja-Gardner, S.; An, J.; Elkon, K.B. Role of the cGAS-STING pathway in systemic and organ-specific diseases. Nat. Rev. Nephrol. 2022, 18, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Shang, G.; Li, J.; Lu, Y.; Bai, X.C.; Zhang, X. Activation of STING by targeting a pocket in the transmembrane domain. Nature 2022, 604, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.P.; Yang, Y.; Wang, Y.Y.; Zhang, X.L.; Shu, H.B. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates Innate Immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small molecules targeting the Innate Immune cGAS-STING-TBK1 signaling pathway. Acta Pharm. Sin. B 2020, 10, 2272–2298. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Ouyang, S.; Song, X.; Wang, Y.; Ru, H.; Shaw, N.; Jiang, Y.; Niu, F.; Zhu, Y.; Qiu, W.; Parvatiyar, K.; et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 2012, 36, 1073–1086. [Google Scholar] [CrossRef]
- Shang, G.; Zhu, D.; Li, N.; Zhang, J.; Zhu, C.; Lu, D.; Liu, C.; Yu, Q.; Zhao, Y.; Xu, S.; et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 2012, 19, 725–727. [Google Scholar] [CrossRef]
- Garland, K.M.; Sheehy, T.L.; Wilson, J.T. Chemical and Biomolecular Strategies for STING Pathway Activation in Cancer Immunotherapy. Chem. Rev. 2022, 122, 5977–6039. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef]
- Shi, H.; Wu, J.; Chen, Z.J.; Chen, C. Molecular basis for the specific recognition of the metazoan cyclic GMP-AMP by the Innate Immune adaptor protein STING. Proc. Natl. Acad. Sci. USA 2015, 112, 8947–8952. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Z.; Xu, Z.; Jin, X.; Gong, Y.; Xia, X.; Yao, Y.; Xu, Z.; Zhou, Y.; Xu, H.; et al. Proteomic Maps of Human Gastrointestinal Stromal Tumor Subgroups. Mol. Cell Proteom. 2019, 18, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Mohr, L.; Toufektchan, E.; von Morgen, P.; Chu, K.; Kapoor, A.; Maciejowski, J. ER-directed TREX1 limits cGAS activation at micronuclei. Mol. Cell 2021, 81, 724–738.e729. [Google Scholar] [CrossRef] [PubMed]
- Ergun, S.L.; Fernandez, D.; Weiss, T.M.; Li, L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019, 178, 290–301.e210. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, P.; Rivara, S.; Liu, C.; Ricci, J.; Ren, X.; Hurley, J.H.; Ablasser, A. Clathrin-associated AP-1 controls termination of STING signalling. Nature 2022, 610, 761–767. [Google Scholar] [CrossRef]
- Kuchitsu, Y.; Mukai, K.; Uematsu, R.; Takaada, Y.; Shinojima, A.; Shindo, R.; Shoji, T.; Hamano, S.; Ogawa, E.; Sato, R.; et al. STING signalling is terminated through ESCRT-dependent microautophagy of vesicles originating from recycling endosomes. Nat. Cell Biol. 2023, 25, 453–466. [Google Scholar] [CrossRef]
- Wu, J.; Yan, N. No Longer A One-Trick Pony: STING Signaling Activity Beyond Interferon. J. Mol. Biol. 2022, 434, 167257. [Google Scholar] [CrossRef]
- Yamamoto, M.; Gohda, J.; Akiyama, T.; Inoue, J.I. TNF receptor-associated factor 6 (TRAF6) plays crucial roles in multiple biological systems through polyubiquitination-mediated NF-κB activation. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 97, 145–160. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e745. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Mann, C.C.; Orzalli, M.H.; King, D.S.; Kagan, J.C.; Lee, A.S.Y.; Kranzusch, P.J. Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-κB Signaling Adaptation. Cell Rep. 2019, 27, 1165–1175.e1165. [Google Scholar] [CrossRef]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Zhu, Y.; Zhang, Q.; Guan, H.; Liu, S.; Chen, S.; Mei, C.; Chen, C.; Liao, Z.; et al. A non-canonical cGAS-STING-PERK pathway facilitates the translational program critical for senescence and organ fibrosis. Nat. Cell Biol. 2022, 24, 766–782. [Google Scholar] [CrossRef]
- Burman, C.; Ktistakis, N.T. Autophagosome formation in mammalian cells. Semin. Immunopathol. 2010, 32, 397–413. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, R.; Tang, D. The STING1 network regulates autophagy and cell death. Signal. Transduct. Target. Ther. 2021, 6, 208. [Google Scholar] [CrossRef]
- Liu, D.; Wu, H.; Wang, C.; Li, Y.; Tian, H.; Siraj, S.; Sehgal, S.A.; Wang, X.; Wang, J.; Shang, Y.; et al. STING directly activates autophagy to tune the Innate Immune response. Cell Death Differ. 2019, 26, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Qian, C.; Wang, Q.; Li, J.; Zhang, H.; Wang, L.; Pu, M.; Huang, Y.; He, Z.; Zhou, T.; et al. STING directly recruits WIPI2 for autophagosome formation during STING-induced autophagy. EMBO J. 2023, e112387. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained Innate Immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Jin, L.; Xu, L.G.; Yang, I.V.; Davidson, E.J.; Schwartz, D.A.; Wurfel, M.M.; Cambier, J.C. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun. 2011, 12, 263–269. [Google Scholar] [CrossRef]
- Patel, S.; Blaauboer, S.M.; Tucker, H.R.; Mansouri, S.; Ruiz-Moreno, J.S.; Hamann, L.; Schumann, R.R.; Opitz, B.; Jin, L. The Common R71H-G230A-R293Q Human TMEM173 Is a Null Allele. J. Immunol. 2017, 198, 776–787. [Google Scholar] [CrossRef]
- Patel, S.; Jin, L. TMEM173 variants and potential importance to human biology and disease. Genes Immun. 2019, 20, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The common HAQ STING variant impairs cGAS-dependent antibacterial responses and is associated with susceptibility to Legionnaires’ disease in humans. PLoS Pathog. 2018, 14, e1006829. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kang, J.A.; Suh, D.I.; Park, E.B.; Lee, C.R.; Choi, S.A.; Kim, S.Y.; Kim, Y.; Park, S.H.; Ye, M.; et al. Tofacitinib relieves symptoms of stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy caused by 2 de novo variants in TMEM173. J. Allergy Clin. Immunol. 2017, 139, 1396–1399.e1312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef]
- Chiliveru, S.; Rahbek, S.H.; Jensen, S.K.; Jørgensen, S.E.; Nissen, S.K.; Christiansen, S.H.; Mogensen, T.H.; Jakobsen, M.R.; Iversen, L.; Johansen, C.; et al. Inflammatory cytokines break down intrinsic immunological tolerance of human primary keratinocytes to cytosolic DNA. J. Immunol. 2014, 192, 2395–2404. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an Innate Immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tuting, T.; Hartmann, G.; Barchet, W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates Innate Immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311.e1. [Google Scholar] [CrossRef]
- Wang, C.; Guan, Y.; Lv, M.; Zhang, R.; Guo, Z.; Wei, X.; Du, X.; Yang, J.; Li, T.; Wan, Y.; et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity 2018, 48, 675–687.e677. [Google Scholar] [CrossRef]
- Gasser, S.; Zhang, W.Y.L.; Tan, N.Y.J.; Tripathi, S.; Suter, M.A.; Chew, Z.H.; Khatoo, M.; Ngeow, J.; Cheung, F.S.G. Sensing of dangerous DNA. Mech. Ageing Dev. 2017, 165, 33–46. [Google Scholar] [CrossRef]
- Choubey, D. Cytosolic DNA sensor IFI16 proteins: Potential molecular integrators of interactions among the aging hallmarks. Ageing Res. Rev. 2022, 82, 101765. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Briard, B.; Place, D.E.; Kanneganti, T.D. DNA Sensing in the Innate Immune Response. Physiology 2020, 35, 112–124. [Google Scholar] [CrossRef]
- Parvatiyar, K.; Zhang, Z.; Teles, R.M.; Ouyang, S.; Jiang, Y.; Iyer, S.S.; Zaver, S.A.; Schenk, M.; Zeng, S.; Zhong, W.; et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 2012, 13, 1155–1161. [Google Scholar] [CrossRef]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef]
- Martin, M.; Hiroyasu, A.; Guzman, R.M.; Roberts, S.A.; Goodman, A.G. Analysis of Drosophila STING Reveals an Evolutionarily Conserved Antimicrobial Function. Cell Rep. 2018, 23, 3537–3550.e3536. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the Innate Immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, R.; Yi, L.; Tang, Y.D.; Zheng, C. UNC93B1 attenuates the cGAS-STING signaling pathway by targeting STING for autophagy-lysosome degradation. J. Med. Virol. 2022, 94, 4490–4501. [Google Scholar] [CrossRef]
- He, Z.; Ye, S.; Xing, Y.; Jiu, Y.; Zhong, J. UNC93B1 curbs cytosolic DNA signaling by promoting STING degradation. Eur. J. Immunol. 2021, 51, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Ding, S. Regulation of cGAS/STING signaling and corresponding immune escape strategies of viruses. Front. Cell. Infect. Microbiol. 2022, 12, 954581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, C.; Senlin, L.; Chen, W. Research advances in the regulation of intrinsic immune signaling pathways by ubiquitination modifications. Chem. Life 2015, 35, 164–175. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting Innate Immune response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, N.; Cui, Y.; Hong, Z.; Liu, X.; Wang, Q.; Li, S.; Liu, H.; Yu, H.; Cai, Y.; et al. The deubiquitinase CYLD is a specific checkpoint of the STING antiviral signaling pathway. PLoS Pathog. 2018, 14, e1007435. [Google Scholar] [CrossRef]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The ubiquitin ligase TRIM56 regulates Innate Immune responses to intracellular double-stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef]
- Balka, K.R.; De Nardo, D. Molecular and spatial mechanisms governing STING signalling. FEBS J. 2021, 288, 5504–5529. [Google Scholar] [CrossRef]
- Ni, G.; Konno, H.; Barber, G.N. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2017, 2, eaah7119. [Google Scholar] [CrossRef]
- Stempel, M.; Chan, B.; Juranić Lisnić, V.; Krmpotić, A.; Hartung, J.; Paludan, S.R.; Füllbrunn, N.; Lemmermann, N.A.; Brinkmann, M.M. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-κB signaling and STING’s dual role during MCMV infection. EMBO J. 2019, 38, e100983. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.L.; Buchan, G.J.; Rühl, M.; Mukai, K.; Salvatore, S.R.; Ogawa, E.; Andersen, S.D.; Iversen, M.B.; Thielke, A.L.; Gunderstofte, C.; et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E7768–E7775. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, E.V.; Zhang, X.; Remillard, D.; Lazar, D.C.; Suciu, R.M.; Wang, Y.; Bianco, G.; Yamashita, Y.; Crowley, V.M.; Schafroth, M.A.; et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182, 1009–1026.e9. [Google Scholar] [CrossRef] [PubMed]
- Siu, T.; Altman, M.D.; Baltus, G.A.; Childers, M.; Ellis, J.M.; Gunaydin, H.; Hatch, H.; Ho, T.; Jewell, J.; Lacey, B.M.; et al. Discovery of a Novel cGAMP Competitive Ligand of the Inactive Form of STING. ACS Med. Chem. Lett. 2019, 10, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Mei, J.; Li, C.; Bai, G.; Maimaiti, M.; Hu, H.; Yu, W.; Sun, L.; Zhang, L.; Cheng, D.; et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc. Natl. Acad. Sci. USA 2021, 118, e2105465118. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, M.; Wu, X.; Zhang, H.; Su, H.; Dang, Y.; Ma, M.; Wang, F.; Xu, J.; Chen, L.; et al. CDK inhibitor Palbociclib targets STING to alleviate autoinflammation. EMBO Rep. 2022, 23, e53932. [Google Scholar] [CrossRef]
- Long, J.; Ying, T.; Zhang, L.; Yu, T.; Wu, J.; Liu, Y.; Li, X.; You, G.; Zhang, L.; Bi, Y. Discovery of fusidic acid derivatives as novel STING inhibitors for treatment of sepsis. Eur. J. Med. Chem. 2022, 244, 114814. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, L.; Ruan, Y.; Deng, B.; Yang, Z.; Ren, Y.; Li, L.; Liu, T.; Zhao, H.; Mai, R.; et al. Novel CRBN-Recruiting Proteolysis-Targeting Chimeras as Degraders of Stimulator of Interferon Genes with In Vivo Anti-Inflammatory Efficacy. J. Med. Chem. 2022, 65, 6593–6611. [Google Scholar] [CrossRef]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Bonacci, G.; Baker, P.R.; Salvatore, S.R.; Shores, D.; Khoo, N.K.; Koenitzer, J.R.; Vitturi, D.A.; Woodcock, S.R.; Golin-Bisello, F.; Cole, M.P.; et al. Conjugated linoleic acid is a preferential substrate for fatty acid nitration. J. Biol. Chem. 2012, 287, 44071–44082. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.; Trostchansky, A.; Schopfer, F.J.; Salvatore, S.R.; Sánchez-Calvo, B.; Vitturi, D.; Valderrama, R.; Barroso, J.B.; Radi, R.; Freeman, B.A.; et al. Olives and olive oil are sources of electrophilic fatty acid nitroalkenes. PLoS ONE 2014, 9, e84884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qin, Z.; Sun, W.; Chu, F.; Zhou, F. Function of Protein S-Palmitoylation in Immunity and Immune-Related Diseases. Front. Immunol. 2021, 12, 661202. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Schön, M.P. Psoriasis. Nat. Rev. Dis. Prim. 2016, 2, 16082. [Google Scholar] [CrossRef]
- Ding, X.L.; Wang, T.L.; Shen, Y.W.; Wang, X.Y.; Zhou, C.; Tian, S. Epidemiological investigation of psoriasis in six provinces and cities in China. Chin. J. Dermatol. Venereol. 2010, 7, 598–601. [Google Scholar] [CrossRef]
- Sawyer, L.M.; Malottki, K.; Sabry-Grant, C.; Yasmeen, N.; Wright, E.; Sohrt, A.; Borg, E.; Warren, R.B. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: A systematic review and network meta-analysis of PASI response. PLoS ONE 2019, 14, e0220868. [Google Scholar] [CrossRef]
- Kong, Y.; Zhang, S.; Wu, R.; Su, X.; Peng, D.; Zhao, M.; Su, Y. New insights into different adipokines in linking the pathophysiology of obesity and psoriasis. Lipids Health Dis. 2019, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Kishimoto, M.; Sugai, J.; Komine, M.; Ohtsuki, M. Risk Factors for the Development of Psoriasis. Int. J. Mol. Sci. 2019, 20, 4347. [Google Scholar] [CrossRef]
- Erkek, E.; Karaduman, A.; Akcan, Y.; Sökmensüer, C.; Bükülmez, G. Psoriasis associated with HCV and exacerbated by interferon alpha: Complete clearance with acitretin during interferon alpha treatment for chronic active hepatitis. Dermatology 2000, 201, 179–181. [Google Scholar] [CrossRef]
- Toussirot, E.; Béreau, M.; Bossert, M.; Malkoun, I.; Lohse, A. Occurrence of Psoriatic Arthritis during Interferon Beta 1a Treatment for Multiple Sclerosis. Case Rep. Rheumatol. 2014, 2014, 949317. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; McCarty, M.F.; Bucana, C.D.; Yuspa, S.H.; Morgan, D.; Arbeit, J.M.; Ellis, L.M.; Cleary, K.R.; Fidler, I.J. Expression of interferon-beta is associated with growth arrest of murine and human epidermal cells. J. Investig. Derm. 1999, 112, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, Q.; Wang, X.; Zhou, H.; Hu, J.; Gu, L.; Hu, Y.; Zeng, F.; Zhao, F.; Yue, C.; et al. Pathogenesis, multi-omics research, and clinical treatment of psoriasis. J. Autoimmun. 2022, 133, 102916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J. Type1 Interferons Potential Initiating Factors Linking Skin Wounds With Psoriasis Pathogenesis. Front. Immunol. 2019, 10, 1440. [Google Scholar] [CrossRef]
- 17th International Congress of Immunology, 19–23 October 2019, Beijing, China. Eur. J. Immunol. 2019, 49 (Suppl. 3), 1–2223. [CrossRef] [PubMed]
- Lebre, M.C.; van der Aar, A.M.; van Baarsen, L.; van Capel, T.M.; Schuitemaker, J.H.; Kapsenberg, M.L.; de Jong, E.C. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J. Investig. Derm. 2007, 127, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.Y. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J. Clin. Investig. 2021, 131, e139333. [Google Scholar] [CrossRef]
- Arican, O.; Aral, M.; Sasmaz, S.; Ciragil, P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediat. Inflamm. 2005, 2005, 273–279. [Google Scholar] [CrossRef]
- Korman, N.J. Management of psoriasis as a systemic disease: What is the evidence. Br. J. Derm. 2020, 182, 840–848. [Google Scholar] [CrossRef]
- Meng, S.; Lin, Z.; Wang, Y.; Wang, Z.; Li, P.; Zheng, Y. Psoriasis therapy by Chinese medicine and modern agents. Chin. Med. 2018, 13, 16. [Google Scholar] [CrossRef]
- Lapteva, M.; Mondon, K.; Möller, M.; Gurny, R.; Kalia, Y.N. Polymeric micelle nanocarriers for the cutaneous delivery of tacrolimus: A targeted approach for the treatment of psoriasis. Mol. Pharm. 2014, 11, 2989–3001. [Google Scholar] [CrossRef]
- Silfvast-Kaiser, A.; Paek, S.Y.; Menter, A. Anti-IL17 therapies for psoriasis. Expert Opin. Biol. 2019, 19, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Chaudhari, K.; Syed, B.A. The psoriasis drugs market. Nat. Rev. Drug Discov. 2015, 14, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schäfer, M.; Coyle, A.J.; Renauld, J.C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef]
- Alwawi, E.A.; Mehlis, S.L.; Gordon, K.B. Treating psoriasis with adalimumab. Clin. Risk Manag. 2008, 4, 345–351. [Google Scholar] [CrossRef]
- Blauvelt, A.; Sofen, H.; Papp, K.; Gooderham, M.; Tyring, S.; Zhao, Y.; Lowry, S.; Mendelsohn, A.; Parno, J.; Reich, K. Tildrakizumab efficacy and impact on quality of life up to 52 weeks in patients with moderate-to-severe psoriasis: A pooled analysis of two randomized controlled trials. J. Eur. Acad. Derm. Venereol. 2019, 33, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, A.C.; Warren, R.B. Brodalumab in psoriasis: Evidence to date and clinical potential. Drugs Context 2019, 8, 212570. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Gong, Z.; Peng, Y.; Li, X.; Zhang, Z.; Zhang, X.; You, X.; Wu, J. Activation of the STING pathway induces peripheral sensitization via neuroinflammation in a rat model of bone cancer pain. Inflamm. Res. 2022. [Google Scholar] [CrossRef]
- Xiaohong, L.; Zhenting, Z.; Yunjie, Y.; Wei, C.; Xiangjin, X.; Kun, X.; Xin, L.; Lu, L.; Jun, L.; Pin, C. Activation of the STING-IRF3 pathway involved in psoriasis with diabetes mellitus. J. Cell Mol. Med. 2022, 26, 2139–2151. [Google Scholar] [CrossRef]
- Inoue, K.; Ishizawa, M.; Kubota, T. Monoclonal anti-dsDNA antibody 2C10 escorts DNA to intracellular DNA sensors in normal mononuclear cells and stimulates secretion of multiple cytokines implicated in lupus pathogenesis. Clin. Exp. Immunol. 2020, 199, 150–162. [Google Scholar] [CrossRef]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2016, 2, 16039. [Google Scholar] [CrossRef]
- Darrah, E.; Andrade, F. NETs: The missing link between cell death and systemic autoimmune diseases. Front. Immunol. 2012, 3, 428. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.M.; Chang, H.C.; Liang, C.C.; Wu, J.J.; Liu, M.F. Deoxyribonuclease-inhibitory antibodies in systemic lupus erythematosus. J. Biomed. Sci 2003, 10, 544–551. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515. [Google Scholar] [CrossRef]
- Gkirtzimanaki, K.; Kabrani, E.; Nikoleri, D.; Polyzos, A.; Blanas, A.; Sidiropoulos, P.; Makrigiannakis, A.; Bertsias, G.; Boumpas, D.T.; Verginis, P. IFNα Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018, 25, 921–933.e5. [Google Scholar] [CrossRef]
- Motwani, M.; McGowan, J.; Antonovitch, J.; Gao, K.M.; Jiang, Z.; Sharma, S.; Baltus, G.A.; Nickerson, K.M.; Marshak-Rothstein, A.; Fitzgerald, K.A. cGAS-STING Pathway Does Not Promote Autoimmunity in Murine Models of SLE. Front. Immunol. 2021, 12, 605930. [Google Scholar] [CrossRef]
- Sharma, S.; Campbell, A.M.; Chan, J.; Schattgen, S.A.; Orlowski, G.M.; Nayar, R.; Huyler, A.H.; Nündel, K.; Mohan, C.; Berg, L.J.; et al. Suppression of systemic autoimmunity by the Innate Immune adaptor STING. Proc. Natl. Acad. Sci. USA 2015, 112, E710–E717. [Google Scholar] [CrossRef]
- Panchanathan, R.; Shen, H.; Zhang, X.; Ho, S.-m.; Choubey, D. Mutually Positive Regulatory Feedback Loop between Interferons and Estrogen Receptor-α in Mice: Implications for Sex Bias in Autoimmunity. PLoS ONE 2010, 5, e10868. [Google Scholar] [CrossRef]
- Panchanathan, R.; Liu, H.; Leung, Y.-K.; Ho, S.-m.; Choubey, D. Bisphenol A (BPA) stimulates the interferon signaling and activates the inflammasome activity in myeloid cells. Mol. Cell. Endocrinol. 2015, 415, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Alimirah, F.; Chen, J.; Xin, H.; Choubey, D. Androgen receptor auto-regulates its expression by a negative feedback loop through upregulation of IFI16 protein. FEBS Lett. 2006, 580, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Congy-Jolivet, N.; Cenac, C.; Dellacasagrande, J.; Puissant-Lubrano, B.; Apoil, P.A.; Guedj, K.; Abbas, F.; Laffont, S.; Sourdet, S.; Guyonnet, S.; et al. Monocytes are the main source of STING-mediated IFN-α production. eBioMedicine 2022, 80, 104047. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Lennard Richard, M.; Brandon, D.; Jones Buie, J.N.; Oates, J.C.; Gilkeson, G.S.; Zhang, X.K. A critical role of the transcription factor fli-1 in murine lupus development by regulation of interleukin-6 expression. Arthritis Rheumatol. 2014, 66, 3436–3444. [Google Scholar] [CrossRef] [PubMed]
- Faco, M.M.; Leone, C.; Campos, L.M.; Febrônio, M.V.; Marques, H.H.; Silva, C.A. Risk factors associated with the death of patients hospitalized for juvenile systemic lupus erythematosus. Braz. J. Med. Biol Res. 2007, 40, 993–1002. [Google Scholar] [CrossRef]
- Barber, C.; Gold, W.L.; Fortin, P.R. Infections in the lupus patient: Perspectives on prevention. Curr. Opin. Rheumatol. 2011, 23, 358–365. [Google Scholar] [CrossRef]
- Caza, T.; Oaks, Z.; Perl, A. Interplay of infections, autoimmunity, and immunosuppression in systemic lupus erythematosus. Int. Rev. Immunol. 2014, 33, 330–363. [Google Scholar] [CrossRef]
- Liu, Z.; Davidson, A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat. Med. 2012, 18, 871–882. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Bernal, C.B.; Zamora, L.D.; Navarra, S.V. Biologic therapies in systemic lupus erythematosus. Int. J. Rheum. Dis. 2015, 18, 146–153. [Google Scholar] [CrossRef]
- Treatment of lupus nephritis with abatacept: The Abatacept and Cyclophosphamide Combination Efficacy and Safety Study. Arthritis Rheumatol. 2014, 66, 3096–3104. [CrossRef] [PubMed]
- Furie, R.; Nicholls, K.; Cheng, T.T.; Houssiau, F.; Burgos-Vargas, R.; Chen, S.L.; Hillson, J.L.; Meadows-Shropshire, S.; Kinaszczuk, M.; Merrill, J.T. Efficacy and safety of abatacept in lupus nephritis: A twelve-month, randomized, double-blind study. Arthritis Rheumatol. Arthritis Rheumatol. 2014, 66, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Cash, H.; Relle, M.; Menke, J.; Brochhausen, C.; Jones, S.A.; Topley, N.; Galle, P.R.; Schwarting, A. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: The IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J. Rheumatol. 2010, 37, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, Y.; Chen, M. Viral strategies for triggering and manipulating mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B., Jr.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- Davidson, S.; Maini, M.K.; Wack, A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J Interferon Cytokine Res. J. Interferon Cytokine Res. 2015, 35, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal. Transduct Target. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Jarai, B.M.; Stillman, Z.; Bomb, K.; Kloxin, A.M.; Fromen, C.A. Biomaterials-Based Opportunities to Engineer the Pulmonary Host Immune Response in COVID-19. ACS Biomater. Sci. Eng. 2021, 7, 1742–1764. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, D.J.; Moore, E.E.; Johnson, J.L.; Burch, J.M.; Cothren, C.C.; Sauaia, A. A 12-year prospective study of postinjury multiple organ failure: Has anything changed? Arch. Surg 2005, 140, 432–438, discussion 438–440. [Google Scholar] [CrossRef]
- Brealey, D.; Singer, M. Multi-organ dysfunction in the critically ill: Effects on different organs. J. R. Coll. Physicians Lond. 2000, 34, 428–431. [Google Scholar]
- Thijs, A.; Thijs, L.G. Pathogenesis of renal failure in sepsis. Kidney Int. Suppl. 1998, 66, S34–S37. [Google Scholar]
- Nuytinck, H.K.; Offermans, X.J.; Kubat, K.; Goris, J.A. Whole-body inflammation in trauma patients. An autopsy study. Arch. Surg. 1988, 123, 1519–1524. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Kopf, M. Balancing protective immunity and immunopathology. Curr. Opin. Immunol. 2002, 14, 413–419. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Williams, D.J.; Hall, M.; Auger, K.A.; Tieder, J.S.; Jerardi, K.E.; Queen, M.A.; Statile, A.M.; Myers, A.L.; Shah, S.S. Association of White Blood Cell Count and C-Reactive Protein with Outcomes in Children Hospitalized for Community-Acquired Pneumonia. Pediatr. Infect. Dis. J. 2015, 34, 792–793. [Google Scholar] [CrossRef]
- Phin, N.; Parry-Ford, F.; Harrison, T.; Stagg, H.R.; Zhang, N.; Kumar, K.; Lortholary, O.; Zumla, A.; Abubakar, I. Epidemiology and clinical management of Legionnaires’ disease. Lancet Infect. Dis. 2014, 14, 1011–1021. [Google Scholar] [CrossRef]
- Mohamed, E.R.; Aly, S.A.; Halby, H.M.; Ahmed, S.H.; Zakaria, A.M.; El-Asheer, O.M. Epidemiological typing of multidrug-resistant Klebsiella pneumoniae, which causes paediatric ventilator-associated pneumonia in Egypt. J. Med. Microbiol. 2017, 66, 628. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef]
- Kausar, S.; Yang, L.; Abbas, M.N.; Hu, X.; Zhao, Y.; Zhu, Y.; Cui, H. Mitochondrial DNA: A Key Regulator of Anti-Microbial Innate Immunity. Genes 2020, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Brissac, T.; Shenoy, A.T.; Patterson, L.A.; Orihuela, C.J. Cell Invasion and Pyruvate Oxidase-Derived H2O2 Are Critical for Streptococcus pneumoniae-Mediated Cardiomyocyte Killing. Infect. Immun. 2018, 86, e00569-17. [Google Scholar] [CrossRef] [PubMed]
- Koppe, U.; Högner, K.; Doehn, J.M.; Müller, H.C.; Witzenrath, M.; Gutbier, B.; Bauer, S.; Pribyl, T.; Hammerschmidt, S.; Lohmeyer, J.; et al. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J. Immunol. 2012, 188, 811–817. [Google Scholar] [CrossRef]
- Parker, D.; Martin, F.J.; Soong, G.; Harfenist, B.S.; Aguilar, J.L.; Ratner, A.J.; Fitzgerald, K.A.; Schindler, C.; Prince, A. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2011, 2, e00016-11. [Google Scholar] [CrossRef]
- Catteau, A.; Roué, G.; Yuste, V.J.; Susin, S.A.; Desprès, P. Expression of dengue ApoptoM sequence results in disruption of mitochondrial potential and caspase activation. Biochimie 2003, 85, 789–793. [Google Scholar] [CrossRef]
- El-Bacha, T.; Midlej, V.; Pereira da Silva, A.P.; Silva da Costa, L.; Benchimol, M.; Galina, A.; Da Poian, A.T. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim. Biophys. Acta 2007, 1772, 1158–1166. [Google Scholar] [CrossRef]
- Yan, J.; Liu, W.; Feng, F.; Chen, L. VDAC oligomer pores: A mechanism in disease triggered by mtDNA release. Cell Biol. Int. 2020, 44, 2178–2181. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs Innate Immun.ity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef]
- Gou, H.; Zhao, M.; Xu, H.; Yuan, J.; He, W.; Zhu, M.; Ding, H.; Yi, L.; Chen, J. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget 2017, 8, 39382–39400. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Benmerzoug, S.; Rose, S.; Bounab, B.; Gosset, D.; Duneau, L.; Chenuet, P.; Mollet, L.; Le Bert, M.; Lambers, C.; Geleff, S.; et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun. 2018, 9, 5226. [Google Scholar] [CrossRef]
- Chau, D.H.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Xie, Y.; Yu, Y.; Liu, G.; Yu, Y.; Yang, X.; Zou, Y.; Ge, J.; Chen, R. The frequency of invariant natural killer T cells correlates with the severity of myocarditis. Viral Immunol. 2014, 27, 88–95. [Google Scholar] [CrossRef]
- Long, Q.; Liao, Y.H.; Xie, Y.; Liang, W.; Cheng, X.; Yuan, J.; Yu, M. Coxsackievirus B3 Directly Induced Th17 Cell Differentiation by Inhibiting Nup98 Expression in Patients with Acute Viral Myocarditis. Front. Cell Infect. Microbiol. 2016, 6, 171. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, T.H.; Park, H.E.; Lee, E.G.; Jung, N.C.; Song, J.Y.; Seo, H.G.; Seung, K.B.; Chang, K.; Lim, D.S. Myosin-primed tolerogenic dendritic cells ameliorate experimental autoimmune myocarditis. Cardiovasc. Res. 2014, 101, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Morosky, S.; Bomberger, J.; Stolz, D.B.; Jackson, W.T.; Coyne, C.B. BPIFB3 regulates autophagy and coxsackievirus B replication through a noncanonical pathway independent of the core initiation machinery. mBio 2014, 5, e02147. [Google Scholar] [CrossRef] [PubMed]
- Darveaux JI, L.R. Infection-related asthma. J. Allergy Clin. Immunol. Pr. 2014, 2, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.F.; Kraft, M. Chronic Infection and Severe Asthma. Immunol. Allergy Clin. N. Am. 2016, 36, 483–502. [Google Scholar] [CrossRef] [PubMed]
- Bergauer, A.; Sopel, N.; Kroß, B.; Vuorinen, T.; Xepapadaki, P.; Weiss, S.T.; Blau, A.; Sharma, H.; Kraus, C.; Springel, R.; et al. IFN-α/IFN-λ responses to respiratory viruses in paediatric asthma. Eur. Respir. J. 2017, 49, 1602489. [Google Scholar] [CrossRef]
- Miller, E.K.; Hernandez, J.Z.; Wimmenauer, V.; Shepherd, B.E.; Hijano, D.; Libster, R.; Serra, M.E.; Bhat, N.; Batalle, J.P.; Mohamed, Y.; et al. A mechanistic role for type III IFN-λ1 in asthma exacerbations mediated by human rhinoviruses. Am. J. Respir. Crit. Care Med. 2012, 185, 508–516. [Google Scholar] [CrossRef]
- Schwantes, E.A.; Manthei, D.M.; Denlinger, L.C.; Evans, M.D.; Gern, J.E.; Jarjour, N.N.; Mathur, S.K. Interferon gene expression in sputum cells correlates with the Asthma Index Score during virus-induced exacerbations. Clin. Exp. Allergy 2014, 44, 813–821. [Google Scholar] [CrossRef]
- Koh, Y.Y.; Park, Y.; Lee, H.J.; Kim, C.K. Levels of interleukin-2, interferon-gamma, and interleukin-4 in bronchoalveolar lavage fluid from patients with Mycoplasma pneumonia: Implication of tendency toward increased immunoglobulin E production. Pediatrics 2001, 107, E39. [Google Scholar] [CrossRef]
- Watanabe, H.; Uruma, T.; Nakamura, H.; Aoshiba, K. The role of Mycoplasma pneumoniae infection in the initial onset and exacerbations of asthma. Allergy Asthma Proc. 2014, 35, 204–210. [Google Scholar] [CrossRef]
- Lai, J.F.; Zindl, C.L.; Duffy, L.B.; Atkinson, T.P.; Jung, Y.W.; van Rooijen, N.; Waites, K.B.; Krause, D.C.; Chaplin, D.D. Critical role of macrophages and their activation via MyD88-NFκB signaling in lung Innate Immun.ity to Mycoplasma pneumoniae. PLoS ONE 2010, 5, e14417. [Google Scholar] [CrossRef]
- Shimizu, T.; Kimura, Y.; Kida, Y.; Kuwano, K.; Tachibana, M.; Hashino, M.; Watarai, M. Cytadherence of Mycoplasma pneumoniae induces inflammatory responses through autophagy and toll-like receptor 4. Infect. Immun. 2014, 82, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Schattenberg, J.M. Non-alcoholic steatohepatitis: Pathogenesis and novel therapeutic approaches. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 68–76. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Zhang, A. Involvement of the STING signaling in COVID-19. Front. Immunol. 2022, 13, 1006395. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Shen, S.; Hu, Y.; Huang, D.; Zheng, W.; Lou, M.; Shi, Y.; Wang, M.; Chen, S.; et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered Innate Immune responses by SARS-CoV-2 proteins. Signal. Transduct. Target. 2021, 6, 123. [Google Scholar] [CrossRef]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- David, C.; Frémond, M.-L. Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI). Cells 2022, 11, 318. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Zhang, X. STING-associated vasculopathy with onset in infancy: A familial case series report and literature review. Ann. Transl. Med. 2021, 9, 176. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef]
- Lin, B.; Torreggiani, S.; Kahle, D.; Rumsey, D.G.; Wright, B.L.; Montes-Cano, M.A.; Silveira, L.F.; Alehashemi, S.; Mitchell, J.; Aue, A.G.; et al. Case Report: Novel SAVI-Causing Variants in STING1 Expand the Clinical Disease Spectrum and Suggest a Refined Model of STING Activation. Front. Immunol. 2021, 12, 636225. [Google Scholar] [CrossRef]
- Frémond, M.L.; Crow, Y.J. STING-Mediated Lung Inflammation and Beyond. J. Clin. Immunol. 2021, 41, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Volpi, S.; Insalaco, A.; Caorsi, R.; Santori, E.; Messia, V.; Sacco, O.; Terheggen-Lagro, S.; Cardinale, F.; Scarselli, A.; Pastorino, C.; et al. Efficacy and Adverse Events During Janus Kinase Inhibitor Treatment of SAVI Syndrome. J. Clin. Immunol. 2019, 39, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.D.; Irizarry-Caro, R.A.; Bennion, B.G.; Ai, T.L.; Smith, A.M.; Miner, C.A.; Sakai, T.; Gonugunta, V.K.; Wu, J.; Platt, D.J.; et al. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 2017, 214, 3279–3292. [Google Scholar] [CrossRef]
- Siedel, H.; Roers, A.; Rösen-Wolff, A.; Luksch, H. Type I interferon-independent T cell impairment in a Tmem173 N153S/WT mouse model of STING associated vasculopathy with onset in infancy (SAVI). Clin. Immunol. 2020, 216, 108466. [Google Scholar] [CrossRef] [PubMed]
- Nazmi, A.; Mukhopadhyay, R.; Dutta, K.; Basu, A. STING mediates neuronal Innate Immune response following Japanese encephalitis virus infection. Sci. Rep. 2012, 2, 347. [Google Scholar] [CrossRef]
- Walko, T.D., 3rd; Bola, R.A.; Hong, J.D.; Au, A.K.; Bell, M.J.; Kochanek, P.M.; Clark, R.S.; Aneja, R.K. Cerebrospinal fluid mitochondrial DNA: A novel DAMP in pediatric traumatic brain injury. Shock 2014, 41, 499–503. [Google Scholar] [CrossRef]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef]
- Abdullah, A.; Zhang, M.; Frugier, T.; Bedoui, S.; Taylor, J.M.; Crack, P.J. STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J. Neuroinflamm. 2018, 15, 323. [Google Scholar] [CrossRef]
- Barrett, J.P.; Henry, R.J.; Shirey, K.A.; Doran, S.J.; Makarevich, O.D.; Ritzel, R.M.; Meadows, V.A.; Vogel, S.N.; Faden, A.I.; Stoica, B.A.; et al. Interferon-β Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration. J. Neurosci. 2020, 40, 2357–2370. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Julius, D.; Basbaum, A.I. Molecular mechanisms of nociception. Nature 2001, 413, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.R.; Chen, O.; Ji, R.R. How Do Sensory Neurons Sense Danger Signals. Trends Neurosci. 2020, 43, 822–838. [Google Scholar] [CrossRef]
- Donnelly, C.R.; Jiang, C.; Andriessen, A.S.; Wang, K.; Wang, Z.; Ding, H.; Zhao, J.; Luo, X.; Lee, M.S.; Lei, Y.L.; et al. STING controls nociception via type I interferon signalling in sensory neurons. Nature 2021, 591, 275–280. [Google Scholar] [CrossRef]
- Wottawa, F.; Bordoni, D.; Baran, N.; Rosenstiel, P.; Aden, K. The role of cGAS/STING in intestinal immunity. Eur. J. Immunol. 2021, 51, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zheng, T.; Gong, W.; Wu, J.; Xie, H.; Li, W.; Zhang, R.; Liu, P.; Liu, J.; Wu, X.; et al. Extracellular vesicles package dsDNA to aggravate Crohn’s disease by activating the STING pathway. Cell Death Dis. 2021, 12, 815. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R.; Blomquist, C.M.; Henare, K.L.; Jirik, F.R. Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci. Rep. 2019, 9, 14281. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Y.; Tao, M.; Zhao, X.; Feng, Q.; Fei, X.; Fu, Y. Atrial Natriuretic Peptide Attenuates Colitis via Inhibition of the cGAS-STING Pathway in Colonic Epithelial Cells. Int. J. Biol. Sci. 2022, 18, 1737–1754. [Google Scholar] [CrossRef]
- Ke, X.; Hu, T.; Jiang, M. cGAS-STING signaling pathway in gastrointestinal inflammatory disease and cancers. FASEB J. 2022, 36, e22029. [Google Scholar] [CrossRef]
- Canesso, M.C.C.; Lemos, L.; Neves, T.C.; Marim, F.M.; Castro, T.B.R.; Veloso, É.S.; Queiroz, C.P.; Ahn, J.; Santiago, H.C.; Martins, F.S.; et al. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol. 2018, 11, 820–834. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, W.; Bilotta, A.J.; Yu, Y.; Zhao, X.; Zhou, Z.; Yao, S.; Xu, J.; Zhou, J.; Dann, S.M.; et al. STING controls intestinal homeostasis through promoting antimicrobial peptide expression in epithelial cells. FASEB J. 2020, 34, 15417–15430. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef]
- Qiao, J.T.; Cui, C.; Qing, L.; Wang, L.S.; He, T.Y.; Yan, F.; Liu, F.Q.; Shen, Y.H.; Hou, X.G.; Chen, L. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 2018, 81, 13–24. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 2018, 155, 1971–1984.e4. [Google Scholar] [CrossRef]
- Grimbacher, B.; Warnatz, K.; Yong, P.F.K.; Korganow, A.S.; Peter, H.H. The crossroads of autoimmunity and immunodeficiency: Lessons from polygenic traits and monogenic defects. J. Allergy Clin. Immunol. 2016, 137, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Mengstie, M.A.; Abebe, E.C.; Teklemariam, A.B.; Mulu, A.T.; Teshome, A.A.; Zewde, E.A.; Muche, Z.T.; Azezew, M.T. Molecular and cellular mechanisms in diabetic heart failure: Potential therapeutic targets. Front. Endocrinol. 2022, 13, 947294. [Google Scholar] [CrossRef]
- Yan, M.; Li, Y.; Luo, Q.; Zeng, W.; Shao, X.; Li, L.; Wang, Q.; Wang, D.; Zhang, Y.; Diao, H.; et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022, 8, 258. [Google Scholar] [CrossRef]
- Lu, Q.B.; Ding, Y.; Liu, Y.; Wang, Z.C.; Wu, Y.J.; Niu, K.M.; Li, K.X.; Zhang, J.R.; Sun, H.J. Metrnl ameliorates diabetic cardiomyopathy via inactivation of cGAS/STING signaling dependent on LKB1/AMPK/ULK1-mediated autophagy. J. Adv. Res. 2022. [Google Scholar] [CrossRef]
- Bai, J.; Liu, F. cGAS-STING signaling and function in metabolism and kidney diseases. J. Mol. Cell Biol. 2021, 13, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [PubMed]
- Zang, N.; Cui, C.; Guo, X.; Song, J.; Hu, H.; Yang, M.; Xu, M.; Wang, L.; Hou, X.; He, Q.; et al. cGAS-STING activation contributes to podocyte injury in diabetic kidney disease. iScience 2022, 25, 105145. [Google Scholar] [CrossRef]
- Feng, Z.; Zang, C.; Zhang, L.; Yin, S.; Zhuang, Q.; Wang, X. STING activation promotes inflammatory response and delays skin wound healing in diabetic mice. Biochem. Biophys. Res. Commun. 2022, 611, 126–131. [Google Scholar] [CrossRef]
- Thomas, C.A.; Tejwani, L.; Trujillo, C.A.; Negraes, P.D.; Herai, R.H.; Mesci, P.; Macia, A.; Crow, Y.J.; Muotri, A.R. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017, 21, 319–331.e8. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskaitė, Ž.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B.; et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent Innate Immune response. EMBO J. 2016, 35, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Costa-Reis, P.; Sullivan, K.E. Monogenic lupus: It’s all new. Curr. Opin. Immunol. 2017, 49, 87–95. [Google Scholar] [CrossRef]
- You, S.; Koh, J.H.; Leng, L.; Kim, W.U.; Bucala, R. The Tumor-Like Phenotype of Rheumatoid Synovium: Molecular Profiling and Prospects for Precision Medicine. Arthritis Rheumatol. 2018, 70, 637–652. [Google Scholar] [CrossRef]
- Wang, Y.; Su, G.-H.; Zhang, F.; Chu, J.-X.; Wang, Y.-S. Cyclic GMP-AMP Synthase Is Required for Cell Proliferation and Inflammatory Responses in Rheumatoid Arthritis Synoviocytes. Mediat. Inflamm. 2015, 2015, 192329. [Google Scholar] [CrossRef]
- Wang, J.; Li, R.; Lin, H.; Qiu, Q.; Lao, M.; Zeng, S.; Wang, C.; Xu, S.; Zou, Y.; Shi, M.; et al. Accumulation of cytosolic dsDNA contributes to fibroblast-like synoviocytes-mediated rheumatoid arthritis synovial inflammation. Int. Immunopharmacol. 2019, 76, 105791. [Google Scholar] [CrossRef]
- Li, R.; Lin, W.; Kuang, Y.; Wang, J.; Xu, S.; Shen, C.; Qiu, Q.; Shi, M.; Xiao, Y.; Liang, L.; et al. cGAS/STING signaling in the regulation of rheumatoid synovial aggression. Ann. Transl. Med. 2022, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.; Neuhoff, M.T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.X.; Sun, Y.; Roh, J.D.; Ng, R.P., Jr.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Choubey, D.; Panchanathan, R. IFI16, an amplifier of DNA-damage response: Role in cellular senescence and aging-associated inflammatory diseases. Ageing Res. Rev. 2016, 28, 27–36. [Google Scholar] [CrossRef]
- Sladitschek-Martens, H.L.; Guarnieri, A.; Brumana, G.; Zanconato, F.; Battilana, G.; Xiccato, R.L.; Panciera, T.; Forcato, M.; Bicciato, S.; Guzzardo, V.; et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature 2022, 607, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Ito, J.; Matsui, N.; Uechi, T.; Onodera, O.; Kakita, A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat. Commun. 2021, 12, 3101. [Google Scholar] [CrossRef]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.H.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD(+) supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 2021, 20, e13329. [Google Scholar] [CrossRef]
- Duan, X.; Ponomareva, L.; Veeranki, S.; Panchanathan, R.; Dickerson, E.; Choubey, D. Differential roles for the interferon-inducible IFI16 and AIM2 Innate Immune sensors for cytosolic DNA in cellular senescence of human fibroblasts. Mol. Cancer Res. 2011, 9, 589–602. [Google Scholar] [CrossRef]
- Ren, C.; Jin, J.; Li, C.; Xiang, J.; Wu, Y.; Zhou, Y.; Sun, L.; Zhang, X.; Tian, N. Metformin inactivates the cGAS-STING pathway through autophagy and suppresses senescence in nucleus pulposus cells. J. Cell Sci. 2022, 135, jcs259738. [Google Scholar] [CrossRef]
- Hamann, L.; Szwed, M.; Mossakowska, M.; Chudek, J.; Puzianowska-Kuznicka, M. First evidence for STING SNP R293Q being protective regarding obesity-associated cardiovascular disease in age-advanced subjects—A cohort study. Immun. Ageing 2020, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Hamann, L.; Ruiz-Moreno, J.S.; Szwed, M.; Mossakowska, M.; Lundvall, L.; Schumann, R.R.; Opitz, B.; Puzianowska-Kuznicka, M. STING SNP R293Q Is Associated with a Decreased Risk of Aging-Related Diseases. Gerontology 2019, 65, 145–154. [Google Scholar] [CrossRef]
- Golbari, N.M.; Porter, M.L.; Kimball, A.B. Current guidelines for psoriasis treatment: A work in progress. Cutis 2018, 101, 10–12. [Google Scholar] [PubMed]
- Masson Regnault, M.; Konstantinou, M.P.; Khemis, A.; Poulin, Y.; Bourcier, M.; Amelot, F.; Bulaï Livideanu, C.; Paul, C. Early relapse of psoriasis after stopping brodalumab: A retrospective cohort study in 77 patients. J. Eur. Acad. Derm. Venereol. 2017, 31, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor [Ref] | Binding Sites | Molecular Mechanism | Biological Activity |
|---|---|---|---|
| Nitro-fatty acid Derivatives [102] | C88, C91 at palmitoylation site and H16 in N-terminus | Covalently bind to the STING cysteines residues, block STING palmitoylation and inhibit STING activation | N.D. |
| C-176/178/170 and H-151 [103] | C91 at palmitoylation site | IC50 (H-151) = 134.4 nM (HFFs cells) | |
| BPK-21/25 [104] | C91 at palmitoylation site | ISRE-Luc activity (BPK-25) = 3.2 μM (THP1 cells) | |
| Astin C [21] | CDN binding site | Compete with cGAMP for the CDNs binding pocket and inhibit STING activation | IC50 = 3.42 ± 0.13 μM (MEFs cells) |
| Compound 18 [105] | CDN binding site | IC50 = 68 nM (STINGHAQ); IC50 = 11 μM (THP1 cells) | |
| SN-011 [106] | CDN binding site | IC50 = 502.8 nM (HFFs cells) | |
| Palbociclib [107] | CDN binding site | Interact with STING CTD and block STING dimerization | IC50 = 0.81 ± 0.93 μM (HEK293 cells) |
| Compound 30 [108] | CDN binding site | Undetermined | IC50 = 1.15 μM (RAW264.7 cells) |
| 6,5-heterocyclic derivatives | Unknown | Undetermined | IC50 ranges from 30 μM to less than 10 nM |
| SP23 [109] | Palmitoylation site | Promote the degradation of STING via ubiquitin-proteasome pathway | DC50 = 3.2 μM (THP1 cells) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Zheng, R.; Pan, Y.; Sun, H. Potential Therapeutic Value of the STING Inhibitors. Molecules 2023, 28, 3127. https://doi.org/10.3390/molecules28073127
Zhang S, Zheng R, Pan Y, Sun H. Potential Therapeutic Value of the STING Inhibitors. Molecules. 2023; 28(7):3127. https://doi.org/10.3390/molecules28073127
Chicago/Turabian StyleZhang, Shangran, Runan Zheng, Yanhong Pan, and Hongbin Sun. 2023. "Potential Therapeutic Value of the STING Inhibitors" Molecules 28, no. 7: 3127. https://doi.org/10.3390/molecules28073127
APA StyleZhang, S., Zheng, R., Pan, Y., & Sun, H. (2023). Potential Therapeutic Value of the STING Inhibitors. Molecules, 28(7), 3127. https://doi.org/10.3390/molecules28073127