


A Density Functional Study on Ethylene Trimerization and Tetramerization Using Real Sasol Cr-PNP Catalysts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
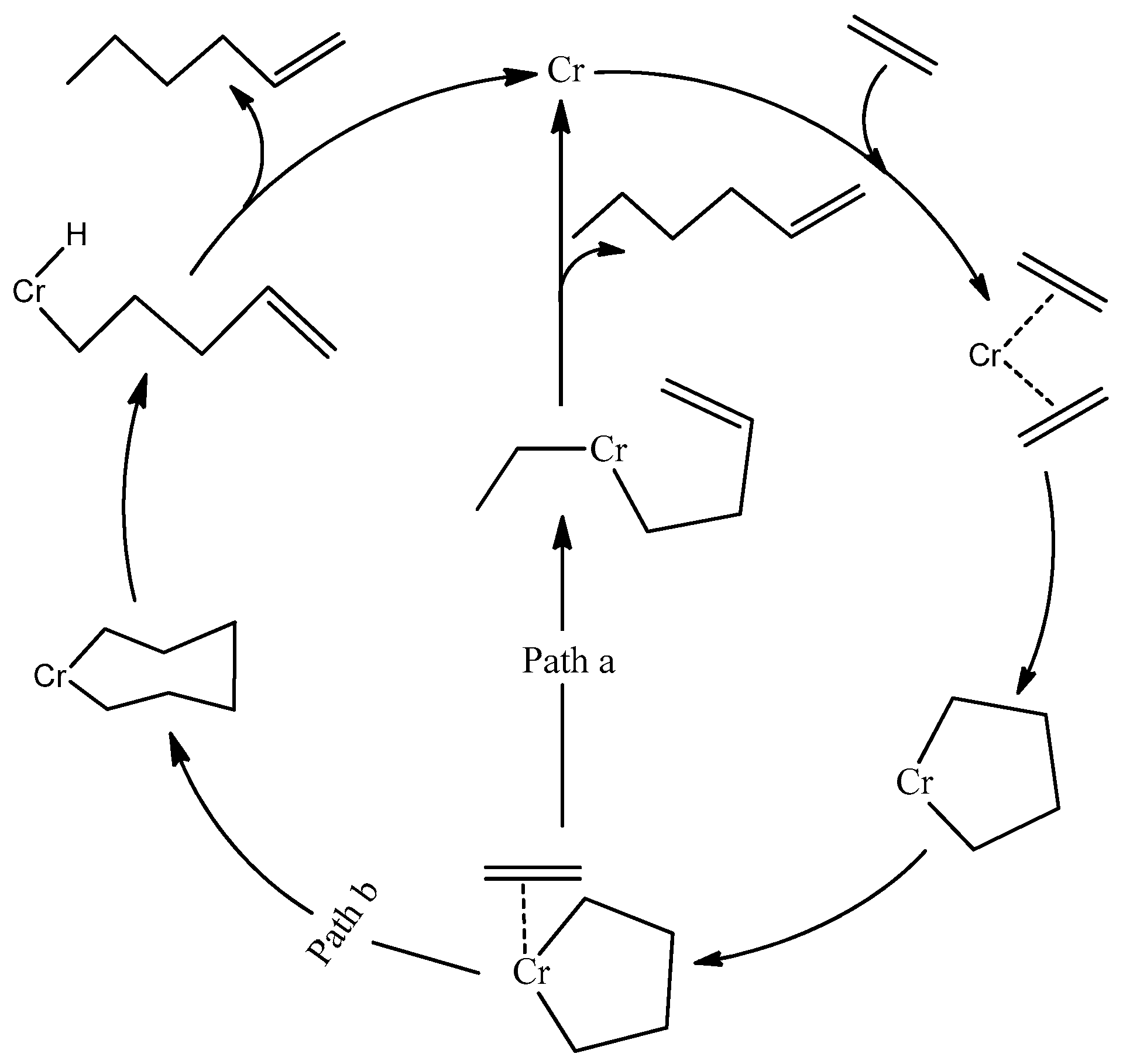
1. Introduction
2. Results and Discussion
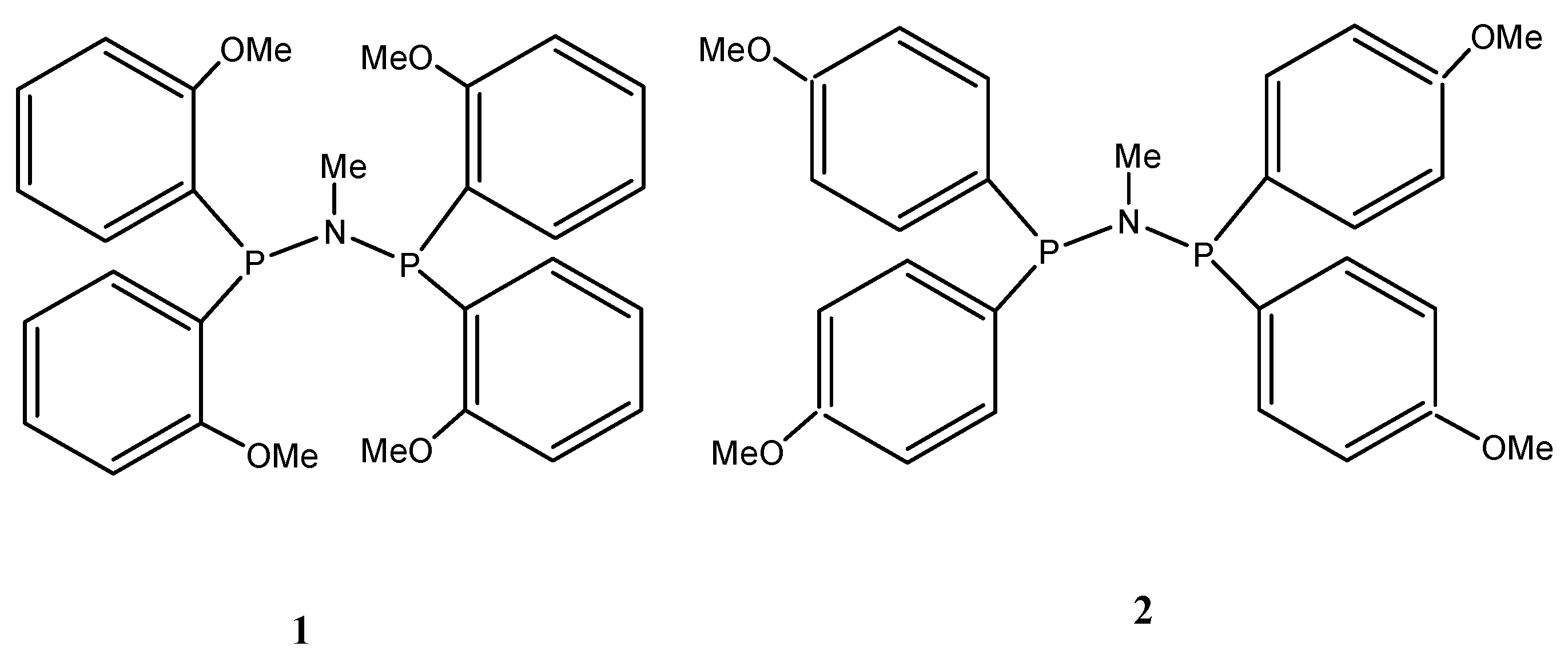
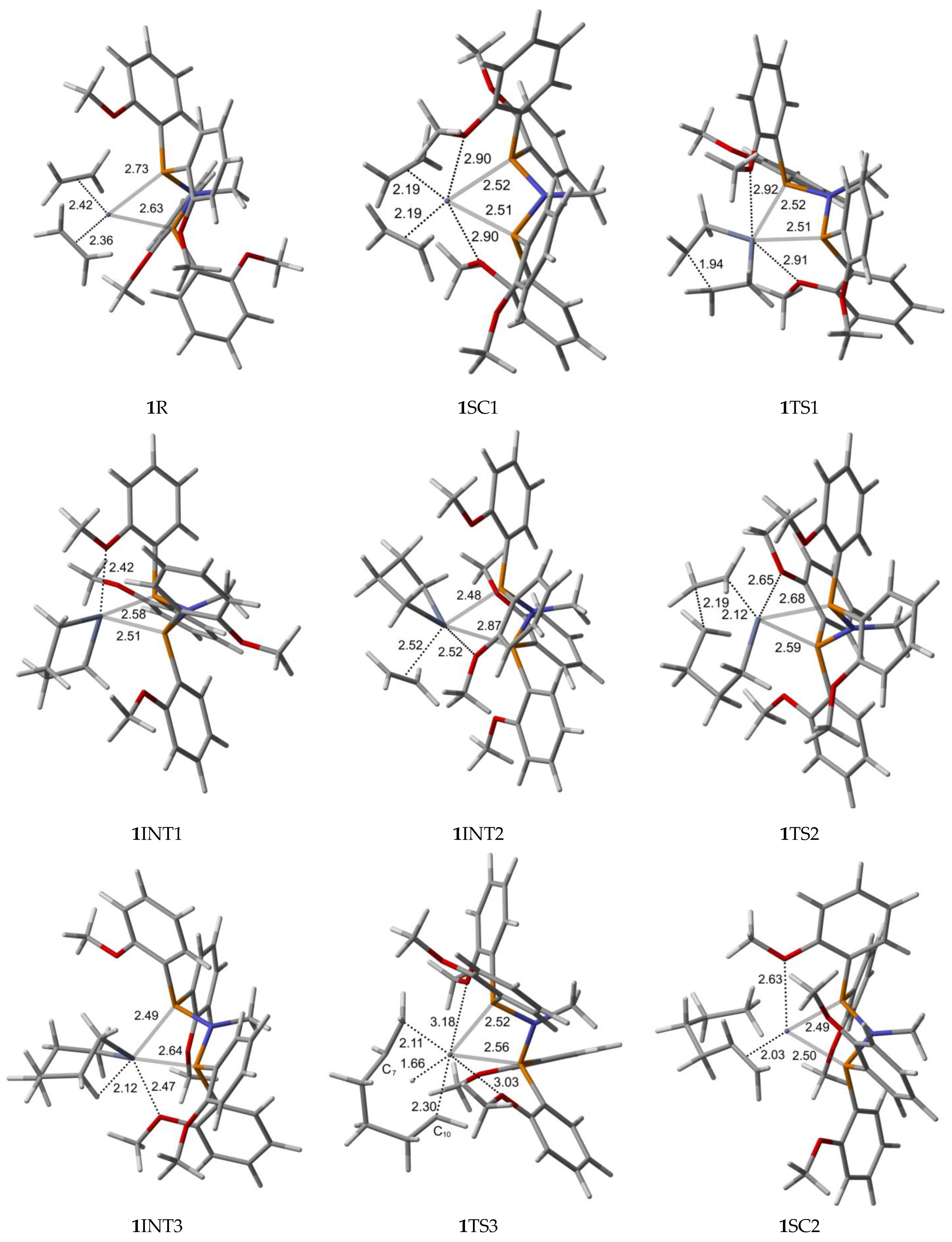
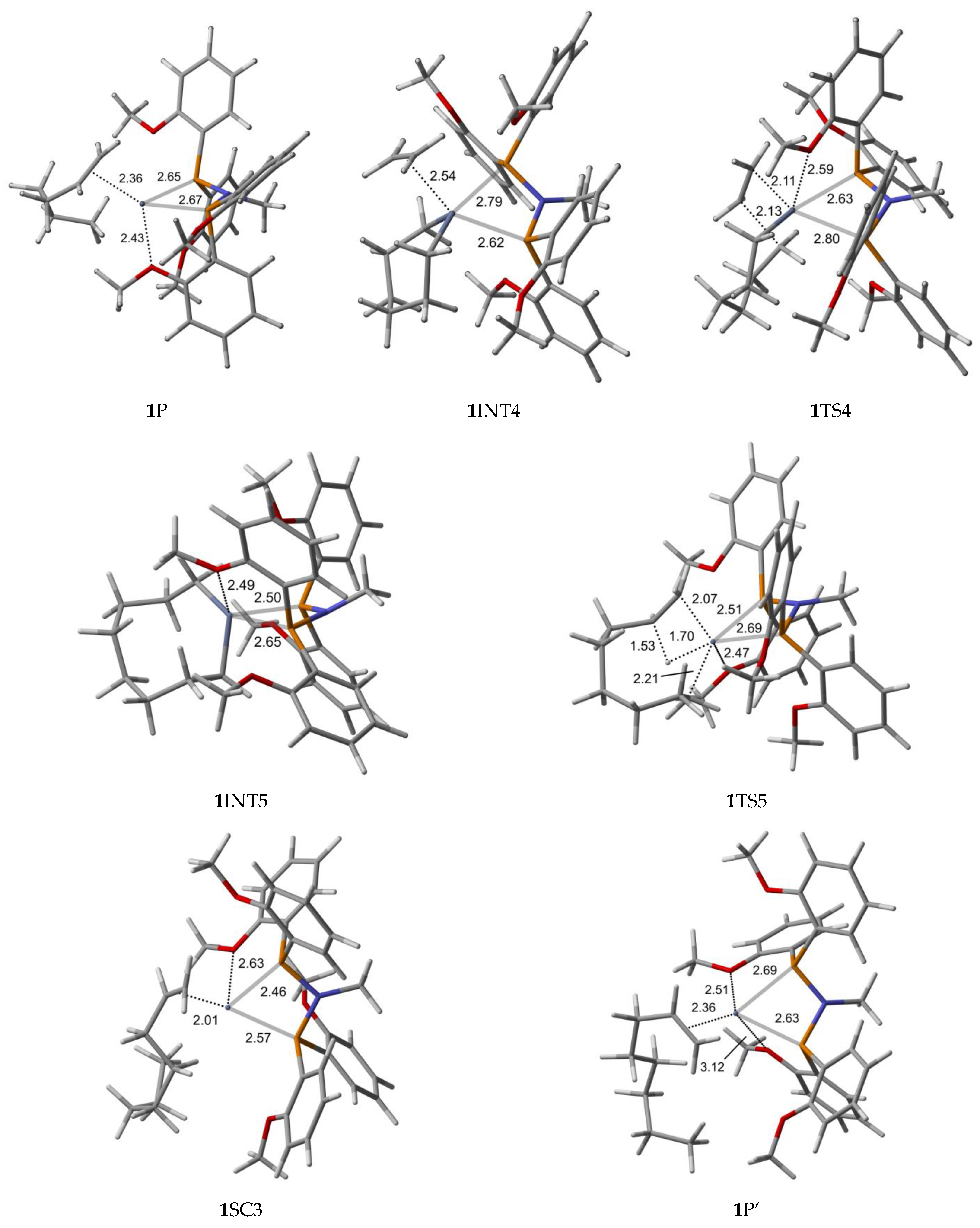
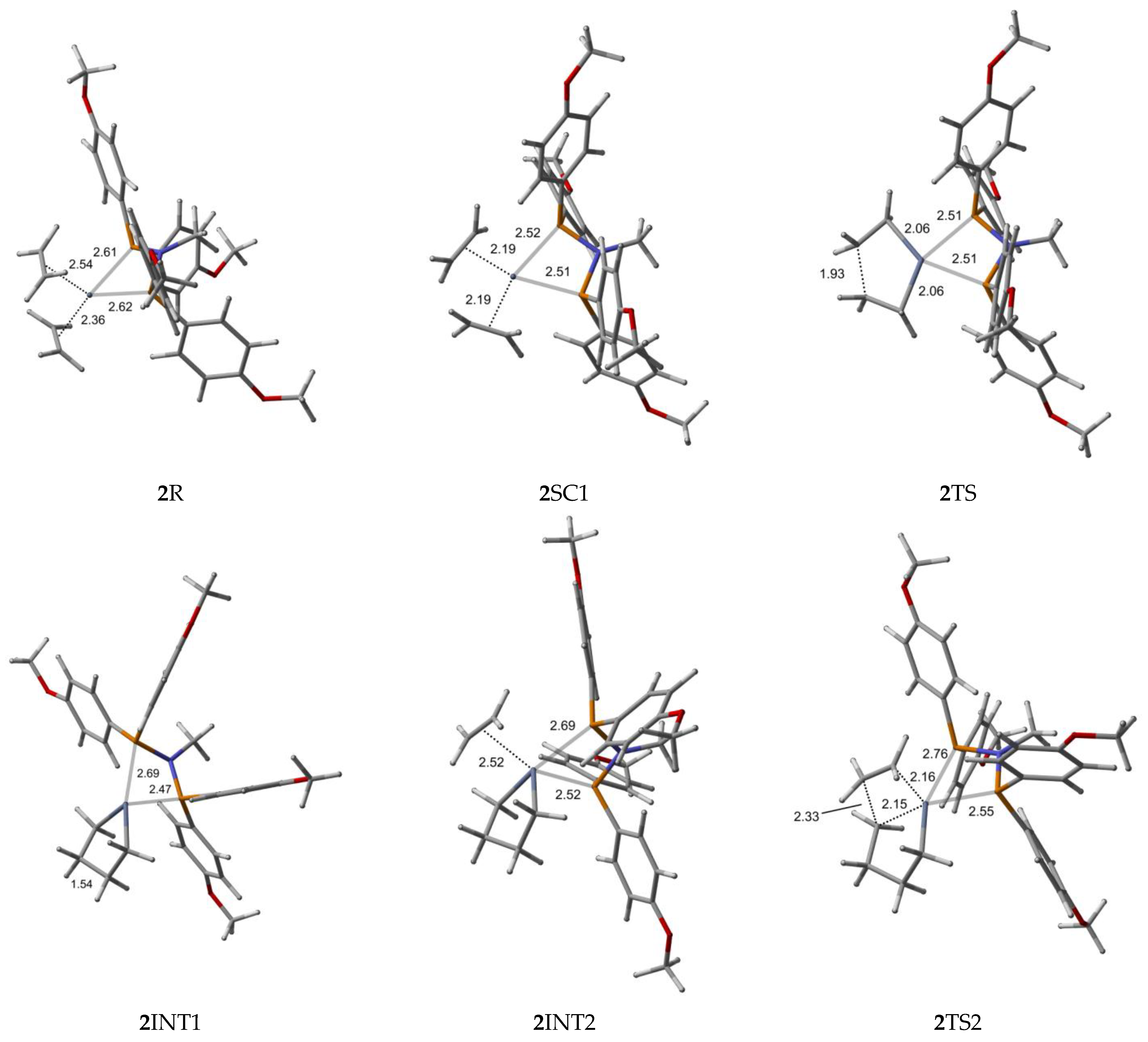
2.1. Structure and Bonding Aspects of the Ortho-Methoxyaryl Cr-PNP Catalyst
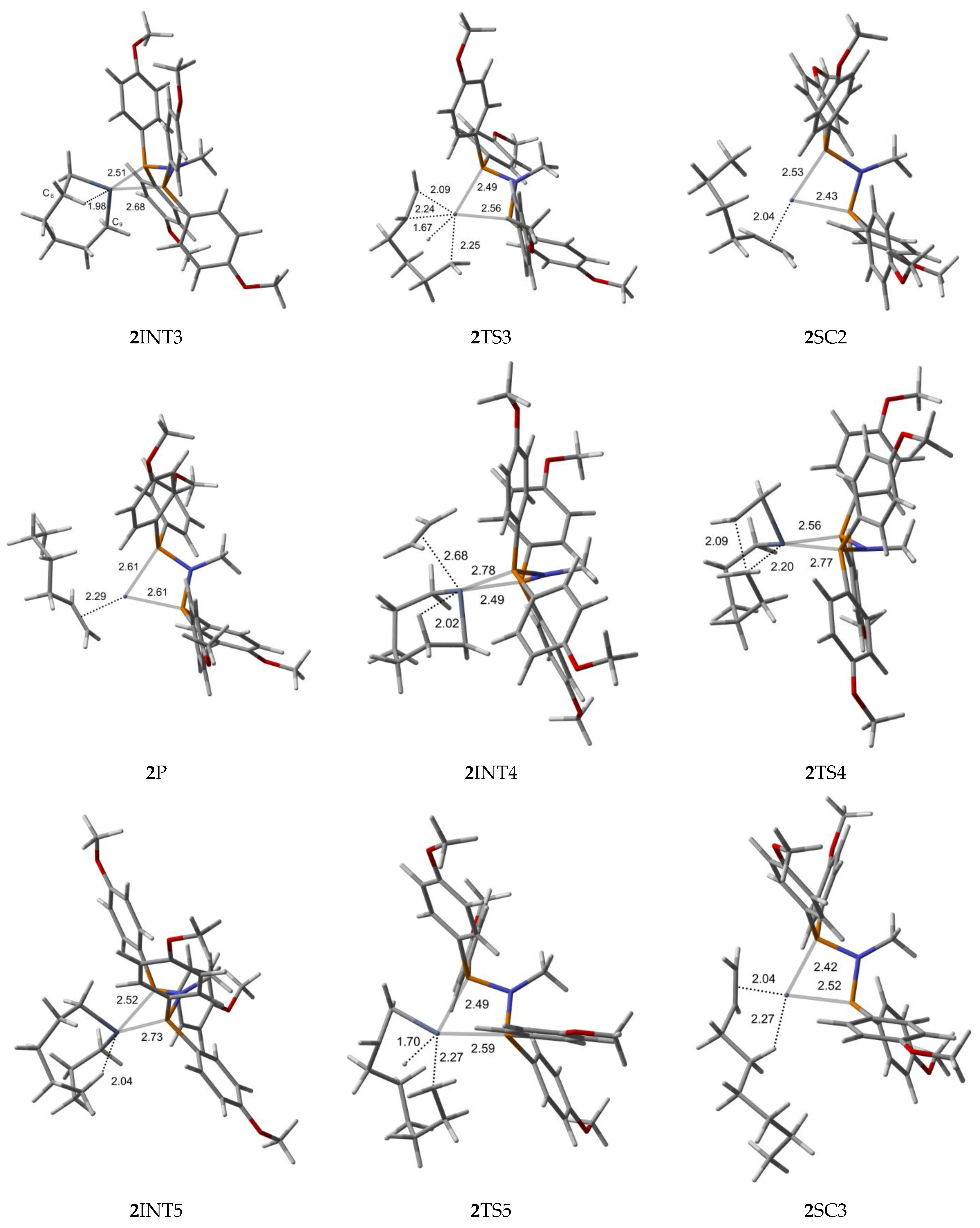
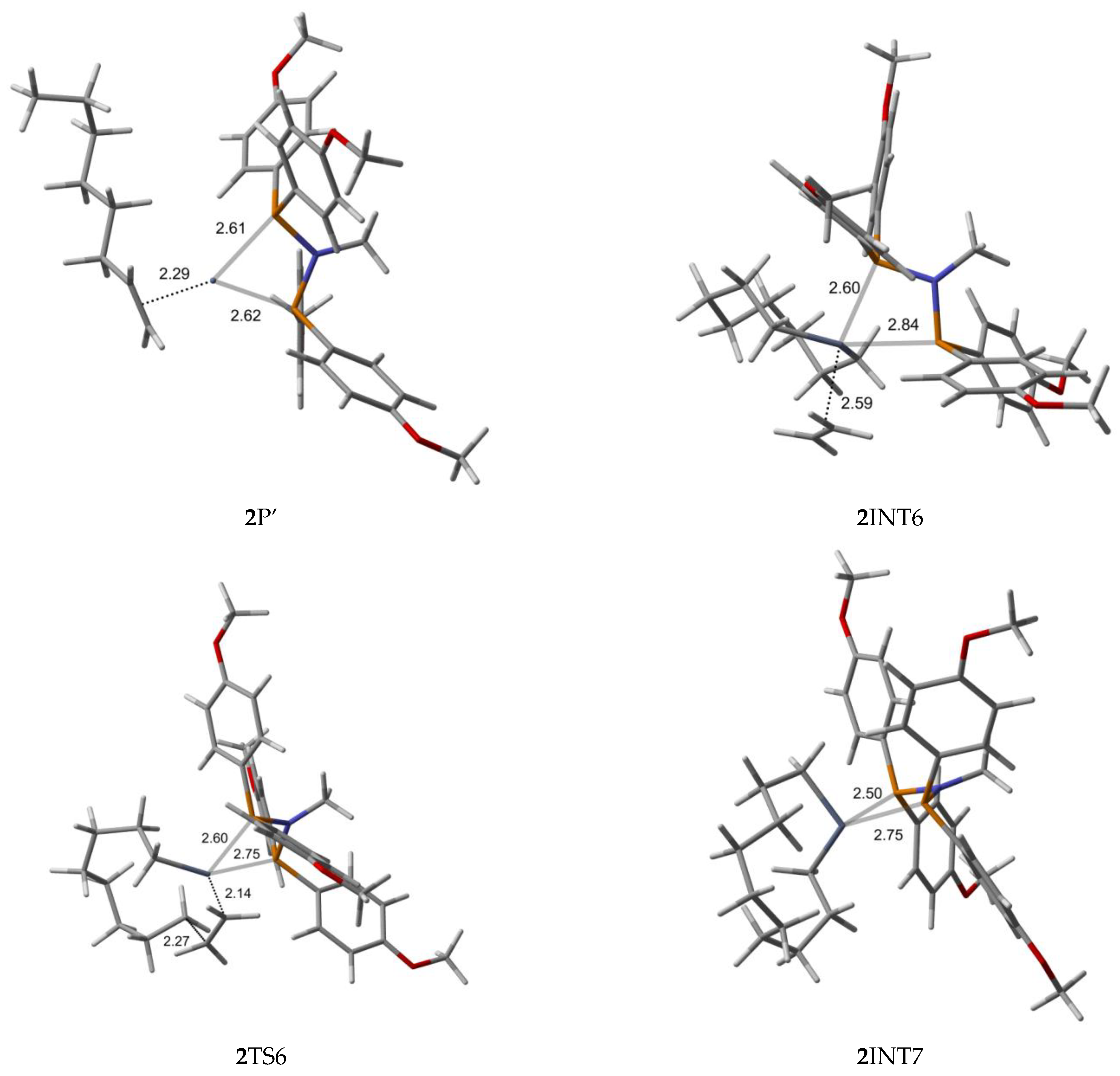
2.2. Structure and Bonding Aspects of the Para-Methoxyaryl Cr-PNP Catalyst
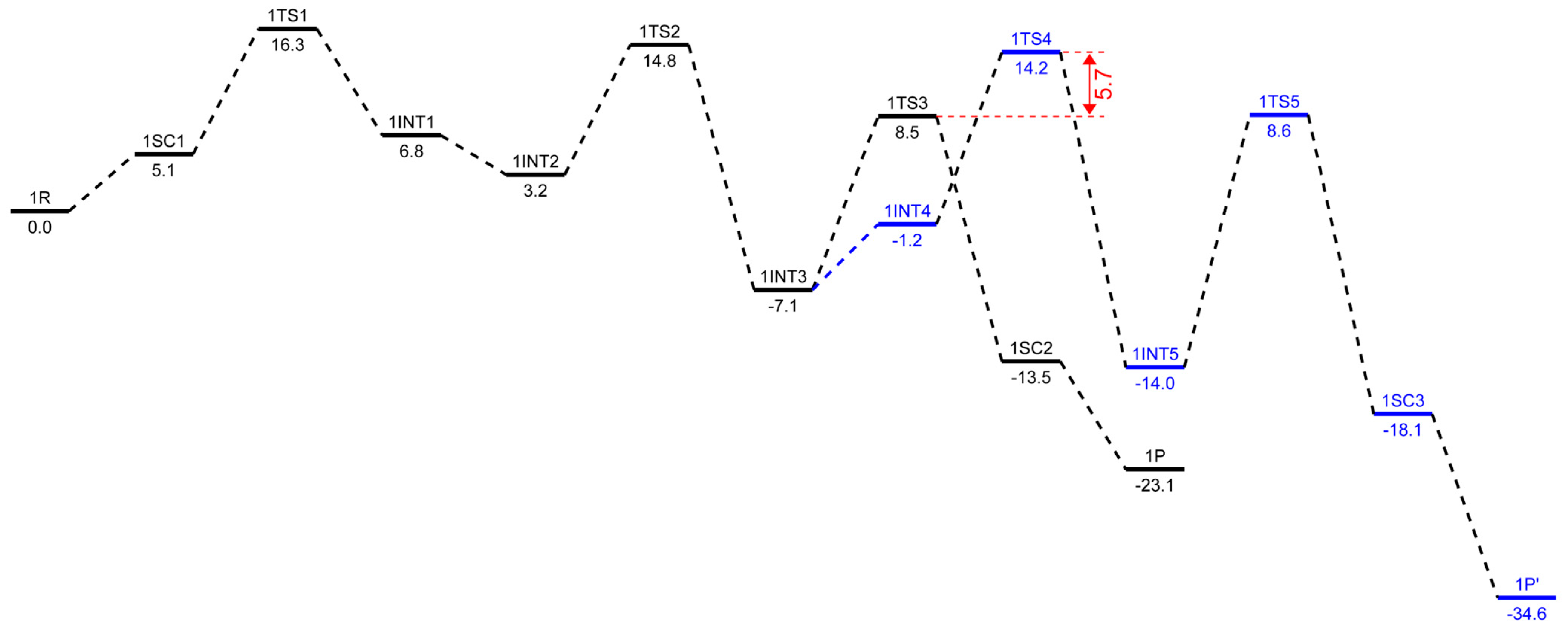
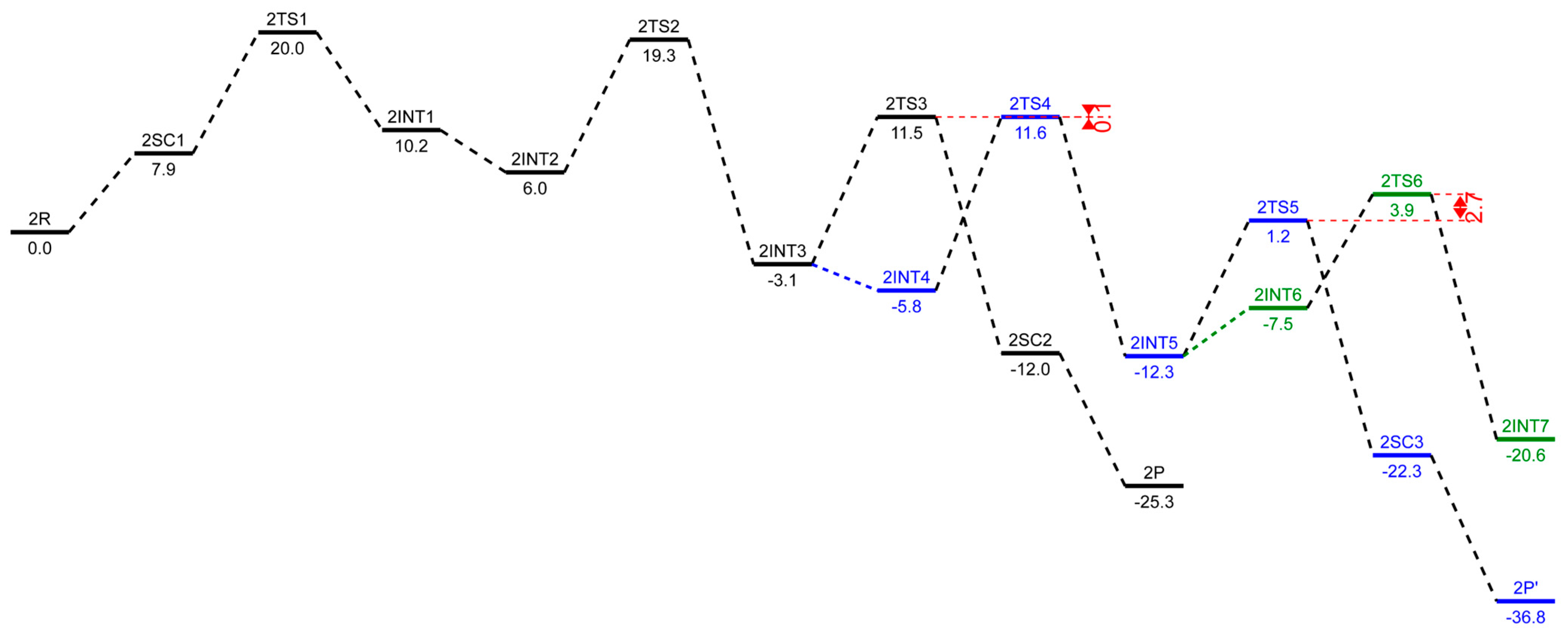
2.3. Energetic Aspects of the Cr-PNP Catalyst
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skupinska, J. Oligomerization of α-olefins to higher oligomers. Chem. Rev. 1991, 91, 613–648. [Google Scholar] [CrossRef]
- Keim, W.; Kowaldt, F.H.; Goddard, R.; Krüger, C. Novel coordination of (benzoylmethylene)triphenylphosphorane in a nickel oligomerization catalyst. Angew. Chem. Int. Ed. Engl. 1978, 17, 466–467. [Google Scholar] [CrossRef]
- Svejda, S.A.; Brookhart, M. Ethylene oligomerization and propylene dimerization using cationic (α-diimine)nickel(II) catalysts. Organometallics 1999, 18, 65–74. [Google Scholar] [CrossRef]
- Killian, C.M.; Johnson, L.K.; Brookhart, M. Preparation of linear α-olefins using cationic nickel(II) α-diimine catalysts. Organometallics 1997, 16, 2005–2007. [Google Scholar] [CrossRef]
- Mecking, S. Cationic nickel and palladium complexes with bidentate ligands for the C-C linkage of olefins. Coord. Chem. Rev. 2000, 203, 325–351. [Google Scholar] [CrossRef]
- Ruther, T.; Braussaud, N.; Cavell, K.J. Novel chromium(III) complexes containing imidazole-based chelate ligands with varying donor sets: synthesis and reactivity. Organometallics 2001, 20, 1247–1250. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Mastroianni, S.; Solan, G.A.; Baugh, S.P.D.; Redshaw, C.; Gibson, V.C.; White, A.J.P.; Williams, D.J.; Elsegood, M.R.J. Oligomerization of ethylene by bis(imino)pyridyliron and -cobalt complexes. Chem. Eur. J. 2000, 6, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Vogt, D. Oligomerization of ethylene to higher linear α-olefins. In Applied Homogeneous Catalysis with Organometallic Compounds; Cornils, B., Herrmann, W.A., Eds.; VCH: Weinheim, Germany, 2000; pp. 245–258. [Google Scholar]
- Reagan, W.K. Phillips Petroleum Company. EU Patent 0417477, 20 March 1991. [Google Scholar]
- McGuinness, D.S.; Brown, D.B.; Tooze, D.B.; Hess, F.M.; Dixon, J.T.; Slawin, A.M.Z. Ethylene trimerization with Cr-PNP and Cr-SNS complexes: Effect of ligand structure, metal oxidation state, and role of activator on catalysis. Organometallics 2006, 25, 3605–3610. [Google Scholar] [CrossRef]
- Bollmann, A.; Blann, K.; Dixon, J.T.; Hess, F.M.; Killan, E.; Maumela, H.; McGuinness, D.; Morgan, D.H.; Neveling, A.; Otto, S.; et al. Ethylene tetramerization: A new route to produce 1-octene in exceptionally high selectivities. J. Am. Chem. Soc. 2004, 126, 14712–14713. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Kim, T.-J.; Chung, J.-H.; Hahn, T.-K.; Chae, S.-S.; Lee, H.-S.; Cheong, M.; Kang, S.O. Bimetallic ethylene tetramerization catalysts derived from chiral DPPDME ligands: Syntheses, structural characterizations, and catalytic performance of [(DPPDME)CrCl3]2 (DPPDME = S,S- and R,R-chiraphos and meso-achiraphos). Organometallics 2010, 29, 5805–5811. [Google Scholar] [CrossRef]
- Son, K.; Waymouth, R.M. Selective ethylene oligomerization in the presence of ZnR2: Synthesis of terminally-functionalized ethylene oligomers. Organometallics 2010, 29, 3515–3520. [Google Scholar] [CrossRef]
- Manyik, R.M.; Walker, W.E.; Wilson, T.P. Union Carbide Corporation. U.S. Patent 3300458, 1967. [Google Scholar]
- Briggs, J.R. The selective trimerization of ethylene to hex-1-ene. Chem. Commun. 1989, 11, 674–675. [Google Scholar] [CrossRef]
- Emrich, R.; Heinemann, O.; Jolly, P.W.; Krueger, C.; Verhovnik, G.P.J. The role of metallacycles in the chromium-catalyzed trimerization of ethylene. Organometallics 1997, 16, 1511–1513. [Google Scholar] [CrossRef]
- Cossee, P. Ziegler-Natta catalysis I. Mechanism of polymerization of α-olefins with Ziegler-Natta catalysts. J. Catal. 1964, 3, 80–88. [Google Scholar] [CrossRef]
- Arlman, E.J.; Cossee, P. Ziegler-Natta catalysis III. Stereospecific polymerization of propene with the catalyst system TiCl3-AlEt3. J. Catal. 1964, 3, 99–104. [Google Scholar] [CrossRef]
- Agapie, T.; Schofer, S.J.; Labinger, J.A.; Bercaw, J.E. Mechanistic studies of the ethylene trimerization reaction with chromium-diphosphine catalysts: Experimental evidence for a mechanism involving metallacyclic intermediates. J. Am. Chem. Soc. 2004, 126, 1304–1305. [Google Scholar] [CrossRef]
- Agapie, T.; Labinger, J.A.; Bercaw, J.E. Mechanistic studies of olefin and alkyne trimerization with chromium catalysts: Deuterium labeling and studies of regiochemistry using a model chromacyclopentane complex. J. Am. Chem. Soc. 2007, 129, 14281–14295. [Google Scholar] [CrossRef]
- Arteaga-Müller, R.; Tsurugi, H.; Saito, T.; Yanagawa, M.; Oda, S.; Mashima, K. New Tantalum ligand-free catalyst system for highly selective trimerization of ethylene affording 1-hexene: New evidence of a metallacycle mechanism. J. Am. Chem. Soc. 2009, 131, 5370–5371. [Google Scholar] [CrossRef]
- Wass, D.F. Chromium-catalysed ethene trimerisation and tetramerisation—Breaking the rules in olefin oligomerisation. Dalton Trans. 2007, 2007, 816–819. [Google Scholar] [CrossRef]
- Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H. Advances in selective ethylene trimerisation—A critical overview. J. Organomet. Chem. 2004, 689, 3641–3668. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, Y.; Ke, Y.; Guo, C.; Zhu, N.; Mi, X.; Ma, Z.; Hu, Y. A new chromium-based catalyst coated with paraffin for ethylene oligomerization and the effect of chromium state on oligomerization selectivity. Appl. Catal. A 2002, 235, 33–38. [Google Scholar] [CrossRef]
- Ban, K.; Hayashi, T.; Suzuki, Y. Mitsui Chemicals Incorporated. JP Patent 11060627, 2 March 1999. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry; John Wiley and Sons, Inc.: New York, NY, USA, 1999; p. 1355. [Google Scholar]
- Poli, R. Open shell organometallics: A general analysis of their electronic structure and reactivity. J. Organomet. Chem. 2004, 689, 4291–4304. [Google Scholar] [CrossRef]
- Poli, R.; Harvey, J.N. Spin forbidden chemical reactions of transition metal compounds. New ideas and new computational challenges. Chem. Soc. Rev. 2003, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Green, J.C.; Harvey, J.N.; Poli, R. Theoretical investigation of the spin crossover transition states of the addition of methane to a series of Group 6 metallocenes using minimum energy crossing points. J. Chem. Soc. Dalton Trans. 2002, 8, 1861–1866. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc. 1998, 99, 95–99. [Google Scholar] [CrossRef]
- Hossain, M.A.; Kim, H.S.; Houk, K.N.; Cheong, M. Spin-crossover in chromium-catalyzed ethylene trimerization: Density functional theory study. Bull. Korean Chem. Soc. 2014, 35, 2835–2838. [Google Scholar] [CrossRef]
- Budzelaar, P.H.M. Ethene trimerization at CrI/CrIII—A density functional theory (DFT) study. Can. J. Chem. 2009, 87, 832–837. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; McGuinness, D.S.; Wierenga, T.S.; Young, C.T. Single- and double-coordination mechanism in ethylene tri- and tetramerization with Cr/PNP catalysts. ACS Catal. 2015, 5, 4152–4166. [Google Scholar] [CrossRef]
- Gong, M.; Liu, Z.; Li, Y.; Ma, Y.; Sun, Q.; Zhang, J.; Liu, B. Selective co-oligomerization of ethylene and 1-hexene by chromium-PNP catalysts: A DFT study. Organometallics 2016, 35, 972–981. [Google Scholar] [CrossRef]
- Köhn, R.D. Reactivity of chromium complexes under spin control. Angew. Chem. Int. Ed. 2008, 47, 245–247. [Google Scholar] [CrossRef]
- Schofer, S.J.; Day, M.W.; Henling, L.M.; Labinger, J.A.; Bercaw, J.E. Ethylene trimerization catalysts based on chromium complexes with a nitrogen-bridged diphosphine ligand having ortho-methoxyaryl or ortho-thiomethoxy substituents: Well-defined catalyst precursors and investigations of the mechanism. Organometallics 2006, 25, 2743–2749. [Google Scholar] [CrossRef]
- Manyik, R.M.; Walker, W.E.; Wilson, T.P. A soluble chromium-based catalyst for ethylene trimerization and polymerization. J. Catal. 1977, 47, 197–209. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, H.; Lee, J.; Paik, H.; Jang, H.G. Roles of chloro compound in homogeneous [Cr(2-ethylhexanoate)3/2,5-dimethylpyrrole/triethylaluminum/chloro compound] catalyst system for ethylene trimerization. Appl. Catal. A 2000, 193, 29–38. [Google Scholar] [CrossRef]
- Wasserscheid, P.; Grimm, S.; Köhn, R.D.; Haufe, M. Synthesis of synthetic lubricants by trimerization of 1-decene and 1-dodecene with homogeneous chromium catalysts. Adv. Synth. Catal. 2001, 343, 814. [Google Scholar] [CrossRef]
- Carter, A.; Cohen, S.A.; Cooley, N.A.; Murphy, A.; Scutt, J.; Wass, D.F. High activity ethylene trimerisation catalysts based on diphosphine ligands. J. Chem. Soc. Chem. Commun. 2002, 8, 858–859. [Google Scholar] [CrossRef]
- Monoi, T.; Sasaki, Y. Silica-supported Cr[N(SiMe3)2]3/isobutylalumoxane catalyst for selective ethylene trimerization. J. Mol. Catal. A Chem. 2002, 187, 135–141. [Google Scholar] [CrossRef]
- McGuinness, D.S.; Wasserscheid, P.; Keim, W.; Hu, C.; Englert, U.; Dixon, J.T.; Grove, C. Novel Cr-PNP complexes as catalysts for the trimerisation of ethylene. J. Chem. Soc. Chem. Commun. 2003, 2003, 334–335. [Google Scholar] [CrossRef]
- McGuinness, D.S.; Wasserscheid, P.; Keim, W.; Morgan, D.; Dixon, J.T.; Bollmann, A.; Maumela, H.; Hess, F.; Englert, U. First Cr(III)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to 1-hexene. J. Am. Chem. Soc. 2003, 125, 5272–5273. [Google Scholar] [CrossRef]
- Morgan, D.H.; Schwikkard, S.L.; Dixon, J.T.; Nair, J.J.; Hunter, R. The effect of aromatic ethers on the trimerisation of ethylene using a chromium catalyst and aryloxy ligands. Adv. Synth. Catal. 2003, 345, 939–942. [Google Scholar] [CrossRef]
- Commereuc, D.; Drochon, S.; Saussine, L. Institut Francais du Petrole. U.S. Patent 6031145, 2000. [Google Scholar]
- Wu, F.-J. Amoco Corp. U.S. Patent 5811618, 1998. [Google Scholar]
- Aoyama, T.; Mimura, H.; Yamamoto, T.; Oguri, M.; Koie, Y. Tosoh Corp. JP Patent 09176229, 1997. [Google Scholar]
- Jolly, P.W. From hein to hexene: Recent advances in the chemistry of organochromium π-complexes. Acc. Chem. Res. 1996, 29, 544–551. [Google Scholar] [CrossRef]
- Deckers, P.J.W.; Hessen, B.; Teuben, J.H. Switching a catalyst System from ethene polymerization to ethene trimerization with a hemilabile ancillary ligand. Angew. Chem. Int. Ed. 2001, 40, 2516–2519. [Google Scholar] [CrossRef]
- Deckers, P.J.W.; Hessen, B.; Teuben, J.H. Catalytic trimerization of ethene with highly active cyclopentadienyl-arene titanium catalysts. Organometallics 2002, 21, 5122–5135. [Google Scholar] [CrossRef]
- Pellecchia, C.; Pappalardo, D.; Oliva, L.; Mazzeo, M.; Gruter, G.-J. Selective co-oligomerization of ethylene and styrenes by half-titanocene catalysts and synthesis of polyethylenes with 4-aryl-1-butyl branches. Macromolecules 2000, 33, 2807–2814. [Google Scholar] [CrossRef]
- Andes, C.; Harkins, S.B.; Murtuza, S.; Oyler, K.; Sen, A. New tantalum-based catalyst system for the selective trimerization of ethene to 1-hexene. J. Am. Chem. Soc. 2001, 123, 7423–7424. [Google Scholar] [CrossRef]
- Santi, R.; Romano, A.M.; Grande, M.; Sommazzi, A.; Masi, F.; Proto, A. ENICHEM S.P.A. WO 0168572. 2001. [Google Scholar]
- Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Hess, F.; Killian, E.; Maumela, H.; Morgan, D.H.; Neveling, A.; Otto, S. Ethylene trimerisation and tetramerisation catalysts with polar-substituted diphosphinoamine ligands. J. Chem. Soc. Chem. Commun. 2005, 5, 622–624. [Google Scholar] [CrossRef]
- Bhaduri, S.; Mukhopadhyay, S.; Kulkarni, S.A. Density functional studies on chromium catalyzed ethylene trimerization. J. Organomet. Chem. 2009, 694, 1297–1307. [Google Scholar] [CrossRef]
- de Bruin, T.J.M.; Magna, L.; Raybaud, P.; Toulhoat, H. Hemilabile ligand induced selectivity: A DFT study on ethylene trimerization catalyzed by titanium complexes. Organometallics 2003, 22, 3404–3413. [Google Scholar] [CrossRef]
- Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Haasbroek, D.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H. Mechanistic investigations of the ethylene tetramerisation reaction. J. Am. Chem. Soc. 2005, 127, 10723–10730. [Google Scholar] [CrossRef]
- Brookhart, M.; Green, M.L.H.; Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 6908–6914. [Google Scholar] [CrossRef]
- Yu, Z.-X.; Houk, K.N. Why trimerization? Computational elucidation of the origin of selective trimerization of ethene catalyzed by [TaCl3(CH3)2] and an agostic-assisted hydride transfer mechanism. Angew. Chem. Int. Ed. 2003, 42, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-fnnctional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Dines, T.J.; Inglis, S. Raman spectroscopic study of supported chromium(vi) oxide catalysts. Phys. Chem. Chem. Phys. 2003, 5, 1320–1328. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1457–1465. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheong, M.; Singh, A. A Density Functional Study on Ethylene Trimerization and Tetramerization Using Real Sasol Cr-PNP Catalysts. Molecules 2023, 28, 3101. https://doi.org/10.3390/molecules28073101
Cheong M, Singh A. A Density Functional Study on Ethylene Trimerization and Tetramerization Using Real Sasol Cr-PNP Catalysts. Molecules. 2023; 28(7):3101. https://doi.org/10.3390/molecules28073101
Chicago/Turabian StyleCheong, Minserk, and Ajeet Singh. 2023. "A Density Functional Study on Ethylene Trimerization and Tetramerization Using Real Sasol Cr-PNP Catalysts" Molecules 28, no. 7: 3101. https://doi.org/10.3390/molecules28073101
APA StyleCheong, M., & Singh, A. (2023). A Density Functional Study on Ethylene Trimerization and Tetramerization Using Real Sasol Cr-PNP Catalysts. Molecules, 28(7), 3101. https://doi.org/10.3390/molecules28073101