
Rethinking Biosynthesis of Aclacinomycin A

Abstract
1. Introduction
2. Biosynthesis of ACM-A
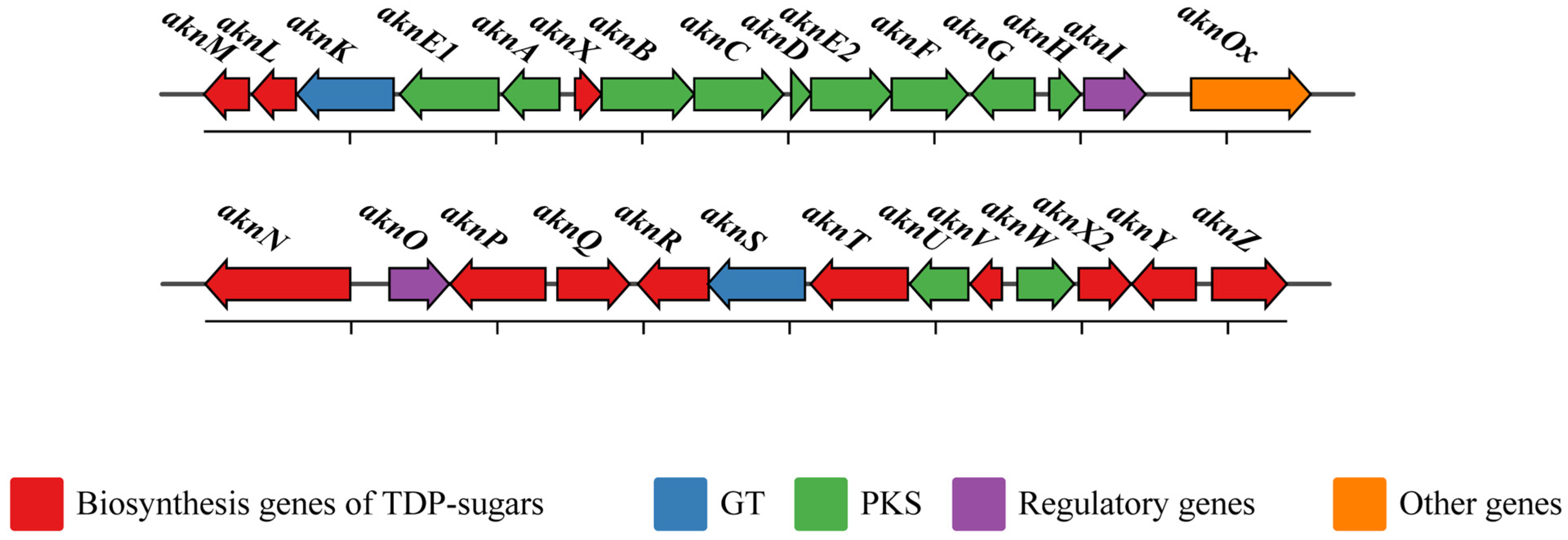
2.1. Gene Cluster for Biosynthesis of Aklavinone in S. galilaeus
2.2. Biosynthesis of TDP-Sugars
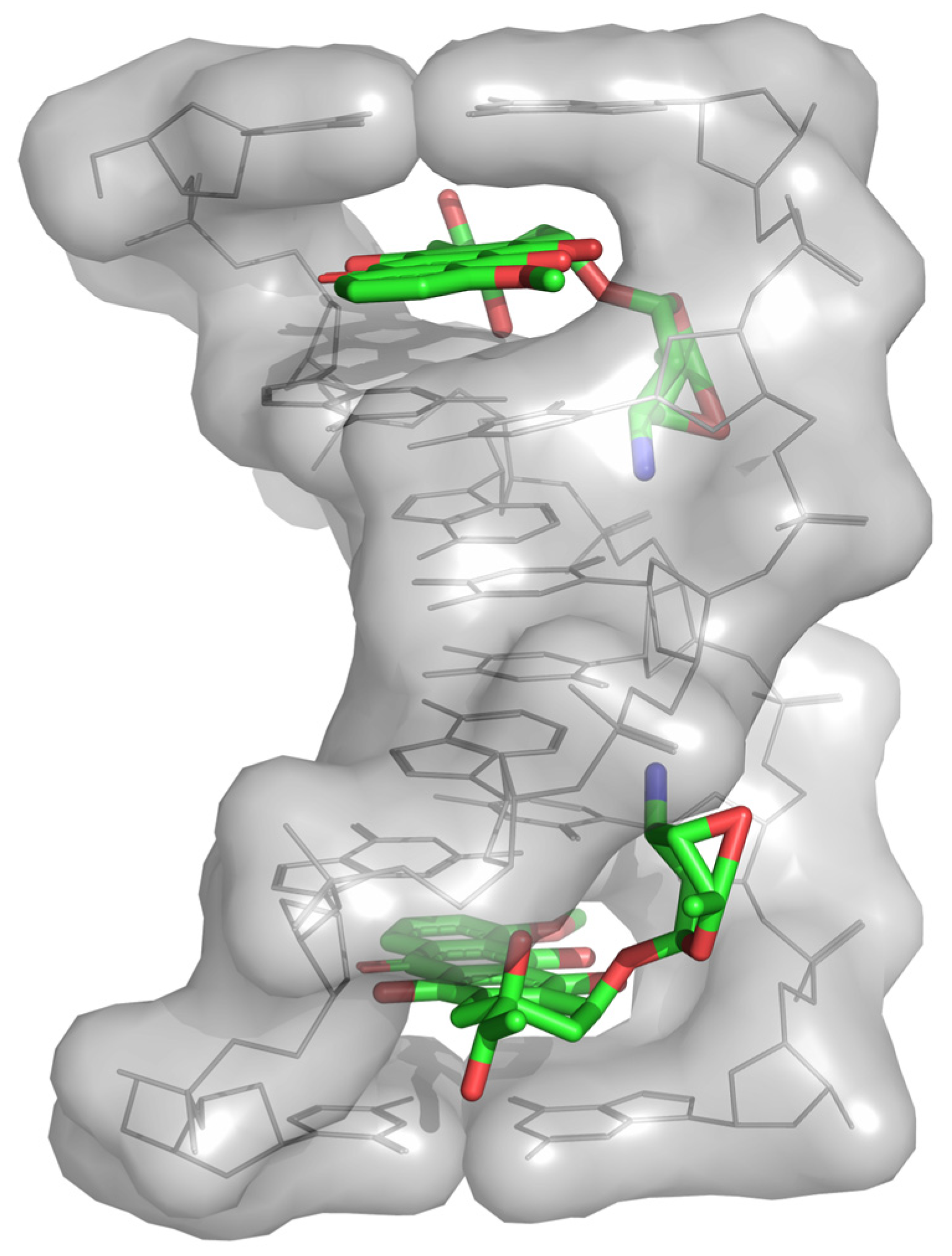
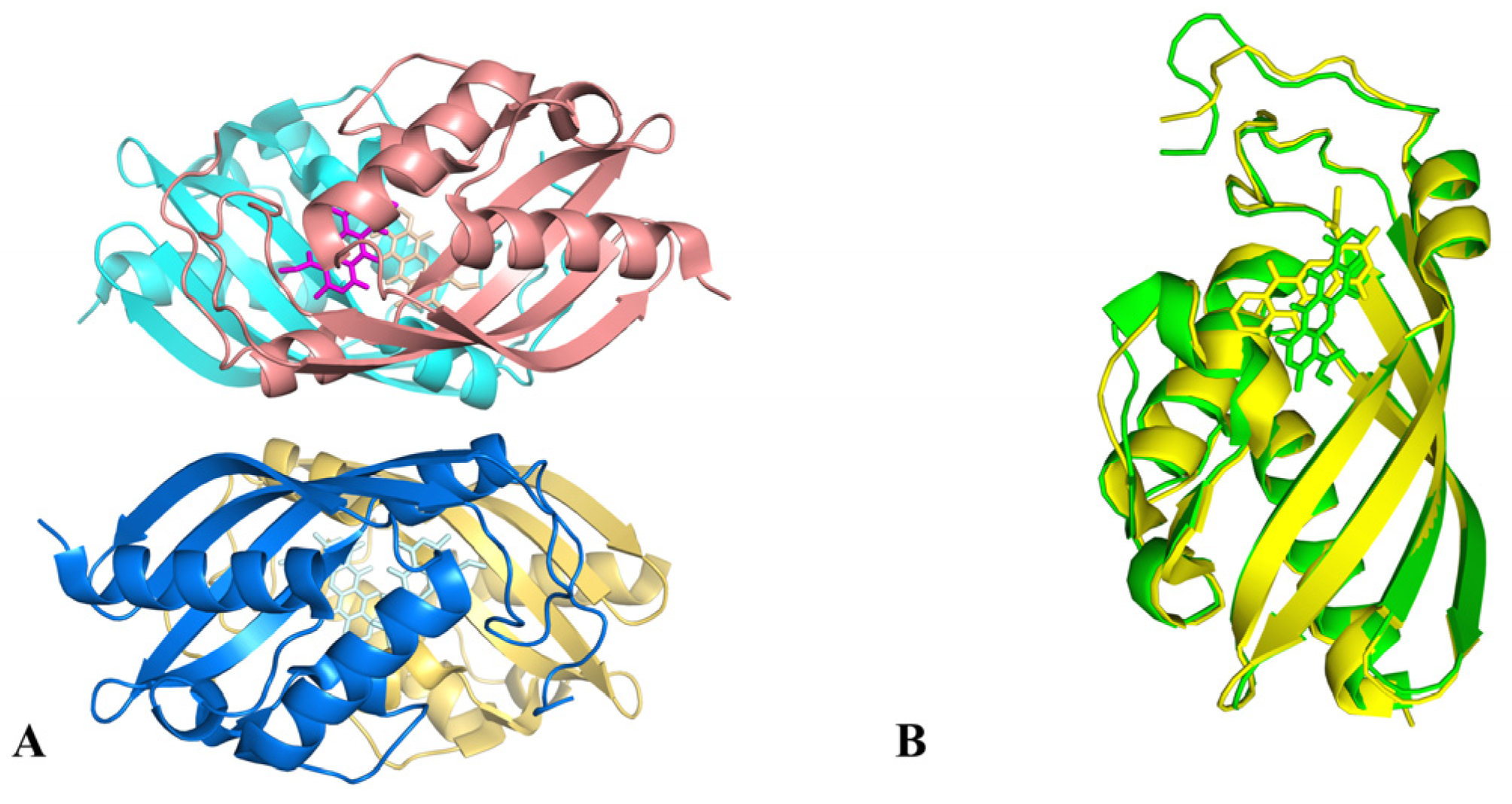
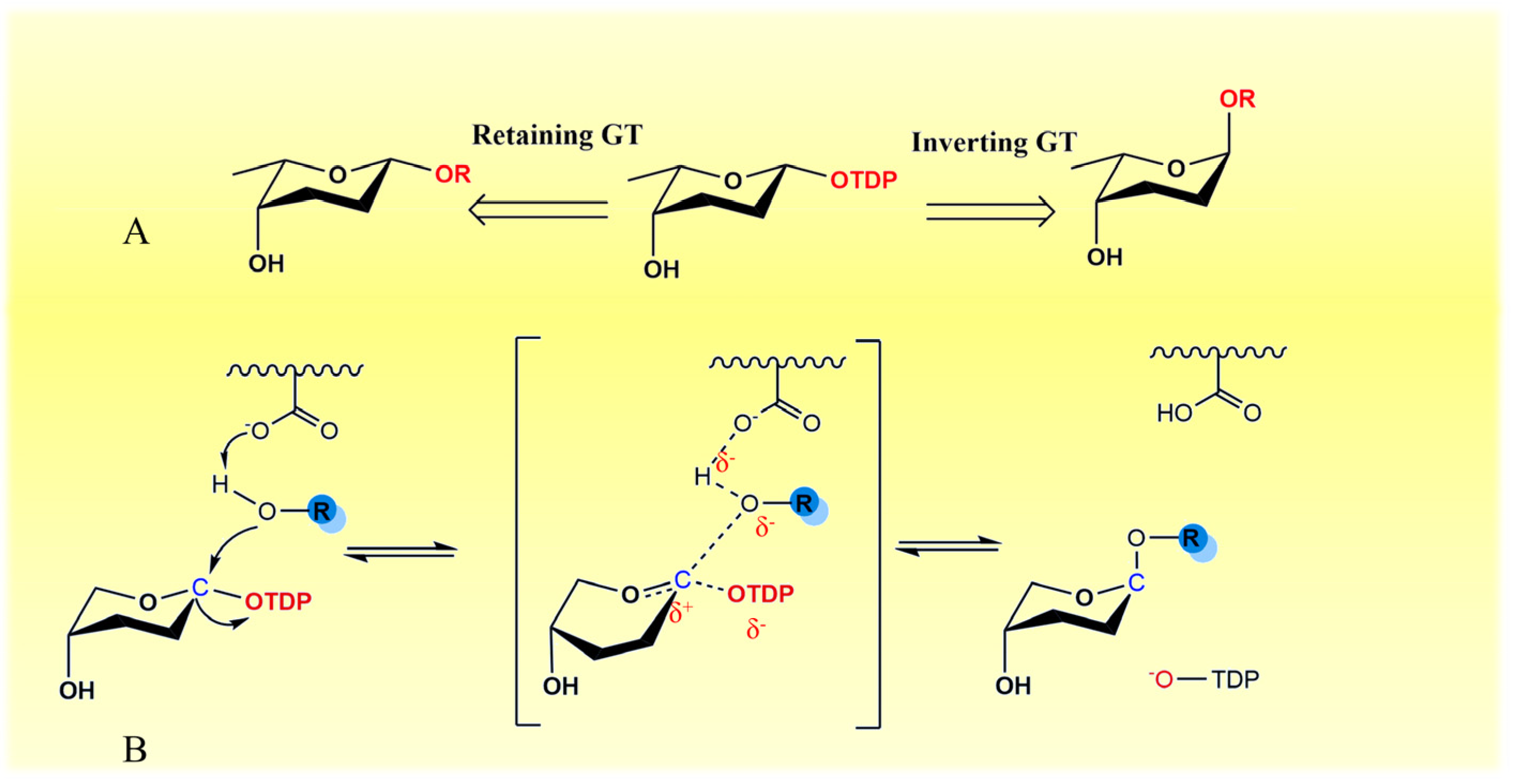
2.3. Transfer of TDP-Sugars
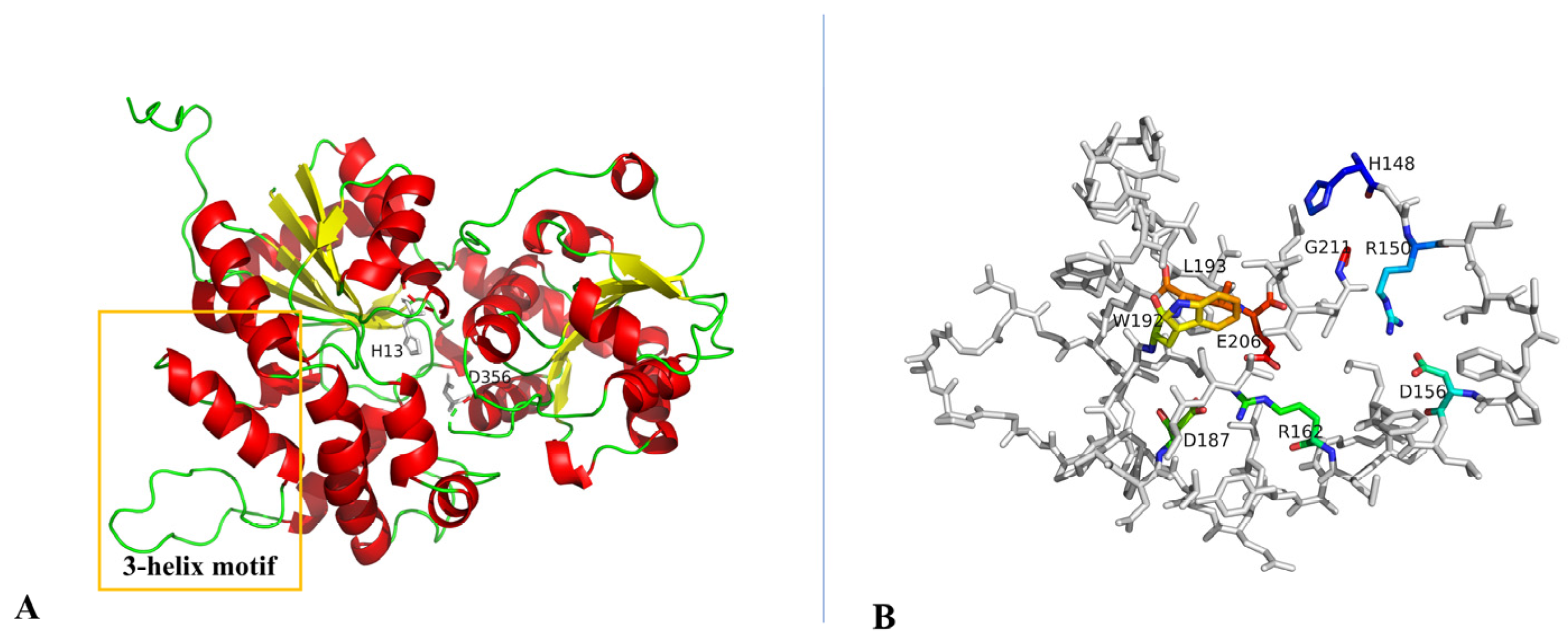
2.4. Transfer of TDP-β-L-Rhodosamine
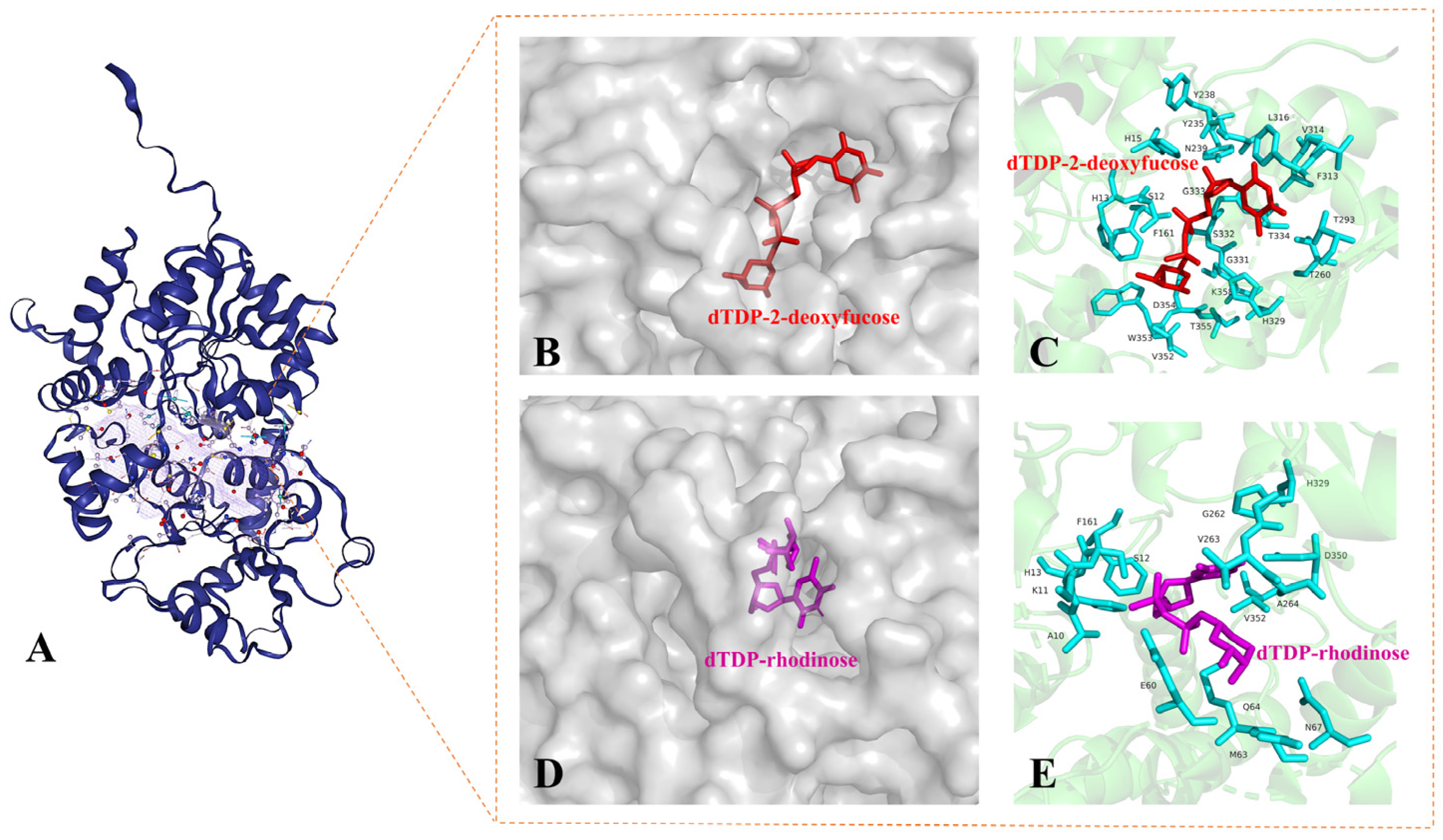
2.5. Transfer of 2-Deoxy-β-L-Fucose and L-Rhodinose
2.6. Optimization of the Catalytic Activity of GTs
2.7. Oxidation of Terminal Sugar Residue
2.8. Regulatory Genes for ACM-A Biosynthesis
3. Strategies for Improving ACM-A Production
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Puyo, S.; Montaudon, D.; Pourquier, P. From old alkylating agents to new minor groove binders. Crit. Rev. Oncol. Hematol. 2014, 89, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Parczyk, K.; Schneider, M.R. The future of antihormone therapy: Innovations based on an established principle. J. Cancer Res. Clin. Oncol. 1996, 122, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Silvis, N.G. Antimetabolites and cytotoxic drugs. Dermatol. Clin. 2001, 19, 105–118, viii–ix. [Google Scholar] [CrossRef] [PubMed]
- Schlager, S.; Drager, B. Exploiting plant alkaloids. Curr. Opin. Biotechnol. 2016, 37, 155–164. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Johnson, L.N. Protein kinase inhibitors: Contributions from structure to clinical compounds. Q. Rev. Biophys. 2009, 42, 1–40. [Google Scholar] [CrossRef]
- Nelson, P.N.; Reynolds, G.M.; Waldron, E.E.; Ward, E.; Giannopoulos, K.; Murray, P.G. Monoclonal antibodies. Mol. Pathol. 2000, 53, 111–117. [Google Scholar] [CrossRef]
- Oki, T.; Matsuzawa, Y.; Yoshimoto, A.; Numata, K.; Kitamura, I. New antitumor antibiotics aclacinomycins A and B. J. Antibiot. 1975, 28, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, Y.H.; Yoo, O.J.; Lee, J.J. Aclacinomycin X, a novel anthracycline antibiotic produced by Streptomyces galilaeus ATCC 31133. Biosci. Biotechnol. Biochem. 1996, 60, 906–908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, H.; Wang, J.Y.; Shen, X.Z.; Deng, Y.H.; Zhang, W. Preparation of magnetic polybutylcyanoacrylate nanospheres encapsulated with aclacinomycin A and its effect on gastric tumor. World J. Gastroenterol. 2004, 10, 2010–2013. [Google Scholar] [CrossRef]
- Lee, Y.L.; Chen, C.W.; Liu, F.H.; Huang, Y.W.; Huang, H.M. Aclacinomycin A sensitizes K562 chronic myeloid leukemia cells to imatinib through p38MAPK-mediated erythroid differentiation. PLoS ONE 2013, 8, e61939. [Google Scholar] [CrossRef]
- Feng, C.; Li, X.; Dong, C.; Zhang, X.; Zhang, X.; Gao, Y. RGD-modified liposomes enhance efficiency of aclacinomycin A delivery: Evaluation of their effect in lung cancer. Drug Des. Devel. Ther. 2015, 9, 4613–4620. [Google Scholar] [PubMed]
- Wang, F.; Xie, M.; Chen, P.; Wang, D.; Yang, M. Homoharringtonine combined with cladribine and aclarubicin (HCA) in acute myeloid leukemia: A new regimen of conventional drugs and its mechanism. Oxid. Med. Cell. Longev. 2022, 2022, 8212286. [Google Scholar] [CrossRef]
- Moore, M.H.; Hunter, W.N.; d’Estaintot, B.L.; Kennard, O. DNA-drug interactions. The crystal structure of d(CGATCG) complexed with daunomycin. J. Mol. Biol. 1989, 206, 693–705. [Google Scholar] [CrossRef]
- Capranico, G.; Binaschi, M.; Borgnetto, M.E.; Zunino, F.; Palumbo, M. A protein-mediated mechanism for the DNA sequence-specific action of topoisomerase II poisons. Trends Pharmacol. Sci. 1997, 18, 323–329. [Google Scholar] [CrossRef]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef]
- Qiao, X.; van der Zanden, S.Y.; Wander, D.P.A.; Borras, D.M.; Song, J.Y.; Li, X.; van Duikeren, S.; van Gils, N.; Rutten, A.; van Herwaarden, T.; et al. Uncoupling DNA damage from chromatin damage to detoxify doxorubicin. Proc. Natl. Acad. Sci. USA 2020, 117, 15182–15192. [Google Scholar] [CrossRef]
- Hori, S.; Shirai, M.; Hirano, S.; Oki, T.; Inui, T.; Tsukagoshi, S.; Ishizuka, M.; Takeuchi, T.; Umezawa, H. Antitumor activity of new anthracycline antibiotics, aclacinomycin-A and its analogs, and their toxicity. Gan 1977, 68, 685–690. [Google Scholar] [PubMed]
- Jung, M.E.; Lowe, J.A. Synthetic approaches to aclacinomycin and pyrromycin antitumour antibiotics via Diels–Alder reactions of 6-alkoxy-2-pyrones: Total synthesis of chrysophanol, helminthosporin and pachybasin. J. Chem. Soc. Chem. Commun. 1978, 3, 95–96. [Google Scholar] [CrossRef]
- Krohn, K. Total Synthesis of Anthracyclinone. Angew. Chem. Int. Ed. 1986, 25, 790–807. [Google Scholar] [CrossRef]
- Monneret, C.; Martin, A.; Pais, M. Synthesis of the Oligosaccharide moieties of musettamycin, marcellomycin and aclacinomycin A, Antitumor antibiotics. J. Carbohydr. Chem. 1988, 7, 417–434. [Google Scholar] [CrossRef]
- Martin, A.; Pais, M.; Monneret, C. Synthesis of a trisaccharide related to the antitumour antibiotic, aclacinomycin A. J. Chem. Soc. Chem. Commun. 1983, 6, 305–306. [Google Scholar] [CrossRef]
- Zhu, S.; Liang, R.; Jiang, H.; Wu, W. An efficient route to polysubstituted tetrahydronaphthols: Silver-catalyzed [4+2] cyclization of 2-alkylbenzaldehydes and alkenes. Angew. Chem. Int. Ed. Engl. 2012, 51, 10861–10865. [Google Scholar] [CrossRef]
- Yao, H.; Vu, M.D.; Liu, X.W. Recent advances in reagent-controlled stereoselective/stereospecific glycosylation. Carbohydr. Res. 2019, 473, 72–81. [Google Scholar] [CrossRef]
- Bennett, C.S.; Galan, M.C. Methods for 2-Deoxyglycoside Synthesis. Chem. Rev. 2018, 118, 7931–7985. [Google Scholar] [CrossRef]
- Wander, D.P.A.; van der Zanden, S.Y.; van der Marel, G.A.; Overkleeft, H.S.; Neefjes, J.; Codee, J.D.C. Doxorubicin and aclarubicin: Shuffling anthracycline glycans for improved anticancer agents. J. Med. Chem. 2020, 63, 12814–12829. [Google Scholar] [CrossRef]
- Cho, W.T.; Kim, W.S.; Kim, M.K.; Park, J.K.; Kim, H.R.; Rhee, S.K.; Domracheva, A.G.; Panichkina, T.B.; Saburoba, L.A.; Nobikoba, L.M.; et al. Method for producing aclacinomycins A, B, Y using Strepomyces lavendofoliae DKRS. US5484712, 16 January 1996. [Google Scholar]
- Oki, T.; Kitamura, I.; Yoshimoto, A.; Matsuzawa, Y.; Shibamoto, N.; Ogasawara, T.; Inui, T.; Takamatsu, A.; Takeuchi, T.; Masuda, T.; et al. Antitumor anthracycline antibiotics, aclacinomycin A and analogues. I. Taxonomy, production, isolation and physicochemical properties. J. Antibiot. 1979, 32, 791–800. [Google Scholar] [CrossRef]
- Kitamura, I.; Tobe, H.; Yoshimoto, A.; Oki, T.; Naganawa, H.; Takeuchi, T.; Umezawa, H. Biosynthesis of aklavinone and aclacinomycins. J. Antibiot. 1981, 34, 1498–1500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Räty, K.; Kantola, J.; Hautala, A.; Hakala, J.; Ylihonko, K.; Mäntsälä, P. Cloning and characterization of Streptomyces galilaeus aclacinomycins polyketide synthase (PKS) cluster. Gene 2002, 293, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Leimkuhler, C.; Gatto, G.J., Jr.; Kruger, R.G.; Oberthur, M.; Kahne, D.; Walsh, C.T. AknT is an activating protein for the glycosyltransferase AknS in L-aminodeoxysugar transfer to the aglycone of aclacinomycin A. Chem. Biol. 2005, 12, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Leimkuhler, C.; Oberthur, M.; Kahne, D.; Walsh, C.T. AknK is an L-2-deoxyfucosyltransferase in the biosynthesis of the anthracycline aclacinomycin A. Biochemistry 2004, 43, 4548–4558. [Google Scholar] [CrossRef]
- Alexeev, I.; Sultana, A.; Mäntsälä, P.; Niemi, J.; Schneider, G. Aclacinomycin oxidoreductase (AknOx) from the biosynthetic pathway of the antibiotic aclacinomycin is an unusual flavoenzyme with a dual active site. Proc. Natl. Acad. Sci. USA 2007, 104, 6170–6175. [Google Scholar] [CrossRef]
- Hoshino, T.; Sekine, Y.; Fujiwara, A. Microbial conversion of anthracycline antibiotics. I. Microbial conversion of aclacinomycin B to aclacinomycin A. J. Antibiot. 1983, 36, 1458–1462. [Google Scholar]
- Gräfe, U.; Dornberger, K.; Fleck, W.F.; Bormann, E.J.; Ihn, W. Bioconversion of aclacinomycin A to Y and B by an intracellular enzyme of Streptomyces spec. AM 33352. Biotechnol. Lett. 1988, 10, 167–170. [Google Scholar] [CrossRef]
- Ridley, C.P.; Lee, H.Y.; Khosla, C. Evolution of polyketide synthases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, R.; Chen, X.; Sun, X.; Yan, Y.; Shen, X.; Yuan, Q. Biosynthesis of aromatic polyketides in microorganisms using type II polyketide synthases. Microb. Cell Fact. 2020, 19, 110. [Google Scholar] [CrossRef]
- Bisang, C.; Long, P.F.; Cortes, J.; Westcott, J.; Crosby, J.; Matharu, A.L.; Cox, R.J.; Simpson, T.J.; Staunton, J.; Leadlay, P.F. A chain initiation factor common to both modular and aromatic polyketide synthases. Nature 1999, 401, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Fujii, I. Heterologous expression systems for polyketide synthases. Nat. Prod. Rep. 2009, 26, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Metsä-Ketelä, M.; Palmu, K.; Kunnari, T.; Ylihonko, K.; Mäntsälä, P. Engineering anthracycline biosynthesis toward angucyclines. Antimicrob. Agents Chemother. 2003, 47, 1291–1296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsai, S.C. The structural enzymology of iterative aromatic polyketide synthases: A critical comparison with fatty acid synthases. Annu. Rev. Biochem. 2018, 87, 503–531. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, N.; Fujii, I.; Ebizuka, Y.; Sankawa, U. Nucleotide sequence of the aknA region of the aklavinone biosynthetic gene cluster of Streptomyces galilaeus. J. Bacteriol. 1994, 176, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Prado, L.; Lombo, F.; Brana, A.F.; Mendez, C.; Rohr, J.; Salas, J.A. Analysis of two chromosomal regions adjacent to genes for a type II polyketide synthase involved in the biosynthesis of the antitumor polyketide mithramycin in Streptomyces argillaceus. Mol. Gen. Genet. 1999, 261, 216–225. [Google Scholar] [CrossRef]
- Chung, J.Y.; Fujii, I.; Harada, S.; Sankawa, U.; Ebizuka, Y. Expression, purification, and characterization of AknX anthrone oxygenase, which is involved in aklavinone biosynthesis in Streptomyces galilaeus. J. Bacteriol. 2002, 184, 6115–6122. [Google Scholar] [CrossRef]
- Räty, K.; Kunnari, T.; Hakala, J.; Mantsala, P.; Ylihonko, K. A gene cluster from Streptomyces galilaeus involved in glycosylation of aclarubicin. Mol. Gen. Genet. 2000, 264, 164–172. [Google Scholar] [CrossRef]
- Kantola, J.; Kunnari, T.; Hautala, A.; Hakala, J.; Ylihonko, K.; Mäntsälä, P. Elucidation of anthracyclinone biosynthesis by stepwise cloning of genes for anthracyclines from three different Streptomyces spp. Microbiology 2000, 146, 155–163. [Google Scholar] [CrossRef]
- Kallio, P.; Sultana, A.; Niemi, J.; Mäntsälä, P.; Schneider, G. Crystal structure of the polyketide cyclase AknH with bound substrate and product analogue: Implications for catalytic mechanism and product stereoselectivity. J. Mol. Biol. 2006, 357, 210–220. [Google Scholar] [CrossRef]
- Kendrew, S.G.; Katayama, K.; Deutsch, E.; Madduri, K.; Hutchinson, C.R. DnrD cyclase involved in the biosynthesis of doxorubicin: Purification and characterization of the recombinant enzyme. Biochemistry 1999, 38, 4794–4799. [Google Scholar] [CrossRef]
- Sultana, A.; Kallio, P.; Jansson, A.; Wang, J.S.; Niemi, J.; Mäntsälä, P.; Schneider, G. Structure of the polyketide cyclase SnoaL reveals a novel mechanism for enzymatic aldol condensation. EMBO J. 2004, 23, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Dickens, M.L.; Ye, J.; Strohl, W.R. Cloning, sequencing, and analysis of aklaviketone reductase from Streptomyces sp. strain C5. J. Bacteriol. 1996, 178, 3384–3388. [Google Scholar] [CrossRef][Green Version]
- Torkkell, S.; Kunnari, T.; Palmu, K.; Mäntsälä, P.; Hakala, J.; Ylihonko, K. The entire nogalamycin biosynthetic gene cluster of Streptomyces nogalater: Characterization of a 20-kb DNA region and generation of hybrid structures. Mol. Genet. Genomics 2001, 266, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Thibodeaux, C.J.; Melancon, C.E., 3rd; Liu, H.W. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew. Chem. Int. Ed. Engl. 2008, 47, 9814–9859. [Google Scholar] [CrossRef]
- Salcedo, R.G.; Olano, C.; Fernandez, R.; Brana, A.F.; Mendez, C.; de la Calle, F.; Salas, J.A. Elucidation of the glycosylation steps during biosynthesis of antitumor macrolides PM100117 and PM100118 and engineering for novel derivatives. Microb. Cell Fact. 2016, 15, 187. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Losey, H.C.; Freel Meyers, C.L. Antibiotic glycosyltransferases. Biochem. Soc. Trans. 2003, 31, 487–492. [Google Scholar] [CrossRef]
- Räty, K.; Hautala, A.; Torkkell, S.; Kantola, J.; Mäntsälä, P.; Hakala, J.; Ylihonko, K. Characterization of mutations in aclacinomycin A-non-producing Streptomyces galilaeus strains with altered glycosylation patterns. Microbiology 2002, 148, 3375–3384. [Google Scholar] [CrossRef][Green Version]
- Otten, S.L.; Gallo, M.A.; Madduri, K.; Liu, X.; Hutchinson, C.R. Cloning and characterization of the Streptomyces peucetius dnmZUV genes encoding three enzymes required for biosynthesis of the daunorubicin precursor thymidine diphospho-L-daunosamine. J. Bacteriol. 1997, 179, 4446–4450. [Google Scholar] [CrossRef][Green Version]
- Hong, L.; Zhao, Z.; Melancon, C.E., 3rd; Zhang, H.; Liu, H.W. In vitro characterization of the enzymes involved in TDP-D-forosamine biosynthesis in the spinosyn pathway of Saccharopolyspora spinosa. J. Am. Chem. Soc. 2008, 130, 4954–4967. [Google Scholar] [CrossRef]
- Han, A.R.; Park, J.W.; Lee, M.K.; Ban, Y.H.; Yoo, Y.J.; Kim, E.J.; Kim, E.; Kim, B.G.; Sohng, J.K.; Yoon, Y.J. Development of a Streptomyces venezuelae-based combinatorial biosynthetic system for the production of glycosylated derivatives of doxorubicin and its biosynthetic intermediates. Appl. Environ. Microbiol. 2011, 77, 4912–4923. [Google Scholar] [CrossRef]
- Palcic, M.M. Glycosyltransferases as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, Z.; Zhang, L.; Wang, J.; Wu, C. Glycosyltransferase GT1 family: Phylogenetic distribution, substrates coverage, and representative structural features. Comput. Struct. Biotechnol. J. 2020, 18, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Gloster, T.M. Advances in understanding glycosyltransferases from a structural perspective. Curr. Opin. Struct. Biol. 2014, 28, 131–141. [Google Scholar] [CrossRef]
- Chang, A.; Singh, S.; Phillips, G.N., Jr.; Thorson, J.S. Glycosyltransferase structural biology and its role in the design of catalysts for glycosylation. Curr. Opin. Biotechnol. 2011, 22, 800–808. [Google Scholar] [CrossRef]
- Brazier-Hicks, M.; Offen, W.A.; Gershater, M.C.; Revett, T.J.; Lim, E.K.; Bowles, D.J.; Davies, G.J.; Edwards, R. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc. Natl. Acad. Sci. USA 2007, 104, 20238–20243. [Google Scholar] [CrossRef]
- Offen, W.; Martinez-Fleites, C.; Yang, M.; Kiat-Lim, E.; Davis, B.G.; Tarling, C.A.; Ford, C.M.; Bowles, D.J.; Davies, G.J. Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 2006, 25, 1396–1405. [Google Scholar] [CrossRef]
- Liang, D.M.; Liu, J.H.; Wu, H.; Wang, B.B.; Zhu, H.J.; Qiao, J.J. Glycosyltransferases: Mechanisms and applications in natural product development. Chem. Soc. Rev. 2015, 44, 8350–8374. [Google Scholar] [CrossRef]
- Breton, C.; Fournel-Gigleux, S.; Palcic, M.M. Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 2012, 22, 540–549. [Google Scholar] [CrossRef]
- Tvaroška, I.; Kozmon, S.; Wimmerova, M.; Koca, J. Substrate-assisted catalytic mechanism of O-GlcNAc transferase discovered by quantum mechanics/molecular mechanics investigation. J. Am. Chem. Soc. 2012, 134, 15563–15571. [Google Scholar] [CrossRef]
- Tvaroška, I.; Kozmon, S.; Wimmerova, M.; Koca, J. A QM/MM investigation of the catalytic mechanism of metal-ion-independent core 2 beta1,6-N-acetylglucosaminyltransferase. Chemistry 2013, 19, 8153–8162. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Song, M.C.; Kim, M.S.; Beom, J.Y.; Jung, J.A.; Cho, H.S.; Yoon, Y.J. One-Pot combinatorial biosynthesis of glycosylated anthracyclines by cocultivation of Streptomyces strains producing aglycones and nucleotide deoxysugars. ACS. Comb. Sci. 2017, 19, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Harrus, D.; Kellokumpu, S.; Glumoff, T. Crystal structures of eukaryote glycosyltransferases reveal biologically relevant enzyme homooligomers. Cell. Mol. Life Sci. 2018, 75, 833–848. [Google Scholar] [CrossRef]
- Isiorho, E.A.; Jeon, B.S.; Kim, N.H.; Liu, H.W.; Keatinge-Clay, A.T. Structural studies of the spinosyn forosaminyltransferase, SpnP. Biochemistry 2014, 53, 4292–4301. [Google Scholar] [CrossRef] [PubMed]
- Moncrieffe, M.C.; Fernandez, M.J.; Spiteller, D.; Matsumura, H.; Gay, N.J.; Luisi, B.F.; Leadlay, P.F. Structure of the glycosyltransferase EryCIII in complex with its activating P450 homologue EryCII. J. Mol. Biol. 2012, 415, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Isiorho, E.A.; Liu, H.W.; Keatinge-Clay, A.T. Structural studies of the spinosyn rhamnosyltransferase, SpnG. Biochemistry 2012, 51, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Otten, S.L.; Liu, X.; Ferguson, J.; Hutchinson, C.R. Cloning and characterization of the Streptomyces peucetius dnrQS genes encoding a daunosamine biosynthesis enzyme and a glycosyl transferase involved in daunorubicin biosynthesis. J. Bacteriol. 1995, 177, 6688–6692. [Google Scholar] [CrossRef]
- Leimkuhler, C.; Fridman, M.; Lupoli, T.; Walker, S.; Walsh, C.T.; Kahne, D. Characterization of rhodosaminyl transfer by the AknS/AknT glycosylation complex and its use in reconstituting the biosynthetic pathway of aclacinomycin A. J. Am. Chem. Soc. 2007, 129, 10546–10550. [Google Scholar] [CrossRef][Green Version]
- Luzhetskyy, A.; Fedoryshyn, M.; Durr, C.; Taguchi, T.; Novikov, V.; Bechthold, A. Iteratively acting glycosyltransferases involved in the hexasaccharide biosynthesis of landomycin A. Chem. Biol. 2005, 12, 725–729. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chang, A.; Singh, S.; Helmich, K.E.; Goff, R.D.; Bingman, C.A.; Thorson, J.S.; Phillips, G.N., Jr. Complete set of glycosyltransferase structures in the calicheamicin biosynthetic pathway reveals the origin of regiospecificity. Proc. Natl. Acad. Sci. USA 2011, 108, 17649–17654. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, K.; Graef, J.; Schoning-Stierand, K.; Rarey, M. GeoMine: Interactive pattern mining of protein-ligand interfaces in the Protein Data Bank. Bioinformatics 2021, 37, 424–425. [Google Scholar] [CrossRef]
- Schoning-Stierand, K.; Diedrich, K.; Fahrrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein-ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef]
- Kim, E.; Moore, B.S.; Yoon, Y.J. Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat. Chem. Biol. 2015, 11, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Schmölzer, K.; Lemmerer, M.; Nidetzky, B. Glycosyltransferase cascades made fit for chemical production: Integrated biocatalytic process for the natural polyphenol C-glucoside nothofagin. Biotechnol. Bioeng. 2018, 115, 545–556. [Google Scholar] [CrossRef]
- Schmölzer, K.; Gutmann, A.; Diricks, M.; Desmet, T.; Nidetzky, B. Sucrose synthase: A unique glycosyltransferase for biocatalytic glycosylation process development. Biotechnol. Adv. 2016, 34, 88–111. [Google Scholar] [CrossRef]
- Mestrom, L.; Marsden, S.R.; Dieters, M.; Achterberg, P.; Stolk, L.; Bento, I.; Hanefeld, U.; Hagedoorn, P.L. Artificial Fusion of mCherry enhances trehalose transferase solubility and stability. Appl. Environ. Microbiol. 2019, 85, e03084-18. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, L.; Zhang, Y.; Zhang, T.; Feng, Y.; Lu, X.; Lan, W.; Wang, J.; Wu, H.; Cao, C.; et al. Highly efficient production of soluble proteins from insoluble inclusion bodies by a two-step-denaturing and refolding method. PLoS ONE 2011, 6, e22981. [Google Scholar] [CrossRef]
- Shu, W.; Zheng, H.; Fu, X.; Zhen, J.; Tan, M.; Xu, J.; Zhao, X.; Yang, S.; Song, H.; Ma, Y. Enhanced heterologous production of glycosyltransferase UGT76G1 by co-expression of endogenous prpD and malK in Escherichia coli and its transglycosylation application in production of rebaudioside. Int. J. Mol. Sci. 2020, 21, 5752. [Google Scholar] [CrossRef]
- Moremen, K.W.; Ramiah, A.; Stuart, M.; Steel, J.; Meng, L.; Forouhar, F.; Moniz, H.A.; Gahlay, G.; Gao, Z.; Chapla, D.; et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 2018, 14, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.A.; Kim, B.G.; Lee, W.J.; Lim, Y.; Chong, Y.; Ahn, J.H. Production of a novel quercetin glycoside through metabolic engineering of Escherichia coli. Appl. Environ. Microbiol. 2012, 78, 4256–4262. [Google Scholar] [CrossRef]
- Hoffmeister, D.; Wilkinson, B.; Foster, G.; Sidebottom, P.J.; Ichinose, K.; Bechthold, A. Engineered urdamycin glycosyltransferases are broadened and altered in substrate specificity. Chem. Biol. 2002, 9, 287–295. [Google Scholar] [CrossRef]
- Kim, H.L.; Kim, A.H.; Park, M.B.; Lee, S.W.; Park, Y.S. Altered sugar donor specificity and catalytic activity of pteridine glycosyltransferases by domain swapping or site-directed mutagenesis. BMB Rep. 2013, 46, 37–40. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, B.G.; Sung, S.; Kim, M.; Mok, H.; Chong, Y.; Ahn, J.H. Engineering flavonoid glycosyltransferases for enhanced catalytic efficiency and extended sugar-donor selectivity. Planta 2013, 238, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Thorson, J.S. A high-throughput fluorescence-based glycosyltransferase screen and its application in directed evolution. Nat. Protoc. 2008, 3, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Gantt, R.W.; Peltier-Pain, P.; Singh, S.; Zhou, M.; Thorson, J.S. Broadening the scope of glycosyltransferase-catalyzed sugar nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 7648–7653. [Google Scholar] [CrossRef]
- Li, C.; Zhang, R.; Wang, J.; Wilson, L.M.; Yan, Y. Protein engineering for improving and diversifying natural product biosynthesis. Trends Biotechnol. 2020, 38, 729–744. [Google Scholar] [CrossRef]
- Kamra, P.; Gokhale, R.S.; Mohanty, D. SEARCHGTr: A program for analysis of glycosyltransferases involved in glycosylation of secondary metabolites. Nucleic Acids Res. 2005, 33, W220–W225. [Google Scholar] [CrossRef]
- Akere, A.; Chen, S.H.; Liu, X.; Chen, Y.; Dantu, S.C.; Pandini, A.; Bhowmik, D.; Haider, S. Structure-based enzyme engineering improves donor-substrate recognition of Arabidopsis thaliana glycosyltransferases. Biochem. J. 2020, 477, 2791–2805. [Google Scholar] [CrossRef]
- Joshi, R.; Trinkl, J.; Haugeneder, A.; Hartl, K.; Franz-Oberdorf, K.; Giri, A.; Hoffmann, T.; Schwab, W. Semirational design and engineering of grapevine glucosyltransferases for enhanced activity and modified product selectivity. Glycobiology 2019, 29, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Schmölzer, K.; Czabany, T.; Luley-Goedl, C.; Pavkov-Keller, T.; Ribitsch, D.; Schwab, H.; Gruber, K.; Weber, H.; Nidetzky, B. Complete switch from alpha-2,3- to alpha-2,6-regioselectivity in Pasteurella dagmatis beta-D-galactoside sialyltransferase by active-site redesign. Chem. Commun. 2015, 51, 3083–3086. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.P.; Bashyal, P.; Parajuli, P.; Yamaguchi, T.; Sohng, J.K. Two Trifunctional leloir glycosyltransferases as biocatalysts for natural products glycodiversification. Org. Lett. 2019, 21, 8058–8064. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Feng, Y. Structural dissection of sterol glycosyltransferase UGT51 from Saccharomyces cerevisiae for substrate specificity. J. Struct. Biol. 2018, 204, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Liu, F.; Shao, W.; Chu, J.; Wu, B.; He, B. Efficient synthesis of crocins from crocetin by a microbial glycosyltransferase from Bacillus subtilis 168. J. Agric. Food Chem. 2018, 66, 11701–11708. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, A.; Ogasawara, T.; Kitamura, I.; Oki, T.; Inui, T.; Takeuchi, T.; Umezawa, H. Enzymatic conversion of aclacinomycin A to Y by a specific oxidoreductase in Streptomyces. J. Antibiot. 1979, 32, 472–481. [Google Scholar] [CrossRef]
- Sultana, A.; Alexeev, I.; Kursula, I.; Mantsala, P.; Niemi, J.; Schneider, G. Structure determination by multiwavelength anomalous diffraction of aclacinomycin oxidoreductase: Indications of multidomain pseudomerohedral twinning. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 149–159. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, H.; Chen, Q.; Luo, M.; Sun, A.; Song, Y.; Ma, J.; Ju, J. Identification of the grincamycin gene cluster unveils divergent roles for GcnQ in different hosts, tailoring the L-rhodinose moiety. Org. Lett. 2013, 15, 3254–3257. [Google Scholar] [CrossRef]
- Leferink, N.G.; Heuts, D.P.; Fraaije, M.W.; van Berkel, W.J. The growing VAO flavoprotein family. Arch. Biochem. Biophys. 2008, 474, 292–301. [Google Scholar] [CrossRef]
- Heuts, D.P.; Scrutton, N.S.; McIntire, W.S.; Fraaije, M.W. What’s in a covalent bond? On the role and formation of covalently bound flavin cofactors. FEBS J. 2009, 276, 3405–3427. [Google Scholar] [CrossRef]
- Winkler, A.; Kutchan, T.M.; Macheroux, P. 6-S-cysteinylation of bi-covalently attached FAD in berberine bridge enzyme tunes the redox potential for optimal activity. J. Biol. Chem. 2007, 282, 24437–24443. [Google Scholar] [CrossRef] [PubMed]
- Winkler, A.; Motz, K.; Riedl, S.; Puhl, M.; Macheroux, P.; Gruber, K. Structural and mechanistic studies reveal the functional role of bicovalent flavinylation in berberine bridge enzyme. J. Biol. Chem. 2009, 284, 19993–20001. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Ho, J.Y.; Huang, C.C.; Lyu, S.Y.; Lee, C.Y.; Huang, Y.T.; Wu, C.J.; Chan, H.C.; Huang, C.J.; Hsu, N.S.; et al. A unique flavin mononucleotide-linked primary alcohol oxidase for glycopeptide A40926 maturation. J. Am. Chem. Soc. 2007, 129, 13384–13385. [Google Scholar] [CrossRef]
- Mo, X.; Huang, H.; Ma, J.; Wang, Z.; Wang, B.; Zhang, S.; Zhang, C.; Ju, J. Characterization of TrdL as a 10-hydroxy dehydrogenase and generation of new analogues from a tirandamycin biosynthetic pathway. Org. Lett. 2011, 13, 2212–2215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, Y.S.; Lyu, S.Y.; Hsu, L.J.; Chen, Y.H.; Huang, Y.T.; Chan, H.C.; Huang, C.J.; Chen, G.H.; Chou, C.C.; et al. Interception of teicoplanin oxidation intermediates yields new antimicrobial scaffolds. Nat. Chem. Biol. 2011, 7, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Berks, B.C.; Sargent, F.; Palmer, T. The Tat protein export pathway. Mol. Microbiol. 2000, 35, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Valverde, J.R.; Gullon, S.; Mellado, R.P. Modelling the metabolism of protein secretion through the Tat route in Streptomyces lividans. BMC Microbiol. 2018, 18, 59. [Google Scholar] [CrossRef]
- Liu, G.; Chater, K.F.; Chandra, G.; Niu, G.; Tan, H. Molecular regulation of antibiotic biosynthesis in streptomyces. Microbiol. Mol. Biol. Rev. 2013, 77, 112–143. [Google Scholar] [CrossRef]
- Wei, J.; He, L.; Niu, G. Regulation of antibiotic biosynthesis in actinomycetes: Perspectives and challenges. Synth. Syst. Biotechnol. 2018, 3, 229–235. [Google Scholar] [CrossRef]
- Krause, J.; Handayani, I.; Blin, K.; Kulik, A.; Mast, Y. Disclosing the potential of the SARP-type regulator papr2 for the activation of antibiotic gene clusters in Streptomycetes. Front. Microbiol. 2020, 11, 225. [Google Scholar] [CrossRef]
- Sheldon, P.J.; Busarow, S.B.; Hutchinson, C.R. Mapping the DNA-binding domain and target sequences of the Streptomyces peucetius daunorubicin biosynthesis regulatory protein, DnrI. Mol. Microbiol. 2002, 44, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ke, M.; Li, J.; Tang, Y.; Wang, N.; Tan, G.; Wang, Y.; Liu, R.; Bai, L.; Zhang, L.; et al. TetR-Type regulator SLCG_2919 is a negative regulator of lincomycin biosynthesis in Streptomyces lincolnensis. Appl. Environ. Microbiol. 2019, 85, e02091-18. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, C.A.; Medema, M.H.; Greenberg, E.P. LuxR homolog-linked biosynthetic gene clusters in proteobacteria. mSystems 2018, 3, e00208-17. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Stock, A.M. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 2009, 63, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Sánchez de la Nieta, R.; Santamaría, R.I.; Díaz, M. Two-Component Systems of Streptomyces coelicolor: An intricate network to be unraveled. Int. J. Mol. Sci. 2022, 23, 15085. [Google Scholar] [CrossRef]
- Kim, W.-S.; Youn, D.J.; Cho, W.; Kim, M.-K.; Kim, H.R.; Rhee, S.K.; Choi, E.-S. Improved production, and purification of aclacinomycin A from Streptomyces lavendofoliae DKRS. J. Microbiol. Biotechnol. 1995, 5, 297–301. [Google Scholar]
- Kim, W.S.; Youn, D.J.; Kim, H.R.; Rhee, S.K.; Choi, E.S. Metabolic conversion of aclacinomycins B and Y to A by pH shift during fermentation with Streptomyces lavendofoliae DKRS. Biotechnol. Tech. 1995, 9, 671–676. [Google Scholar] [CrossRef]
- Ylihonko, K.; Hakala, J.; Niemi, J.; Lundell, J.; Mantsala, P. Isolation and characterization of aclacinomycin A-non-producing Streptomyces galilaeus (ATCC 31615) mutants. Microbiology 1994, 140, 1359–1365. [Google Scholar] [CrossRef]
- Lee, T.S.; Khosla, C.; Tang, Y. Engineered biosynthesis of aklanonic acid analogues. J. Am. Chem. Soc. 2005, 127, 12254–12262. [Google Scholar] [CrossRef]
- Wang, R.; Nguyen, J.; Hecht, J.; Schwartz, N.; Brown, K.V.; Ponomareva, L.V.; Niemczura, M.; van Dissel, D.; van Wezel, G.P.; Thorson, J.S.; et al. A biobricks metabolic engineering platform for the biosynthesis of anthracyclinones in Streptomyces coelicolor. ACS Synth. Biol. 2022, 11, 4193–4209. [Google Scholar] [CrossRef]
- Belknap, K.C.; Park, C.J.; Barth, B.M.; Andam, C.P. Genome mining of biosynthetic and chemotherapeutic gene clusters in Streptomyces bacteria. Sci. Rep. 2020, 10, 2003. [Google Scholar] [CrossRef] [PubMed]
- Bundale, S.; Begde, D.; Pillai, D.; Gangwani, K.; Nashikkar, N.; Kadam, T.; Upadhyay, A. Novel aromatic polyketides from soil Streptomyces spp.: Purification, characterization and bioactivity studies. World J. Microbiol. Biotechnol. 2018, 34, 67. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Li, X.; Li, Z.; Zhan, X.; Mao, X.; Li, Y. The application of regulatory cascades in Streptomyces: Yield enhancement and metabolite mining. Front. Microbiol. 2020, 11, 406. [Google Scholar] [CrossRef] [PubMed]
- Khokhlov, A.S.; Tovarova, I.I.; Borisova, L.N.; Pliner, S.A.; Shevchenko, L.; Kornitskaia, E.I.; Ivkina, N.S.; Rapoport, I.A. The A-factor, responsible for streptomycin biosynthesis by mutant strains of Actinomyces streptomycini. Dok. Akad. Nauk SSSR 1967, 177, 232–325. [Google Scholar]
- Horinouchi, S.; Beppu, T. Hormonal control by A-factor of morphological development and secondary metabolism in Streptomyces. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2007, 83, 277–295. [Google Scholar] [CrossRef]
- Tan, G.Y.; Bai, L.; Zhong, J.J. Exogenous 1,4-butyrolactone stimulates A-factor-like cascade and validamycin biosynthesis in Streptomyces hygroscopicus 5008. Biotechnol. Bioeng. 2013, 110, 2984–2993. [Google Scholar] [CrossRef]
- Misaki, Y.; Yamamoto, S.; Suzuki, T.; Iwakuni, M.; Sasaki, H.; Takahashi, Y.; Inada, K.; Kinashi, H.; Arakawa, K. SrrB, a pseudo-receptor protein, acts as a negative regulator for lankacidin and lankamycin production in Streptomyces rochei. Front. Microbiol. 2020, 11, 1089. [Google Scholar] [CrossRef]
- Ma, D.; Wang, C.; Chen, H.; Wen, J. Manipulating the expression of SARP family regulator BulZ and its target gene product to increase tacrolimus production. Appl. Microbiol. Biotechnol. 2018, 102, 4887–4900. [Google Scholar] [CrossRef]
- Pokhrel, A.R.; Chaudhary, A.K.; Nguyen, H.T.; Dhakal, D.; Le, T.T.; Shrestha, A.; Liou, K.; Sohng, J.K. Overexpression of a pathway specific negative regulator enhances production of daunorubicin in bldA deficient Streptomyces peucetius ATCC 27952. Microbiol. Res. 2016, 192, 96–102. [Google Scholar] [CrossRef]
- Malla, S.; Niraula, N.P.; Liou, K.; Sohng, J.K. Improvement in doxorubicin productivity by overexpression of regulatory genes in Streptomyces peucetius. Res. Microbiol. 2010, 161, 109–117. [Google Scholar] [CrossRef]
- Nah, H.J.; Park, J.; Choi, S.; Kim, E.S. WblA, a global regulator of antibiotic biosynthesis in Streptomyces. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab007. [Google Scholar] [CrossRef] [PubMed]
- Merrick, M.J. A morphological and genetic mapping study of bald colony mutants of Streptomyces coelicolor. J. Gen. Microbiol. 1976, 96, 299–315. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, S.H.; Lee, H.N.; Lee, S.Y.; Kim, E.S. Isolation and genetic manipulation of the antibiotic down-regulatory gene, wblA ortholog for doxorubicin-producing Streptomyces strain improvement. Appl. Microbiol. Biotechnol. 2010, 86, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular mechanisms of two-component signal transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed]
- Recio, E.; Colinas, A.; Rumbero, A.; Aparicio, J.F.; Martin, J.F. PI factor, a novel type quorum-sensing inducer elicits pimaricin production in Streptomyces natalensis. J. Biol. Chem. 2004, 279, 41586–41593. [Google Scholar] [CrossRef] [PubMed]
- Kitani, S.; Miyamoto, K.T.; Takamatsu, S.; Herawati, E.; Iguchi, H.; Nishitomi, K.; Uchida, M.; Nagamitsu, T.; Omura, S.; Ikeda, H.; et al. Avenolide, a Streptomyces hormone controlling antibiotic production in Streptomyces avermitilis. Proc. Natl. Acad. Sci. USA 2011, 108, 16410–16415. [Google Scholar] [CrossRef]
- Matselyukh, B.; Mohammadipanah, F.; Laatsch, H.; Rohr, J.; Efremenkova, O.; Khilya, V. N-methylphenylalanyl-dehydrobutyrine diketopiperazine, an A-factor mimic that restores antibiotic biosynthesis and morphogenesis in Streptomyces globisporus 1912-B2 and Streptomyces griseus 1439. J. Antibiot. 2015, 68, 9–14. [Google Scholar] [CrossRef]
- Takano, E. Gamma-butyrolactones: Streptomyces signalling molecules regulating antibiotic production and differentiation. Curr. Opin. Microbiol. 2006, 9, 287–294. [Google Scholar] [CrossRef]
- Tian, J.; Yang, G.; Gu, Y.; Sun, X.; Lu, Y.; Jiang, W. Developing an endogenous quorum-sensing based CRISPRi circuit for autonomous and tunable dynamic regulation of multiple targets in Streptomyces. Nucleic Acids Res. 2020, 48, 8188–8202. [Google Scholar] [CrossRef]
- Liu, X.; Li, J.; Li, Y.; Li, J.; Sun, H.; Zheng, J.; Zhang, J.; Tan, H. A visualization reporter system for characterizing antibiotic biosynthetic gene clusters expression with high-sensitivity. Commun. Biol. 2022, 5, 901. [Google Scholar] [CrossRef]
- Adhikari, S.; Curtis, P.D. DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 2016, 40, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Veeraraghavan, B.; Elangovan, R.; Vivekanandan, P. Antibiotic resistance and epigenetics: More to it than meets the eye. Antimicrob. Agents Chemother. 2020, 64, e02225-19. [Google Scholar] [CrossRef] [PubMed]
- Na, D.; Yoo, S.M.; Chung, H.; Park, H.; Park, J.H.; Lee, S.Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 2013, 31, 170–174. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Pokhrel, A.R.; Hue, N.T.; Yoo, J.C.; Sohng, J.K. Paired-termini antisense RNA mediated inhibition of DoxR in Streptomyces peucetius ATCC 27952. Biotechnol. Bioprocess Eng. 2015, 20, 381–388. [Google Scholar] [CrossRef]
- Galm, U.; Hager, M.H.; Van Lanen, S.G.; Ju, J.; Thorson, J.S.; Shen, B. Antitumor antibiotics: Bleomycin, enediynes, and mitomycin. Chem. Rev. 2005, 105, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Malla, S.; Niraula, N.P.; Liou, K.; Sohng, J.K. Self-resistance mechanism in Streptomyces peucetius: Overexpression of drrA, drrB and drrC for doxorubicin enhancement. Microbiol. Res. 2010, 165, 259–267. [Google Scholar] [CrossRef]
- Yin, S.; Wang, X.; Shi, M.; Yuan, F.; Wang, H.; Jia, X.; Yuan, F.; Sun, J.; Liu, T.; Yang, K.; et al. Improvement of oxytetracycline production mediated via cooperation of resistance genes in Streptomyces rimosus. Sci. China Life Sci. 2017, 60, 992–999. [Google Scholar] [CrossRef]
- Yu, L.; Yan, X.; Wang, L.; Chu, J.; Zhuang, Y.; Zhang, S.; Guo, M. Molecular cloning and functional characterization of an ATP-binding cassette transporter OtrC from Streptomyces rimosus. BMC Biotechnol. 2012, 12, 52. [Google Scholar] [CrossRef]
- Yao, H.; Shen, Z.; Wang, Y.; Deng, F.; Liu, D.; Naren, G.; Dai, L.; Su, C.C.; Wang, B.; Wang, S.; et al. Emergence of a potent multidrug efflux pump variant that enhances campylobacter resistance to multiple antibiotics. mBio 2016, 7, e01543-16. [Google Scholar] [CrossRef]
- Nag, A.; Mehra, S. A major facilitator superfamily (MFS) efflux pump, SCO4121, from Streptomyces coelicolor with roles in multidrug resistance and oxidative stress tolerance and its regulation by a MarR regulator. Appl. Environ. Microbiol. 2021, 87, e02238-20. [Google Scholar] [CrossRef]
- Ochi, K.; Okamoto, S.; Tozawa, Y.; Inaoka, T.; Hosaka, T.; Xu, J.; Kurosawa, K. Ribosome engineering and secondary metabolite production. Adv. Appl. Microbiol. 2004, 56, 155–184. [Google Scholar] [PubMed]
- Hosoya, Y.; Okamoto, S.; Muramatsu, H.; Ochi, K. Acquisition of certain streptomycin-resistant (str) mutations enhances antibiotic production in bacteria. Antimicrob. Agents Chemother. 1998, 42, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Shentu, X.; Liu, N.; Tang, G.; Tanaka, Y.; Ochi, K.; Xu, J.; Yu, X. Improved antibiotic production and silent gene activation in Streptomyces diastatochromogenes by ribosome engineering. J. Antibiot. 2016, 69, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Tian, Y.; Tan, H. Enhancement of salinomycin production by ribosome engineering in Streptomyces albus. Sci. China Life Sci. 2019, 62, 276–279. [Google Scholar] [CrossRef]
- Lv, X.A.; Jin, Y.Y.; Li, Y.D.; Zhang, H.; Liang, X.L. Genome shuffling of Streptomyces viridochromogenes for improved production of avilamycin. Appl. Microbiol. Biotechnol. 2013, 97, 641–648. [Google Scholar] [CrossRef]
- Payne, D.J.; Warren, P.V.; Holmes, D.J.; Ji, Y.; Lonsdale, J.T. Bacterial fatty-acid biosynthesis: A genomics-driven target for antibacterial drug discovery. Drug Discov. Today 2001, 6, 537–544. [Google Scholar] [CrossRef]
- Craney, A.; Ozimok, C.; Pimentel-Elardo, S.M.; Capretta, A.; Nodwell, J.R. Chemical perturbation of secondary metabolism demonstrates important links to primary metabolism. Chem. Biol. 2012, 19, 1020–1027. [Google Scholar] [CrossRef]
- Lomovskaya, N.; Otten, S.L.; Doi-Katayama, Y.; Fonstein, L.; Liu, X.C.; Takatsu, T.; Inventi-Solari, A.; Filippini, S.; Torti, F.; Colombo, A.L.; et al. Doxorubicin overproduction in Streptomyces peucetius: Cloning and characterization of the dnrU ketoreductase and dnrV genes and the doxA cytochrome P-450 hydroxylase gene. J. Bacteriol. 1999, 181, 305–318. [Google Scholar] [CrossRef]
- Malla, S.; Niraula, N.P.; Liou, K.; Sohng, J.K. Enhancement of doxorubicin production by expression of structural sugar biosynthesis and glycosyltransferase genes in Streptomyces peucetius. J. Biosci. Bioeng. 2009, 108, 92–98. [Google Scholar] [CrossRef]
- Wong, M.; Badri, A.; Gasparis, C.; Belfort, G.; Koffas, M. Modular optimization in metabolic engineering. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 587–602. [Google Scholar] [CrossRef]
- Zhao, M.; Zhao, Y.; Yao, M.; Iqbal, H.; Hu, Q.; Liu, H.; Qiao, B.; Li, C.; Skovbjerg, C.A.S.; Nielsen, J.C.; et al. Pathway engineering in yeast for synthesizing the complex polyketide bikaverin. Nat. Commun. 2020, 11, 6197. [Google Scholar] [CrossRef] [PubMed]
- Beites, T.; Mendes, M.V. Chassis optimization as a cornerstone for the application of synthetic biology based strategies in microbial secondary metabolism. Front. Microbiol. 2015, 6, 906. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Deng, Z.; Liu, T. Streptomyces species: Ideal chassis for natural product discovery and overproduction. Metab. Eng. 2018, 50, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, Y.; Tang, Y. Engineered biosynthesis of bacterial aromatic polyketides in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 20683–20688. [Google Scholar] [CrossRef]
- Liu, X.; Hua, K.; Liu, D.; Wu, Z.L.; Wang, Y.; Zhang, H.; Deng, Z.; Pfeifer, B.A.; Jiang, M. Heterologous biosynthesis of type ii polyketide products using E. coli. ACS Chem. Biol. 2020, 15, 1177–1183. [Google Scholar] [CrossRef]
- Sun, D.; Liu, C.; Zhu, J.; Liu, W. Connecting metabolic pathways: Sigma factors in Streptomyces spp. Front. Microbiol. 2017, 8, 2546. [Google Scholar] [CrossRef]
- Zhuo, Y.; Zhang, W.; Chen, D.; Gao, H.; Tao, J.; Liu, M.; Gou, Z.; Zhou, X.; Ye, B.C.; Zhang, Q.; et al. Reverse biological engineering of hrdB to enhance the production of avermectins in an industrial strain of Streptomyces avermitilis. Proc. Natl. Acad. Sci. USA 2010, 107, 11250–11254. [Google Scholar] [CrossRef]
- Van Brempt, M.; Clauwaert, J.; Mey, F.; Stock, M.; Maertens, J.; Waegeman, W.; De Mey, M. Predictive design of sigma factor-specific promoters. Nat. Commun. 2020, 11, 5822. [Google Scholar] [CrossRef]
- Chen, D.; Arkin, A.P. Sequestration-based bistability enables tuning of the switching boundaries and design of a latch. Mol. Syst. Biol. 2012, 8, 620. [Google Scholar] [CrossRef]
- Costello, A.; Badran, A.H. Synthetic biological circuits within an orthogonal central dogma. Trends Biotechnol. 2021, 39, 59–71. [Google Scholar] [CrossRef]
- Li, W.; Ma, L.; Shen, X.; Wang, J.; Feng, Q.; Liu, L.; Zheng, G.; Yan, Y.; Sun, X.; Yuan, Q. Targeting metabolic driving and intermediate influx in lysine catabolism for high-level glutarate production. Nat. Commun. 2019, 10, 3337. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, C.; Bai, L. Conversion of the high-yield salinomycin producer Streptomyces albus BK3-25 into a surrogate host for polyketide production. Sci. China Life Sci. 2017, 60, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, X.; Jiang, M.; Bai, L. Enhanced salinomycin production by adjusting the supply of polyketide extender units in Streptomyces albus. Metab. Eng. 2016, 35, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.S.; Ye, C.; Zhang, Z.; Lu, Y.; Jing, K. Daptomycin antibiotic production processes in fed-batch fermentation by Streptomyces roseosporus NRRL11379 with precursor effect and medium optimization. Bioprocess Biosyst. Eng. 2014, 37, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Zabala, D.; Brana, A.F.; Salas, J.A.; Mendez, C. Increasing antibiotic production yields by favoring the biosynthesis of precursor metabolites glucose-1-phosphate and/or malonyl-CoA in Streptomyces producer strains. J. Antibiot. 2016, 69, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Myronovskyi, M.; Rosenkranzer, B.; Nadmid, S.; Pujic, P.; Normand, P.; Luzhetskyy, A. Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab. Eng. 2018, 49, 316–324. [Google Scholar] [CrossRef]
- Ajikumar, P.K.; Xiao, W.H.; Tyo, K.E.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Duan, J.; Qin, Y.; Yang, X.; Ren, J.; Li, G. Improving the yield of xenocoumacin 1 enabled by in situ product removal. ACS Omega 2020, 5, 20391–20398. [Google Scholar] [CrossRef]
- Malla, S.; Niraula, N.P.; Singh, B.; Liou, K.; Sohng, J.K. Limitations in doxorubicin production from Streptomyces peucetius. Microbiol. Res. 2010, 165, 427–435. [Google Scholar] [CrossRef]
- Chen, X.; Li, S.; Zhang, B.; Sun, H.; Wang, J.; Zhang, W.; Meng, W.; Chen, T.; Dyson, P.; Liu, G. A new bacterial tRNA enhances antibiotic production in Streptomyces by circumventing inefficient wobble base-pairing. Nucleic Acids Res. 2022, 50, 7084–7096. [Google Scholar] [CrossRef]
- Hwang, C.K.; Kim, H.S.; Hong, Y.S.; Kim, Y.H.; Hong, S.K.; Kim, S.J.; Lee, J.J. Expression of Streptomyces peucetius genes for doxorubicin resistance and aklavinone 11-hydroxylase in Streptomyces galilaeus ATCC 31133 and production of a hybrid aclacinomycin. Antimicrob. Agents Chemother. 1995, 39, 1616–1620. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Yin, Y.; Tang, G.L. Directed biosynthesis of iso-aclacinomycins with improved anticancer activity. Org. Lett. 2020, 22, 150–154. [Google Scholar] [CrossRef]
- Hadicke, O.; von Kamp, A.; Aydogan, T.; Klamt, S. OptMDFpathway: Identification of metabolic pathways with maximal thermodynamic driving force and its application for analyzing the endogenous CO2 fixation potential of Escherichia coli. PLoS Comput. Biol. 2018, 14, e1006492. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, G.; Chen, J.; Ge, M.; Jiang, W.; Lu, Y. Multiplexed site-specific genome engineering for overproducing bioactive secondary metabolites in actinomycetes. Metab. Eng. 2017, 40, 80–92. [Google Scholar] [CrossRef]
- Wang, H.H.; Kim, H.; Cong, L.; Jeong, J.; Bang, D.; Church, G.M. Genome-scale promoter engineering by coselection MAGE. Nat. Methods 2012, 9, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Tong, Y.; Weber, T.; Lee, S.Y. CRISPR/Cas-based genome engineering in natural product discovery. Nat. Prod. Rep. 2019, 36, 1262–1280. [Google Scholar] [CrossRef]
- Rogers, J.K.; Taylor, N.D.; Church, G.M. Biosensor-based engineering of biosynthetic pathways. Curr. Opin. Biotechnol. 2016, 42, 84–91. [Google Scholar] [CrossRef]
- Kannan, S.; Altae-Tran, H.; Jin, X.; Madigan, V.J.; Oshiro, R.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Compact RNA editors with small Cas13 proteins. Nat. Biotechnol. 2022, 40, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Product | Number of Amino Acid Residues | Uniprot | Functions |
|---|---|---|---|
| AknA | 261 | Q9L553 | β-ketoreductase |
| AknB | 423 | Q9L551 | KSα |
| AknC | 407 | Q9L550 | KSβ |
| AknD | 91 | Q9L549 | Acyl carrier protein (ACP) |
| AknE1 | 450 | Q9L554 | Aromatase (ARO) |
| AknE2 | 368 | Q9L548 | Determines the starter unit |
| AknF | 347 | Q9L547 | Malonyl-CoA: ACP transacylase (MAT) |
| AknW | 259 | Q9L4U2 | Cyclase |
| AknX | 122 | Q9L552 | Mono-oxygenase (OXY) |
| AknG | 286 | Q9L546 | Methyl transferase (MET) |
| AknH | 144 | O52646 | Aklanonic acid methyl ester cyclase (AAME-cyclase) |
| AknU | 267 | Q9L4U4 | Aklaviketone reductase (KRII) |
| Gene Product | Number of Amino Acid Residues | Uniprot | Functions | Isozyme |
|---|---|---|---|---|
| AknY | 291 | Q9L4U0 | Glucose-1-phosphate thymidylyltransferase | DnmL |
| AknR | 323 | Q9L4U7 | dTDP-Glucose 4,6-dehydratase | DnmM |
| AknN | 662 | Q9L4V1 | 2,3-Dehydratase | DnmT, |
| AknQ | 329 | Q9L4U8 | 3-Ketoreductase | SpnN |
| AknP | 434 | Q9L4U9 | 3-Dehydratase | SpnQ |
| AknZ | 341 | Q9L4T9 | Aminotransferase | DnmJ, |
| AknL | 201 | Q9L556 | 3,5-Epimerase | DnmU, |
| AknM | 206 | Q9L557 | Reductase | DnmV |
| AknX2 | 238 | Q9L4U1 | N-methyltransferase | – |
| Organism | Secondary Metabolite | Strategy | Production Improved | Ref. |
|---|---|---|---|---|
| S. lavendofoliae | ACM-A | NTG mutagenesis | 300% | [127] |
| S. coelicolor | aklavinone | optimization of promoters, vectors, and chassis strains | >100% | [131] |
| S. galilaeus | ACM-A | adjusting broth pH | Data not shown | [128] |
| S. galilaeus | ACM-A | NTG mutagenesis | Data not shown | [129] |
| S. coelicolor | DXR/DNR/akavinone | disruption of global downregulator gene | 30% | [144] |
| S. peucetius | daunorubicin | overexpression of resistance genes | 410% | [157] |
| S. peucetius | daunorubicin | overexpression of global regulator | 45.7% | [140] |
| S. peucetius | doxorubicin | overexpression of rate-limiting enzymes | 86% | [169] |
| S. peucetius | doxorubicin | expression of structural sugar biosynthesis and glycosyltransferase genes | 460% | [170] |
| S. avermitilis | avermectin | ribosomal engineering | 50% | [178] |
| S. rochei | Lankacidin/lankamycin | deletion of GBL receptors | >300% | [138] |
| S. hygroscopicus | validamycin | exogenous addition of A-Factor analogues | 30% | [137] |
| S. tsukubaensis | tacrolimus | expression of GBL synthetase | 36% | [139] |
| S. rimosus | oxytetracycline | overexpression of transporters | 60% | [159] |
| S. coelicolor | germicidins | exogenous addition of ARC2 | Data not shown | [167] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Tian, P. Rethinking Biosynthesis of Aclacinomycin A. Molecules 2023, 28, 2761. https://doi.org/10.3390/molecules28062761
Xu Z, Tian P. Rethinking Biosynthesis of Aclacinomycin A. Molecules. 2023; 28(6):2761. https://doi.org/10.3390/molecules28062761
Chicago/Turabian StyleXu, Ziling, and Pingfang Tian. 2023. "Rethinking Biosynthesis of Aclacinomycin A" Molecules 28, no. 6: 2761. https://doi.org/10.3390/molecules28062761
APA StyleXu, Z., & Tian, P. (2023). Rethinking Biosynthesis of Aclacinomycin A. Molecules, 28(6), 2761. https://doi.org/10.3390/molecules28062761