
Impact of the Fukushima Accident on 3H and 14C Environmental Levels: A Review of Ten Years of Investigation

Abstract
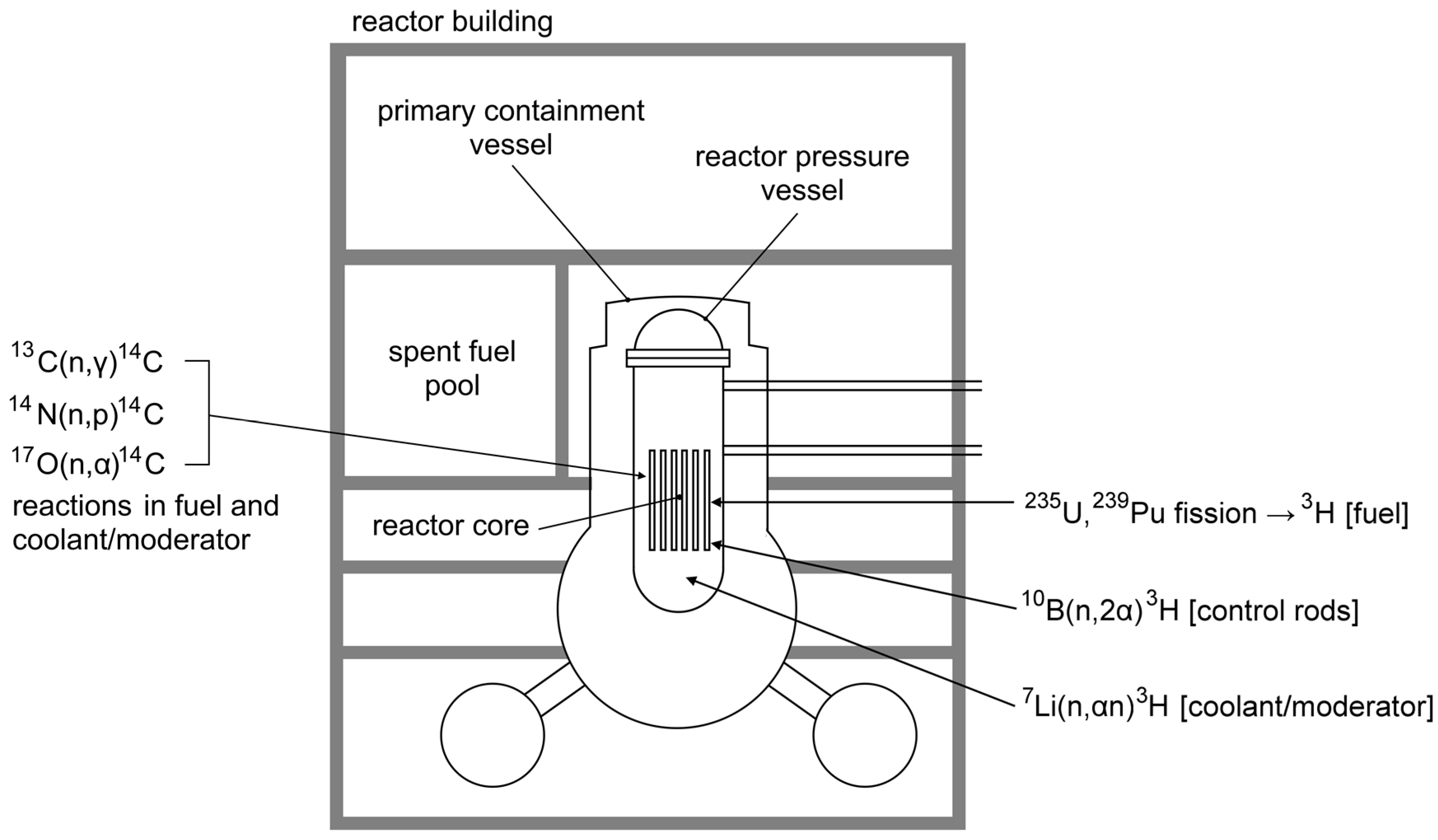
1. Introduction
2. Methods for the Determination of 3H and 14C
2.1. Liquid Scintillation Counting (LSC)
2.2. 3H-3He in-growth Mass Spectrometry
2.3. Accelerator Mass Spectrometry (AMS)
3. FDNPP Impact on Tritium Environmental Levels
3.1. The Concentration of 3H in Atmospheric Precipitation and Water Vapor
3.2. The Concentration of 3H in the Pacific Ocean and Coastal Seawater
3.3. The Concentration of 3H in Freshwater Systems
3.4. The Concentration of 3H in Biota and Other Samples
4. FDNPP Impact on Radiocarbon Environmental Levels
4.1. The Activity of 14C in the North Pacific Ocean and Coastal Seawater
4.2. The Activity of 14C in Tree Rings and Other Samples
5. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Povinec, P.P.; Hirose, K.; Aoyama, M.; Tateda, Y. Fukushima Accident: 10 Years After; Elsevier: New York, NY, USA, 2021. [Google Scholar]
- IAEA. The Fukushima Daiichi Accident; IAEA: Vienna, Austria, 2015. [Google Scholar]
- Steinhauser, G. Fukushima’s Forgotten Radionuclides: A Review of the Understudied Radioactive Emissions. Environ. Sci. Technol. 2014, 48, 4649–4663. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, G.; Brandl, A.; Johnson, T.E. Comparison of the Chernobyl and Fukushima Nuclear Accidents: A Review of the Environmental Impacts. Sci. Total Environ. 2014, 470–471, 800–817. [Google Scholar] [CrossRef] [PubMed]
- Masson, O.; Baeza, A.; Bieringer, J.; Brudecki, K.; Bucci, S.; Cappai, M.; Carvalho, F.P.; Connan, O.; Cosma, C.; Dalheimer, A.; et al. Tracking of Airborne Radionuclides from the Damaged Fukushima Dai-Ichi Nuclear Reactors by European Networks. Environ. Sci. Technol. 2011, 45, 7670–7677. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M. Long-Term Behavior of 137Cs and 3H Activities from TEPCO Fukushima NPP1 Accident in the Coastal Region off Fukushima, Japan. J. Radioanal. Nucl. Chem. 2018, 316, 1243–1252. [Google Scholar] [CrossRef]
- Tsumune, D.; Tsubono, T.; Aoyama, M.; Uematsu, M.; Misumi, K.; Maeda, Y.; Yoshida, Y.; Hayami, H. One-Year, Regional-Scale Simulation of 137Cs Radioactivity in the Ocean Following the Fukushima Dai-Ichi Nuclear Power Plant Accident. Biogeosciences 2013, 10, 5601–5617. [Google Scholar] [CrossRef]
- Aoyama, M.; Uematsu, M.; Tsumune, D.; Hamajima, Y. Surface Pathway of Radioactive Plume of TEPCO Fukushima NPP1 Released 134Cs and 137Cs. Biogeosciences 2013, 10, 3067–3078. [Google Scholar] [CrossRef]
- Kumamoto, Y.; Aoyama, M.; Hamajima, Y.; Murata, A.; Kawano, T. Impact of Fukushima-Derived Radiocesium in the Western North Pacific Ocean about Ten Months after the Fukushima Dai-Ichi Nuclear Power Plant Accident. J. Environ. Radioact. 2015, 140, 114–122. [Google Scholar] [CrossRef]
- UNSCEAR. Sources and Effects of Ionizing Radiation. Report of the United Nations Scientific Committee on the Effects of Atomic Radiation to the General Assembly; UNSCEAR: New York, NY, USA, 2008. [Google Scholar]
- IAEA. Worldwide Marine Radioactivity Studies (WOMARS): Radionuclide Levels in Oceans and Seas; IAEA: Vienna, Austria, 2005. [Google Scholar]
- Akata, N.; Kakiuchi, H.; Shima, N.; Iyogi, T.; Momoshima, N.; Hisamatsu, S. Tritium Concentrations in the Atmospheric Environment at Rokkasho, Japan before the Final Testing of the Spent Nuclear Fuel Reprocessing Plant. J. Environ. Radioact. 2011, 102, 837–842. [Google Scholar] [CrossRef]
- Muranaka, T.; Yamashita, J.; Shima, N. Variation of Tritium Concentration in Coastal Seawater Collected along the Pacific Coast in Aomori Prefecture. Fusion Sci. Technol. 2011, 60, 1264–1267. [Google Scholar] [CrossRef]
- Kokubon, Y.; Fujita, H.; Nkano, M.; Sumiya, S. Tritium Concentration and Diffusion in Seawater Discharged from Tokai Reprocessing Plant. Prog. Nucl. Sci. Technol. 2011, 1, 384–387. [Google Scholar] [CrossRef]
- Diabate, S.; Strack, S. Organically Bound Tritium. Health Phys. 1993, 65, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Workman, W.G.; Korolevych, V.; Davis, P.A. Field Measurements of Key Parameters Associated with Nocturnal OBT Formation in Vegetables Grown under Canadian Conditions. J. Environ. Radioact. 2012, 104, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Baglan, N.; Davis, P.A. Current Understanding of Organically Bound Tritium (OBT) in the Environment. J. Environ. Radioact. 2013, 126, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, J.; Bronić, I.K.; Todorović, N.; Stojković, I.; Barešić, J.; Petrović-Pantić, T. Tritium in Water: Hydrology and Health Implications. In Tritium: Advances in Research and Applications; Janković, M.M., Ed.; Nova Science Publishers: New York, NY, USA, 2018; pp. 157–212. [Google Scholar]
- ICRP. Age-Dependent Doses to Members of the Public from Intake of Radionuclides—Part 1. ICRP Publication 56; ICRP: Oxford, UK, 1990. [Google Scholar]
- Saljoughian, M.; Williams, P. Recent Developments in Tritium Incorporation for Radiotracer Studies. Curr. Pharm. Des. 2000, 6, 1029–1056. [Google Scholar] [CrossRef] [PubMed]
- Eastoe, C.J.; Watts, C.J.; Ploughe, M.; Wright, W.E. Future Use of Tritium in Mapping Pre-Bomb Groundwater Volumes. Ground Water 2012, 50, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Broecker, W.S.; Peng, T.H.; Ostlund, G. The Distribution of Bomb Tritium in the Ocean. J. Geophys. Res. 1986, 91, 14331. [Google Scholar] [CrossRef]
- Key, R.M.; Kozyr, A.; Sabine, C.L.; Lee, K.; Wanninkhof, R.; Bullister, J.L.; Feely, R.A.; Millero, F.J.; Mordy, C.; Peng, T.H. A Global Ocean Carbon Climatology: Results from Global Data Analysis Project (GLODAP). Global Biogeochem. Cycles 2004, 18, GB4031. [Google Scholar] [CrossRef]
- Zazzeri, G.; Yeomans, E.A.; Graven, H.D.; Acuña Yeomans, E.; Graven, H.D. Global and Regional Emissions of Radiocarbon from Nuclear Power Plants from 1972 to 2016. Radiocarbon 2018, 60, 1068–1081. [Google Scholar] [CrossRef]
- Rubin, S.I.; Key, R.M. Separating Natural and Bomb-Produced Radiocarbon in the Ocean: The Potential Alkalinity Method. Glob. Biogeochem. Cycles 2002, 16, 1105. [Google Scholar] [CrossRef]
- Sweeney, C.; Gloor, E.; Jacobson, A.R.; Key, R.M.; McKinley, G.; Sarmiento, J.L.; Wanninkhof, R. Constraining Global Air-Sea Gas Exchange for CO2 with Recent Bomb 14C Measurements. Glob. Biogeochem. Cycles 2007, 21, GB2015. [Google Scholar] [CrossRef]
- Broecker, W.S.; Sutherland, S.; Smethie, W.; Peng, T.-H.; Ostlund, G. Oceanic Radiocarbon: Separation of the Natural and Bomb Components. Glob. Biogeochem. Cycles 1995, 9, 263–288. [Google Scholar] [CrossRef]
- Peacock, S. Debate over the Ocean Bomb Radiocarbon Sink: Closing the Gap. Glob. Biogeochem. Cycles 2004, 18, GB2022. [Google Scholar] [CrossRef]
- Tauchi, H.; Toyoshima-Sasatani, M.; Nagashima, H.; Shimura, T.; Umata, T.; Tachibana, A. Tritium Biology in Japan: A Search for a New Approach. Fusion Eng. Des. 2018, 128, 28–32. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of Ionizing Radiation-Induced DNA Double-Strand Breaks by Non-Homologous End-Joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Stuiver, M.; Polach, H.A. Discussion; Reporting of 14C Data. Radiocarbon 1977, 19, 355–363. [Google Scholar] [CrossRef]
- Hou, X. Rapid Analysis of 14C and 3H in Graphite and Concrete for Decommissioning of Nuclear Reactor. Appl. Radiat. Isot. 2005, 62, 871–882. [Google Scholar] [CrossRef]
- Kakiuchi, H.; Momoshima, N.; Okai, T.; Maeda, Y. Tritium Concentration in Ocean. J. Radioanal. Nucl. Chem. 1999, 239, 523–526. [Google Scholar] [CrossRef]
- Nishihara, K.; Yamagishi, I.; Yasuda, K.; Ishimori, K.; Tanaka, K.; Kuno, T.; Inada, S.; Gotoh, Y. Radionuclide Release to Stagnant Water in the Fukushima-1 Nuclear Power Plant1. J. Nucl. Sci. Technol. 2015, 52, 301–307. [Google Scholar] [CrossRef]
- Xu, S.; Cook, G.T.; Cresswell, A.J.; Dunbar, E.; Freeman, S.P.H.T.; Hou, X.; Jacobsson, P.; Kinch, H.R.; Naysmith, P.; Sanderson, D.C.W.; et al. Radiocarbon Releases from the 2011 Fukushima Nuclear Accident. Sci. Rep. 2016, 6, 36947. [Google Scholar] [CrossRef]
- Povinec, P.P.; Hirose, K.; Aoyama, M. Fukushima Accident: Radioactivity Impact on the Environment; Elsevier: New York, NY, USA, 2013. [Google Scholar]
- Ješkovský, M.; Kaizer, J.; Kontuĺ, I.; Lujaniené, G.; Müllerová, M.; Povinec, P.P. Analysis of Environmental Radionuclides. In Handbook of Radioactivity Analysis: Volume 2; L’Annunziata, M.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 137–261. [Google Scholar]
- Hou, X.; Roos, P. Critical Comparison of Radiometric and Mass Spectrometric Methods for the Determination of Radionuclides in Environmental, Biological and Nuclear Waste Samples. Anal. Chim. Acta 2008, 608, 105–139. [Google Scholar] [CrossRef]
- Jakonić, I.; Todorović, N.; Nikolov, J.; Bronić, I.K.; Tenjović, B.; Vesković, M. Optimization of Low-Level LS Counter Quantulus 1220 for Tritium Determination in Water Samples. Radiat. Phys. Chem. 2014, 98, 69–76. [Google Scholar] [CrossRef]
- Lehto, J.; Hou, X. Chemistry and Analysis of Radionuclides: Laboratory Techniques and Methodology; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Vokal, B.; Kobal, I. Radiocarbon Releases at the Krško Nuclear Power Plant. Radiocarbon 1997, 39, 285–292. [Google Scholar] [CrossRef]
- Horvatinčić, N.; Barešić, J.; Krajcar Bronić, I.; Obelić, B. Measurement of Low 14 C Activities in a Liquid Scintillation Counter in the Zagreb Radiocarbon Laboratory. Radiocarbon 2004, 46, 105–116. [Google Scholar] [CrossRef]
- Tamers, M.A. Chemical Yield Optimization of the Benzene Synthesis for Radiocarbon Dating. Int. J. Appl. Radiat. Isot. 1975, 26, 676–682. [Google Scholar] [CrossRef]
- Horvatinčić, N.; Obelić, B.; Krajcar Bronić, I.; Srdoč, D.; Bistrović, R. Sources of Radon Contamination in 14C Dating. Radiocarbon 1995, 37, 749–757. [Google Scholar] [CrossRef]
- Hood, D.; Hatfield, R.; Patrick, C.; Stipp, J.; Tamers, M.; Leidl, R.; Lyons, B.; Polach, H.; Robertson, S.; Zhou, W. Radon Elimination During Benzene Preparation for Radiocarbon Dating by Liquid Scintillation Spectrometry. Radiocarbon 1989, 31, 254–259. [Google Scholar] [CrossRef][Green Version]
- Theodórsson, P. Simultaneously Measuring 14C and Radon in Benzene Dating Samples. Radiocarbon 2005, 47, 231–234. [Google Scholar] [CrossRef]
- Hou, X. Tritium and 14C in the Environment and Nuclear Facilities: Sources and Analytical Methods. J. Nucl. Fuel Cycle Waste Technol. 2018, 16, 11–39. [Google Scholar] [CrossRef]
- Clarke, W.B.; Jenkins, W.J.; Top, Z. Determination of Tritium by Mass Spectrometric Measurement of 3He. Int. J. Appl. Radiat. Isot. 1976, 27, 515–522. [Google Scholar] [CrossRef]
- Surano, K.A.; Hudson, G.B.; Failor, R.A.; Sims, J.M.; Holland, R.C.; MacLean, S.C.; Garrison, J.C. Heliuim-3 Mass Spectrometry for Low-Level Tritium Analysis of Environmental Samples. J. Radioanal. Nucl. Chem. Artic. 1992, 161, 443–453. [Google Scholar] [CrossRef]
- Jean-Baptiste, P.; Mantisi, F.; Dapoigny, A.; Stievenard, M. Design and Performance of a Mass Spectrometric Facility for Measuring Helium Isotopes in Natural Waters and for Low-Level Tritium Determination by the 3He Ingrowth Method. Int. J. Radiat. Appl. Instrum. Part 1992, 43, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Palcsu, L.; Major, Z.; Köllő, Z.; Papp, L. Using an Ultrapure 4He Spike in Tritium Measurements of Environmental Water Samples by the 3He-Ingrowth Method. Rapid Commun. Mass Spectrom. 2010, 24, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.; Palcsu, L.; Major, Z.; Rinyu, L.; Tóth, I. A Mass Spectrometric Line for Tritium Analysis of Water and Noble Gas Measurements from Different Water Amounts in the Range of Microlitres and Millilitres. Isotopes Environ. Health Stud. 2012, 48, 494–511. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.J.; McElroy, R.G.C.; Surette, R.A.; Brown, R.M. Tritium Sampling and Measurement. Health Phys. 1993, 65, 610–627. [Google Scholar] [CrossRef]
- Love, A.H.; Hunt, J.R.; Vogel, J.S.; Knezovich, J.P. Improving Tritium Exposure Reconstructions Using Accelerator Mass Spectrometry. Anal. Bioanal. Chem. 2004, 379, 198–203. [Google Scholar] [CrossRef][Green Version]
- Jull, A.J.T.; Burr, G.S. Accelerator Mass Spectrometry: Is the Future Bigger or Smaller? Earth Planet. Sci. Lett. 2006, 243, 305–325. [Google Scholar] [CrossRef]
- Povinec, P.P.; Litherland, A.E.; von Reden, K.F. Developments in Radiocarbon Technologies: From the Libby Counter to Compound-Specific AMS Analyses. Radiocarbon 2009, 51, 45–78. [Google Scholar] [CrossRef]
- Bronk Ramsey, C.; Ditchfield, P.; Humm, M. Using a Gas Ion Source for Radiocarbon AMS and GC-AMS. Radiocarbon 2004, 46, 25–32. [Google Scholar] [CrossRef]
- Fahrni, S.M.; Wacker, L.; Synal, H.A.; Szidat, S. Improving a Gas Ion Source for 14C AMS. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2013, 294, 320–327. [Google Scholar] [CrossRef]
- Povinec, P.P.; Masarik, J.; Ješkovský, M.; Kaizer, J.; Šivo, A.; Breier, R.; Pánik, J.; Staníček, J.; Richtáriková, M.; Zahoran, M.; et al. Development of the Accelerator Mass Spectrometry Technology at the Comenius University in Bratislava. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2015, 361, 87–94. [Google Scholar] [CrossRef]
- Slota, P.J.; Jull, A.J.T.; Linick, T.W.; Toolin, L.J. Preparation of Small Samples for 14C Accelerator Targets by Catalytic Reduction of CO. Radiocarbon 1987, 29, 303–306. [Google Scholar] [CrossRef]
- Xu, X.; Trumbore, S.E.; Zheng, S.; Southon, J.R.; McDuffee, K.E.; Luttgen, M.; Liu, J.C. Modifying a Sealed Tube Zinc Reduction Method for Preparation of AMS Graphite Targets: Reducing Background and Attaining High Precision. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2007, 259, 320–329. [Google Scholar] [CrossRef]
- Orsovszki, G.; Rinyu, L. Flame-Sealed Tube Graphitization Using Zinc as the Sole Reduction Agent: Precision Improvement of EnvironMICADAS 14C Measurements on Graphite Targets. Radiocarbon 2015, 57, 979–990. [Google Scholar] [CrossRef]
- Xu, X.; Gao, P.; Salamanca, E.G. Ultra Small-Mass Graphitization by Sealed Tube Zinc Reduction Method for AMS 14C Measurements. Radiocarbon 2013, 55, 608–616. [Google Scholar] [CrossRef]
- Matsumoto, T.; Maruoka, T.; Shimoda, G.; Obata, H.; Kagi, H.; Suzuki, K.; Yamamoto, K.; Mitsuguchi, T.; Hagino, K.; Tomioka, N.; et al. Tritium in Japanese Precipitation Following the March 2011 Fukushima Daiichi Nuclear Plant Accident. Sci. Total Environ. 2013, 445–446, 365–370. [Google Scholar] [CrossRef]
- Peterson, H.T.; Baker, D.A. Tritium Production, Releases and Population Doses at Nuclear Power Reactors. Fusion Technol. 1985, 8, 2544–2550. [Google Scholar] [CrossRef]
- Maruoka, T.; Kawamuto, T.; Ohno, T.; Muramatsu, Y.; Matsuzaki, H.; Matsumoto, T.; Aggarwal, P. Tritium and Iodine-129 Concentrations in Precipitation at Tsukuba, Japan, after the Fukushima Daiichi Nuclear Power Plant Accident. Geochem. J. 2017, 51, 449–455. [Google Scholar] [CrossRef]
- Sakakibara, K.; Iwagami, S.; Tsujimura, M.; Abe, Y.; Hada, M.; Pun, I.; Onda, Y. Groundwater Age and Mixing Process for Evaluation of Radionuclide Impact on Water Resources Following the Fukushima Dai-Ichi Nuclear Power Plant Accident. J. Contam. Hydrol. 2019, 223, 103474. [Google Scholar] [CrossRef]
- Kaizer, J.; Aoyama, M.; Kumamoto, Y.; Molnár, M.; Palcsu, L.; Povinec, P.P. Tritium and Radiocarbon in the Western North Pacific Waters: Post-Fukushima Situation. J. Environ. Radioact. 2018, 184–185, 83–94. [Google Scholar] [CrossRef]
- Querfeld, R.; Pasi, A.E.; Shozugawa, K.; Vockenhuber, C.; Synal, H.A.; Steier, P.; Steinhauser, G. Radionuclides in Surface Waters around the Damaged Fukushima Daiichi NPP One Month after the Accident: Evidence of Significant Tritium Release into the Environment. Sci. Total Environ. 2019, 689, 451–456. [Google Scholar] [CrossRef]
- Takahata, N.; Tomonaga, Y.; Kumamoto, Y.; Yamada, M.; Sano, Y. Direct Tritium Emissions to the Ocean from the Fukushima Dai-Ichi Nuclear Accident. Geochem. J. 2018, 52, 211–217. [Google Scholar] [CrossRef]
- Povinec, P.P.; Aoyama, M.; Biddulph, D.; Breier, R.; Buesseler, K.; Chang, C.C.; Golser, R.; Hou, X.L.; Ješkovský, M.; Jull, A.J.T.; et al. Cesium, Iodine and Tritium in NW Pacific Waters-a Comparison of the Fukushima Impact with Global Fallout. Biogeosciences 2013, 10, 5481–5496. [Google Scholar] [CrossRef]
- Ueda, S.; Hasegawa, H.; Kakiuchi, H.; Ochiai, S.; Akata, N.; Hisamatsu, S. Nuclear Accident-Derived 3H in River Water of Fukushima Prefecture during 2011–2014. J. Environ. Radioact. 2015, 146, 102–109. [Google Scholar] [CrossRef]
- Hirao, S.; Kakiuchi, H. Investigation of Atmospheric Tritiated Water Vapor Level around the Fukushima Daiichi Nuclear Power Plant. Fusion Eng. Des. 2021, 171, 112556. [Google Scholar] [CrossRef]
- Hirao, S.; Kakiuchi, H.; Akata, N.; Tamari, T.; Sugihara, S.; Shima, N.; Yokoyama, S.; Tanaka, M. Characterization of Atmospheric Tritiated Water Concentration in the Vicinity of the Fukushima Daiichi Nuclear Power Plant. J. Radioanal. Nucl. Chem. 2022, 331, 3077–3083. [Google Scholar] [CrossRef]
- Machida, M.; Iwata, A.; Yamada, S.; Otosaka, S.; Kobayashi, T.; Funasaka, H.; Takami, M. Estimation of Temporal Variation of Discharged Tritium from Port of Fukushima Dai-Ichi Nuclear Power Plant: Analysis of the Temporal Variation and Comparison with Released Tritium from Japan and Major Nuclear Facilities Worldwide. Trans. At. Energy Soc. Japan 2022, 21, J20.036. [Google Scholar] [CrossRef]
- Povinec, P.P.; Liong Wee Kwong, L.; Kaizer, J.; Molnár, M.; Nies, H.; Palcsu, L.; Papp, L.; Pham, M.K.; Jean-Baptiste, P. Impact of the Fukushima Accident on Tritium, Radiocarbon and Radiocesium Levels in Seawater of the Western North Pacific Ocean: A Comparison with Pre-Fukushima Situation. J. Environ. Radioact. 2017, 166, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Kajino, M.; Tanaka, T.Y.; Sekiyama, T.T.; Tsumune, D.; Tsubono, T.; Hamajima, Y.; Inomata, Y.; Gamo, T. 134Cs and 137Cs in the North Pacific Ocean Derived from the March 2011 TEPCO Fukushima Dai-Ichi Nuclear Power Plant Accident, Japan. Part Two: Estimation of 134Cs and 137Cs Inventories in the North Pacific Ocean. J. Oceanogr. 2016, 72, 67–76. [Google Scholar] [CrossRef]
- Shirotani, Y.; Inatomi, N.; Miyamoto, K.; Yamada, M.; Kusakabe, M. Distributions of Tritium in the Coastal Waters off Aomori and Iwate Prefectures. Fusion Eng. Des. 2021, 172, 112738. [Google Scholar] [CrossRef]
- Kumamoto, Y.; Murata, A.; Kawano, T.; Aoyama, M. Fukushima-Derived Radiocesium in the Northwestern Pacific Ocean in February 2012. Appl. Radiat. Isot. 2013, 81, 335–339. [Google Scholar] [CrossRef]
- Kaizer, J.; Kumamoto, Y.; Molnár, M.; Palcsu, L.; Povinec, P.P. Temporal Changes in Tritium and Radiocarbon Concentrations in the Western North Pacific Ocean (1993–2012). J. Environ. Radioact. 2020, 218, 106238. [Google Scholar] [CrossRef]
- Kashiwaya, K.; Muto, Y.; Kubo, T.; Ikawa, R.; Nakaya, S.; Koike, K.; Marui, A. Spatial Variations of Tritium Concentrations in Groundwater Collected in the Southern Coastal Region of Fukushima, Japan, after the Nuclear Accident. Sci. Rep. 2017, 7, 12578. [Google Scholar] [CrossRef] [PubMed]
- Shozugawa, K.; Hori, M.; Johnson, T.E.; Takahata, N.; Sano, Y.; Kavasi, N.; Sahoo, S.K.; Matsuo, M. Landside Tritium Leakage over through Years from Fukushima Dai-Ichi Nuclear Plant and Relationship between Countermeasures and Contaminated Water. Sci. Rep. 2020, 10, 19925. [Google Scholar] [CrossRef]
- Aoyama, M.; Thébault, H.; Hamajima, Y.; Charmasson, S.; Arnaud, M.; Duffa, C. 137Cs and Tritium Concentrations in Seawater off the Fukushima Prefecture: Results from the SOSO 5 Rivers Cruise (October 2014). In Oceanography Challenges to Future Earth; Springer International Publishing: Cham, Switzerland, 2019; pp. 407–409. [Google Scholar]
- Sakuma, K.; Machida, M.; Kurikami, H.; Iwata, A.; Yamada, S.; Iijima, K. A Modeling Approach to Estimate 3H Discharge from Rivers: Comparison of Discharge from the Fukushima Dai-Ichi and Inventory in Seawater in the Fukushima Coastal Region. Sci. Total Environ. 2022, 806, 151344. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, T.; Kakiuchi, H.; Tauchi, H.; Yamamoto, T.; Yamamoto, I. Discussions on Tritiated Water Treatment for Fukushima Daiichi Nuclear Power Station. Fusion Sci. Technol. 2020, 76, 430–438. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimada, A.; Hoshi, A.; Yasuda, M.; Ozawa, M.; Kameo, Y. Radiochemical Analysis of Rubble and Trees Collected from Fukushima Daiichi Nuclear Power Station. J. Nucl. Sci. Technol. 2014, 51, 1032–1043. [Google Scholar] [CrossRef]
- Kuwata, H.; Misono, T.; Fujiwara, K.; Takeishi, M.; Manabe, S.; Kitamura, A. Rapid Tritium Analysis for Marine Products in the Coastal Area of Fukushima. Radiat. Environ. Med. 2020, 9, 28–34. [Google Scholar]
- Tatebe, H.; Yasuda, I. Oyashio Southward Intrusion and Cross-Gyre Transport Related to Diapycnal Upwelling in the Okhotsk Sea. J. Phys. Oceanogr. 2004, 34, 2327–2341. [Google Scholar] [CrossRef]
- Ding, L.; Ge, T.; Gao, H.; Luo, C.; Xue, Y.; Druffel, E.R.M.; Wang, X. Large Variability of Dissolved Inorganic Radiocarbon in the Kuroshio Extension of the Northwest North Pacific. Radiocarbon 2018, 60, 691–704. [Google Scholar] [CrossRef]
- Xu, S.; Cook, G.T.; Cresswell, A.J.; Dunbar, E.; Freeman, S.P.H.T.; Hastie, H.; Hou, X.; Jacobsson, P.; Naysmith, P.; Sanderson, D.C.W.; et al. 14C Levels in the Vicinity of the Fukushima Dai-Ichi Nuclear Power Plant Prior to the 2011 Accident. J. Environ. Radioact. 2016, 157, 90–96. [Google Scholar] [CrossRef]
- Chen, B.; Xu, S.; Cook, G.T.; Freeman, S.P.H.T.; Hou, X.; Philip, C.L.; Katsuhiko, N. Local Variance of Atmospheric 14C Concentrations around Fukushima Dai-Ichi Nuclear Power Plant from 2010 to 2012. J. Radioanal. Nucl. Chem. 2017, 314, 1001–1007. [Google Scholar] [CrossRef]
- Varga, T.; Palcsu, L.; Ohta, T.; Mahara, Y.; Jull, A.J.T.; Molnár, M. Variation of 14C in Japanese Tree Rings Related to the Fukushima Nuclear Accident. Radiocarbon 2019, 61, 1029–1040. [Google Scholar] [CrossRef]
- Aramaki, T.; Nakaoka, S.; Terao, Y.; Kushibashi, S.; Kobayashi, T.; Osonoi, Y.; Mukai, H.; Tohjima, Y. Variation of Surface Radiocarbon in the North Pacific During Summer Season 2004–2016. Radiocarbon 2019, 61, 1367–1375. [Google Scholar] [CrossRef]
- Key, R.M.; Quay, P.D.; Schlosser, P.; McNichol, A.P.; von Reden, K.; Schneider, R.J.; Elder, K.L.; Stuiver, M.; Göte Östlund, H. Woce Radiocarbon IV: Pacific Ocean Results; P10, P13N, P14C, P18, P19 & S4P. Radiocarbon 2002, 44, 239–392. [Google Scholar]
- Kumamoto, Y.; Murata, A.; Kawano, T.; Watanabe, S.; Fukasawa, M. Decadal Changes in Bomb-Produced Radiocarbon in the Pacific Ocean from the 1990s to 2000s. Radiocarbon 2013, 55, 1641–1650. [Google Scholar] [CrossRef]
- Xu, S.; Cook, G.T.; Cresswell, A.J.; Dunbar, E.; Freeman, S.P.H.T.; Hastie, H.; Hou, X.; Jacobsson, P.; Naysmith, P.; Sanderson, D.C.W. Radiocarbon Concentration in Modern Tree Rings from Fukushima, Japan. J. Environ. Radioact. 2015, 146, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Matsunaka, T.; Sasa, K.; Takahashi, T.; Matsumura, M.; Satou, Y.; Shen, H.; Sueki, K.; Matsuzaki, H. Pre-and Post-Accident 14C Activities in Tree Rings near the Fukushima Dai-Ichi Nuclear Power Plant. Radiocarbon 2019, 61, 1633–1642. [Google Scholar] [CrossRef]
- Paterne, M.; Evrard, O.; Hatté, C.; Laceby, P.J.; Nouet, J.; Onda, Y. Radiocarbon and Radiocesium in Litter Fall at Kawamata, ~45 Km NW from the Fukushima Dai-Ichi Nuclear Power Plant (Japan). J. Radioanal. Nucl. Chem. 2019, 319, 1093–1101. [Google Scholar] [CrossRef]
- Buesseler, K.O. Opening the floodgates at Fukushima. Science 2020, 369, 621–622. [Google Scholar] [CrossRef]
- Men, W. Discharge of contaminated water from the Fukushima Daiichi Nuclear Power Plant Accident into the Northwest Pacific: What is known and what needs to be known. Mar. Pollut. Bull. 2021, 173, 112984. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, G.; Zhang, M.; Wang, G.; de With, G.; Bezhenar, R.; Maderich, V.; Xia, C.; Zhao, B.; Jung, K.T.; et al. Transport and dispersion of tritium from the radioactive water of the Fukushima Daiichi nuclear plant. Mar. Pollut. Bull. 2021, 169, 112515. [Google Scholar] [CrossRef] [PubMed]
- Bezhenar, R.; Takata, H.; de With, G.; Maderich, V. Planned release of contaminated water from the Fukushima storage tanks into the ocean: Simulation scenarios of radiological impact for aquatic biota and human from seafood consumption. Mar. Pollut. Bull. 2021, 173, 112969. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Unit Number | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Reactor type | BWR-3 | BWR-4 | BWR-4 | BWR-4 |
| Type of fuel | LEU | LEU | MOX | LEU |
| Moderator | Light water | |||
| Coolant | Light water | |||
| Start of operation | March 1971 | July 1974 | March 1976 | October 1978 |
| Thermal output (MWt) | 1380 | 2381 | 2381 | 2381 |
| Electric output (MWe) | 460 | 784 | 784 | 784 |
| No. of fuel assemblies | 400 | 548 | 548 | 548 |
| Status prior to the accident | Operational | Operational | Operational | Shutdown |
| Estimated total core inventory (EBq) | 14.4 | 24.4 | 24.5 | - |
| Location | Direction from the FDNPP | Distance from the FDNPP | Δ14C (‰) | 14C activity (Bq kg−1 C) | Reference |
|---|---|---|---|---|---|
| Okuma | SW | 1 km | 40.3 | 233.4 | [95] |
| 37.5 | 232.8 | [97] | |||
| 256.5 | 284.0 | [97] | |||
| 266.8 | 285.4 | [97] | |||
| Futaba | NW | 2.5 km | 152.4 | 258.2 | [35] |
| Namie | N | 8 km | 23.3 | 230.7 | [35] |
| Tomioka | S | 9 km | 48.6 | 236.3 | [97] |
| 73.5 | 241.9 | [97] | |||
| Takase | NW | 11 km | 65.7 | 240.0 | [35] |
| Ogaki | NW | 14 km | 57.2 | 237.8 | [35] |
| Shimotsushima | NW | 32 km | 38.4 | 232.7 | [35] |
| Yamakiya | NW | 38 km | 28.9 | 232.9 | [35] |
| Iwaki | SW | 50 km | 32.9 | 232.2 | [35] |
| Koriyama | W | 60 km | 36.3 | 233.1 | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaizer, J.; Kontuľ, I.; Povinec, P.P. Impact of the Fukushima Accident on 3H and 14C Environmental Levels: A Review of Ten Years of Investigation. Molecules 2023, 28, 2548. https://doi.org/10.3390/molecules28062548
Kaizer J, Kontuľ I, Povinec PP. Impact of the Fukushima Accident on 3H and 14C Environmental Levels: A Review of Ten Years of Investigation. Molecules. 2023; 28(6):2548. https://doi.org/10.3390/molecules28062548
Chicago/Turabian StyleKaizer, Jakub, Ivan Kontuľ, and Pavel P. Povinec. 2023. "Impact of the Fukushima Accident on 3H and 14C Environmental Levels: A Review of Ten Years of Investigation" Molecules 28, no. 6: 2548. https://doi.org/10.3390/molecules28062548
APA StyleKaizer, J., Kontuľ, I., & Povinec, P. P. (2023). Impact of the Fukushima Accident on 3H and 14C Environmental Levels: A Review of Ten Years of Investigation. Molecules, 28(6), 2548. https://doi.org/10.3390/molecules28062548