3. Material and Methods
3.1. Synthesis—General
All commercial reagents were used without purification. NMR spectra were recorded using a Bruker DPX-300 spectrometer in CDCl
3 (
1H: 300 MHz;
13C: 75 MHz). Chemical shifts are reported as parts per million (δ, ppm). The residual solvent peak (CHCl
3 or DMSO-
d6) was used as the internal standard: 7.28 or 2.51 for
1H and 77.07 or 40.00 ppm for
13C. Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet/doublets of doublets (see
Supplementary Materials). Coupling constants, J, are reported in Hz. Mass spectra were recorded using a Bruker microTOF spectrometer (ionization by electrospray, positive ion detection). Melting points were determined in open capillary tubes using a Stuart SMP50 Automatic Melting Point Apparatus. Analytical thin-layer chromatography was carried out on UV-254 silica gel plates using appropriate eluents. Compounds were visualized with short-wavelength UV light. Column chromatography was performed using silica gel Merk grade 60 (0.040–0.063 mm) 230–400 mesh. All reactions were conducted under an atmosphere of argon.
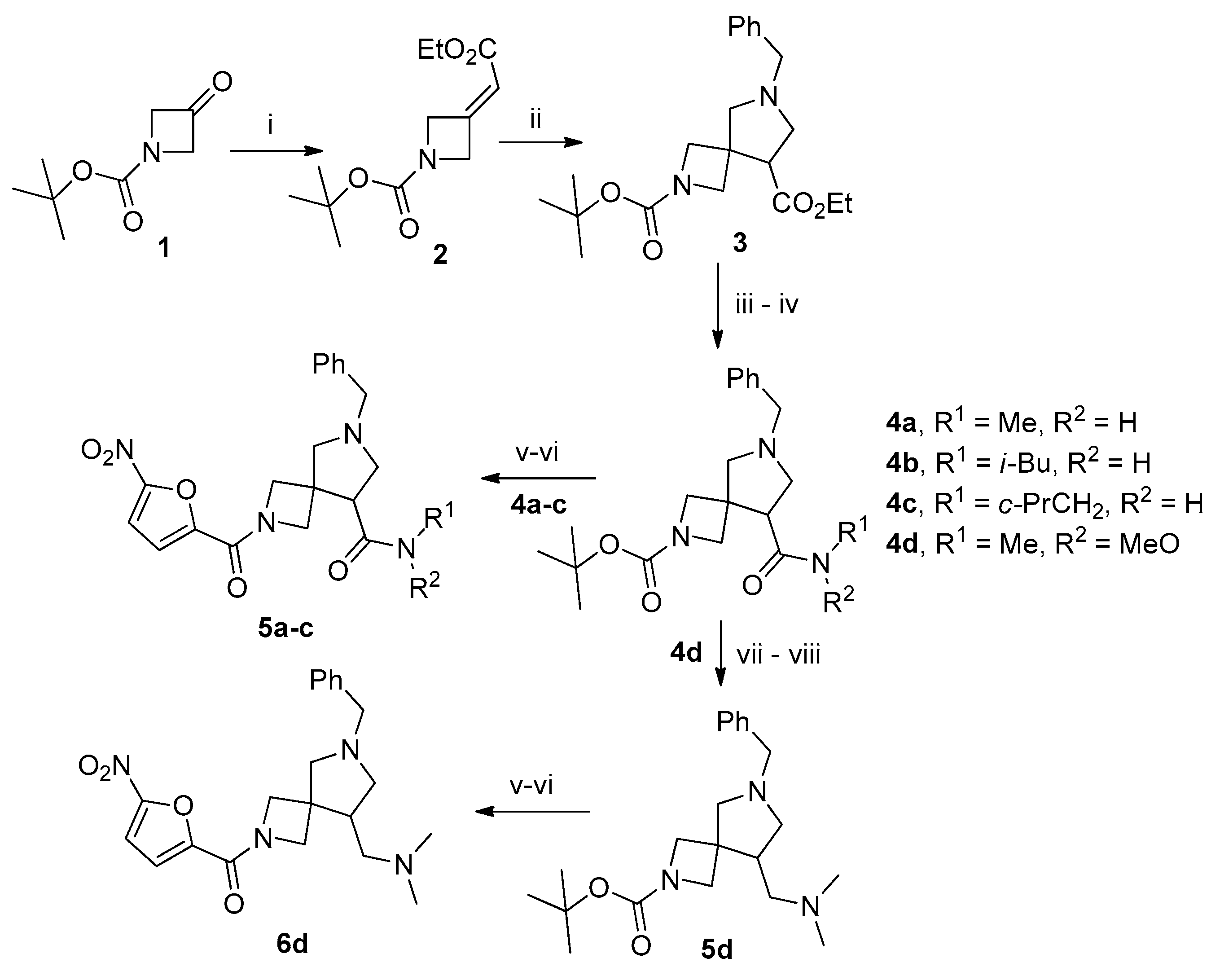
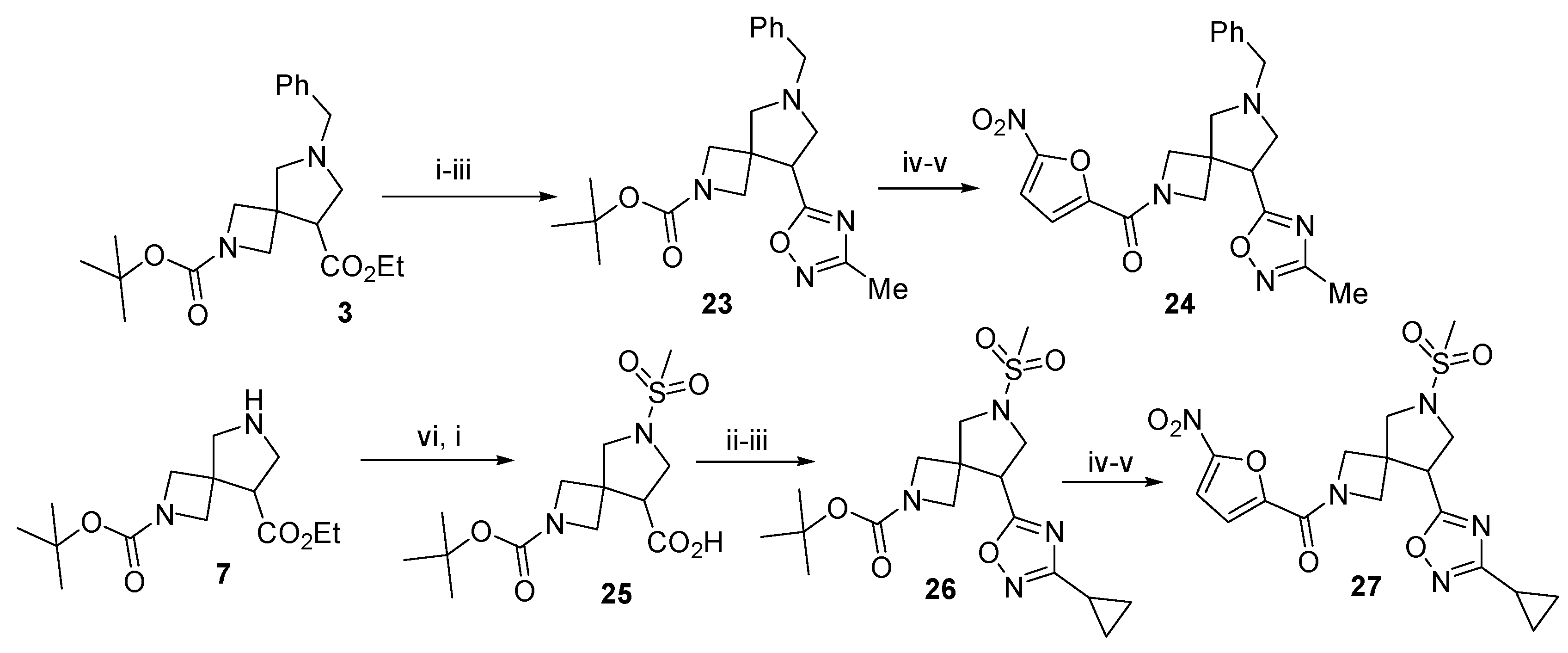
3.2. 2-(tert-Butyl) 8-ethyl 6-benzyl-2,6-diazaspiro[3.4]octane-2,8-dicarboxylate (3)
Prepared according to the literature procedure [
10] with minor modifications. To a suspension of NaH (60% dispersion in mineral oil, 1.88 g, 0.047 mol, 1.15 equiv.) in THF (150 mL) at 0 °C, triethylphosphonoacetate (11 g, 0.054 mol, 1.2 equiv.) was added. The resulting mixture was allowed to warm up to r. t. and stirred at that temperature for 30 min. It was cooled down to 0 °C, at which point
tert-butyl 3-oxazetidine-1-carboxylate (7 g, 0.041 mol, 1.0 equiv.) in THF (50 mL) was added. The reaction mixture was allowed to reach r. t. again and stirred at that temperature overnight. It was then diluted with ethyl acetate and washed with sat. aq. NaHCO
3, water, and brine. The organic phase was separated and dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica, eluting with 0→10% ethyl acetate in hexane. Yield of
2—9 g (92%), colorless oil. The spectral data of compound
2 were in accordance with those reported in the literature:
1H NMR (300 MHz, CDCl
3) δ 5.74 (dd,
J = 4.5, 2.2 Hz, 1H), 4.80 (dd,
J = 6.3, 2.9 Hz, 2H), 4.57 (dt,
J = 5.3, 2.7 Hz, 2H), 4.31–4.02 (m, 2H), 1.45 (s, 9H), 1.26 (t,
J = 7.1 Hz, 3H); LCMS (ESI):
m/z (M + H) calcd, 242.3; found, 242.2.
To a solution of 2 (5 g, 20.75 mmol, 1 equiv.) in acetonitrile (50 mL), LiF (2.15 g, 83 mmol, 4 equiv.) and (methoxymethyl)-1-phenyl-N-(trimethylsilylmethyl)methanamine (6.25 g, 25 mmol, 1.2 equiv.) were added and the resulting mixture was stirred at 60 °C overnight. The solvent was removed in vacuo and the residue was dissolved in ethyl acetate (50 mL). The solution was washed with sat. aq. citric acid (3 × 25 mL). The combined aqueous phases were extracted with ethyl acetate (2 × 100 mL). The pH of the aqueous phase was brought to 8 with sat. aq. K2CO3 and it was extracted with ethyl acetate (2 × 100 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give 4.3 g (56%) of the title product as a colorless oil. This product was used in subsequent steps without further purification. The spectral data of compound 3 were in accordance with those reported in the literature: 1H NMR (300 MHz, DMSO-d6) δ 7.46–7.14 (m, 5H), 4.22–4.00 (m, 2H), 3.94 (dd, J = 16.6, 8.4 Hz, 2H), 3.78–3.51 (m, 5H), 3.12 (t, J = 7.6 Hz, 1H), 2.83–2.66 (m, 4H), 1.36 (s, 9H), 1.19 (t, J = 7.1 Hz, 3H); LCMS (ESI): m/z (M + H+) calcd, 375.5; found, 375.4.
3.3. 2-tert-Butoxycarbonyl-6-benzyl-8-(methylcarbamoyl)-2,6-diazaspiro[3.4]octane (4a)
General Procedure A. To a solution of 3 (1 g, 3.35 mmol, 1.0 equiv.) in 1,4-dioxane (10 mL) was added aqueous solution (1 mL) of LiOH∙H2O (0.175 g, 4.2 mmol, 1.25 equiv.) and the resulting mixture was stirred for 12 h. HOBt (0.56 g, 4.2 mmol,1.25 equiv.), EDC∙HCl (0.81 g, 4.2 mmol, 1.25 equiv.), and methylamine (1.4 equiv.) were added and the resulting mixture was stirred for 12 h. The solvent was removed in vacuo and the residue was dissolved in ethyl acetate (50 mL). The solution was washed with 10% aqueous K2CO3, brine, dried over anhydrous Na2SO4,, filtered, and concentrated in vacuo to obtain 0.68 g (66%) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.38–7.19 (m, 5H), 7.00 (s, 1H), 3.88 (d, J = 4.4 Hz, 1H), 3.85 (d, J = 5.0 Hz, 1H), 3.79 (d, J = 8.4 Hz, 1H), 3.69 (d, J = 9.1 Hz, 1H), 3.63 (s, 1H), 3.01 (d, J = 9.7 Hz, 1H), 2.89–2.62 (m, 6H), 1.39 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 173.52, 156.08, 138.07, 128.53, 128.45, 127.33, 79.56, 65.33, 59.44, 57.16, 53.85, 42.07, 28.28, 25.99; HRMS (ESI) m/z calcd for C20H30N3O3 [M + H+] 360.2287, found 360.2291.
3.4. 2-tert-Butoxycarbonyl-6-benzyl-8-(isobutylcarbamoyl)-2,6-diazaspiro[3.4]octane (4b)
This compound was synthesized according to general procedure A from 3 (0.5 g, 1.33 mmol). Yield 0.36 g (68%), colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.40–7.10 (m, 6H), 3.90 (d, J = 8.9 Hz, 2H), 3.80 (d, J = 8.5 Hz, 1H), 3.69 (d, J = 9.1 Hz, 1H), 3.63 (s, 2H), 3.15–2.98 (m, 3H), 2.92 (dd, J = 9.6, 2.7 Hz, 1H), 2.80 (dd, J = 6.5, 2.7 Hz, 1H), 2.73–2.52 (m, 2H), 1.82–1.60 (m, 1H), 1.39 (s, 9H), 0.87 (dd, J = 6.7, 1.0 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 178.85, 167.17, 155.96, 138.18, 128.53, 128.37, 127.24, 79.69, 64.33, 59.41, 57.47, 43.93, 43.22, 29.65, 28.26, 11.60, −0.05; HRMS (ESI) m/z calcd for C23H36N3O3 [M + H+] 402.2756, found 402.2761.
3.5. 2-tert-Butoxycarbonyl-6-benzyl-N-(cyclopropylmethyl)-2,6-diazaspiro[3.4]octane-8-carboxamide (4c)
This compound was synthesized according to general procedure A from 3 (0.5 g, 1.33 mmol). Yield 0.38 g (72%), colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.40–7.14 (m, 5H), 3.91 (d, J = 9.2 Hz, 2H), 3.81 (d, J = 8.5 Hz, 1H), 3.70 (d, J = 9.0 Hz, 1H), 3.64 (d, J = 1.8 Hz, 1H), 3.14–3.02 (m, 2H), 2.92 (dd, J = 9.5, 2.8 Hz, 1H), 2.80 (dd, J = 6.6, 2.9 Hz, 1H), 2.72–2.57 (m, 2H), 1.03–0.77 (m, 1H), 0.57–0.35 (m, 2H), 0.25–0.08 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 172.72, 138.13, 128.48, 128.44, 127.35, 79.52, 65.67, 59.49, 57.17, 54.05, 43.98, 41.98, 28.27, 10.71, 3.39; HRMS (ESI) m/z calcd for C23H34N3O3 [M + H+] 400.2600, found 400.2597.
3.6. 2-tert-Butoxycarbonyl-1-(6-benzyl-2,6-diazaspiro[3.4]oct-8-yl)-N,N-dimethylmethanamine (5d)
Compound 4d was synthesized according to general procedure A from 3 (1 g, 2.66 mmol) and used in the next step without further purification. A solution of this compound (0.7 g, 1.8 mmol, 1 equiv.) in absolute THF (3 mL) was added to a suspension of LAH (0.07 g, 1.8 mmol, 1 equiv.) in THF (10 mL) at −70 °C. The reaction mixture was stirred at that temperature for 30 min and allowed to reach −5 °C. Then, it was cooled to −30 °C and decomposed by adding water (0.1 mL), 15% aq. NaOH (0.1 mL), and water (0.3 mL). The resulting precipitate was filtered off and the filtrate was concentrated in vacuo. The resulting aldehyde was used immediately in the next step. It was dissolved in CH2Cl2 (10 mL) and the solution was treated, on vigorous stirring, with 33% aq. dimethylamine (0.5 mL). Sodium triacetoxyborohydride (0.96 g, 4.5 mmol, 2.5 equiv.) was added in portions and the resulting mixture was stirred overnight. Sat. aq. NaHCO3 was added and the resulting mixture was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica, eluting with 10% methanol in CH2Cl2 to give 0.25 g (72%) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.47–7.06 (m, 5H), 4.07 (d, J = 8.8 Hz, 1H), 3.78 (s, 2H), 3.66–3.48 (m, 3H), 3.01–2.91 (m, 1H), 2.89 (d, J = 9.3 Hz, 1H), 2.55 (d, J = 9.3 Hz, 1H), 2.45–2.30 (m, 1H), 2.30–2.15 (m, 9H), 1.42 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 156.38, 138.61, 128.65, 128.22, 126.97, 79.28, 65.56, 61.35, 60.07, 59.18, 45.67, 41.99, 41.73, 28.33; HRMS (ESI) m/z calcd for C21H34N3O2 [M + H+] 360.2651, found 360.2655.
3.7. 2-tert-Butoxycarbonyl-8-(1-benzyl-5-methyl-1H-imidazol-2-yl)-6-methyl-2,6-diazaspiro[3.4]octane (10)
To a solution of 3 (2 g, 5.3 mmol) in ethanol (25 mL) was added 10% Pd/C (0.25 g) and the mixture was hydrogenated in an autoclave at a start pressure of 100 atm and r. t. for 12 h. The reaction mixture was filtered through a plug of Celite and the filtrate was concentrated in vacuo. The yield of compound 7 was 1.6 g (quantitative, assuming analytical purity), obtained as a colorless oil. It was used directly in the next step without further purification.
To a vigorously stirred solution of 7 (1.6 g, 5.3 mmol, 1 equiv.) in CH2Cl2 (25 mL), 37% aq. formaldehyde solution (1 mL) was added followed by portion-wise addition of sodium triacetoxyborohydride (2.8 g, 13.25 mmol, 2.5 equiv.). The resulting mixture was stirred overnight. Sat. aq. NaHCO3 (10 mL) was added, whereupon the mixture was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The resulting product 8 (1.2 g, 72%, assuming analytical purity) was used in the next step without further purification.
To a solution of 8 (1 g, 3.35 mmol, 1.0 equiv.) in 1,4-dioxane (10 mL), aq. solution of LiOH∙H2O (1 mL, 0.175 g, 4.2 mmol, 1.25 equiv.) was added and the resulting mixture was stirred for 12 h. Thereupon HOBt (0.56 g, 4.2 mmol, 1.25 equiv.), EDC∙HCl (0.81 g, 4.2 mmol, 1.25 equiv.), and propargylamine (0.29 g, 4.7 mmol, 1.4 equiv.) were added. The resulting mixture was stirred for 12 h. The solvent was removed in vacuo and the residue was dissolved in ethyl acetate (50 mL). The solution was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give 0.68 g (66%, assuming analytical purity) of product 9 as a colorless oil, which was used in the next step without further purification.
To a solution of compound 9 (0.3 g, 0.97 mmol, 1 equiv.) in toluene (10 mL), benzylamine (0.13 g, 0.24 mmol, 1.25 equiv.) was added followed by Zn(OTf)2 (0.088 g, 1.21 mmol, 0.25 equiv.). The mixture was heated under reflux with a Dean-Stark trap over 8 h. Upon cooling to r. t., the reaction mixture was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (0.29 g, 75%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.35–7.19 (m, 3H), 6.96–6.86 (m, 2H), 6.83 (s, 1H), 5.19–4.93 (m, 2H), 3.77 (d, J = 8.6 Hz, 1H), 3.60 (d, J = 9.5 Hz, 1H), 3.47–3.29 (m, 3H), 3.11 (d, J = 9.3 Hz, 1H), 3.05 (t, J = 8.6 Hz, 1H), 2.33 (s, 3H), 2.15 (d, J = 0.7 Hz, 3H), 1.39 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 155.97, 147.40, 136.58, 129.02, 127.85, 127.40, 125.84, 125.47, 79.24, 68.07, 62.57, 46.39, 44.59, 44.36, 41.55, 28.31, 9.70; HRMS (ESI) m/z calcd for C23H33N4O2 [M + H+] 397.2603, found 397.2598.
3.8. 2-tert-Butoxycarbonyl-6-methyl-8-(5-methyloxazol-2-yl)-2,6-diazaspiro[3.4]octane (11)
To a solution of compound 9 (0.3 g, 0.97 mmol, 1 equiv.) in toluene (10 mL), Zn(OTf)2 (0.088 g, 1.21 mmol, 0.25 equiv.) was added and the mixture was heated under reflux with a Dean-Stark trap over 16 h. Upon cooling to r. t., the mixture was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (0.14 g, 47%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.63 (d, J = 1.2 Hz, 1H), 4.00 (d, J = 8.6 Hz, 1H), 3.89 (d, J = 8.6 Hz, 1H), 3.60 (d, J = 9.2 Hz, 1H), 3.51 (t, J = 8.0 Hz, 1H), 3.41 (d, J = 9.2 Hz, 1H), 3.18–3.10 (m, 1H), 3.06 (d, J = 9.4 Hz, 1H), 2.82–2.70 (m, 2H), 2.39 (s, 3H), 2.27 (s, 3H), 1.40 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 162.33, 156.09, 148.93, 122.69, 79.38, 67.35, 59.58, 46.23, 43.93, 41.86, 28.26, 10.79; HRMS (ESI) m/z calcd for C16H26N3O3 [M + H+] 308.1974, found 308.1980.
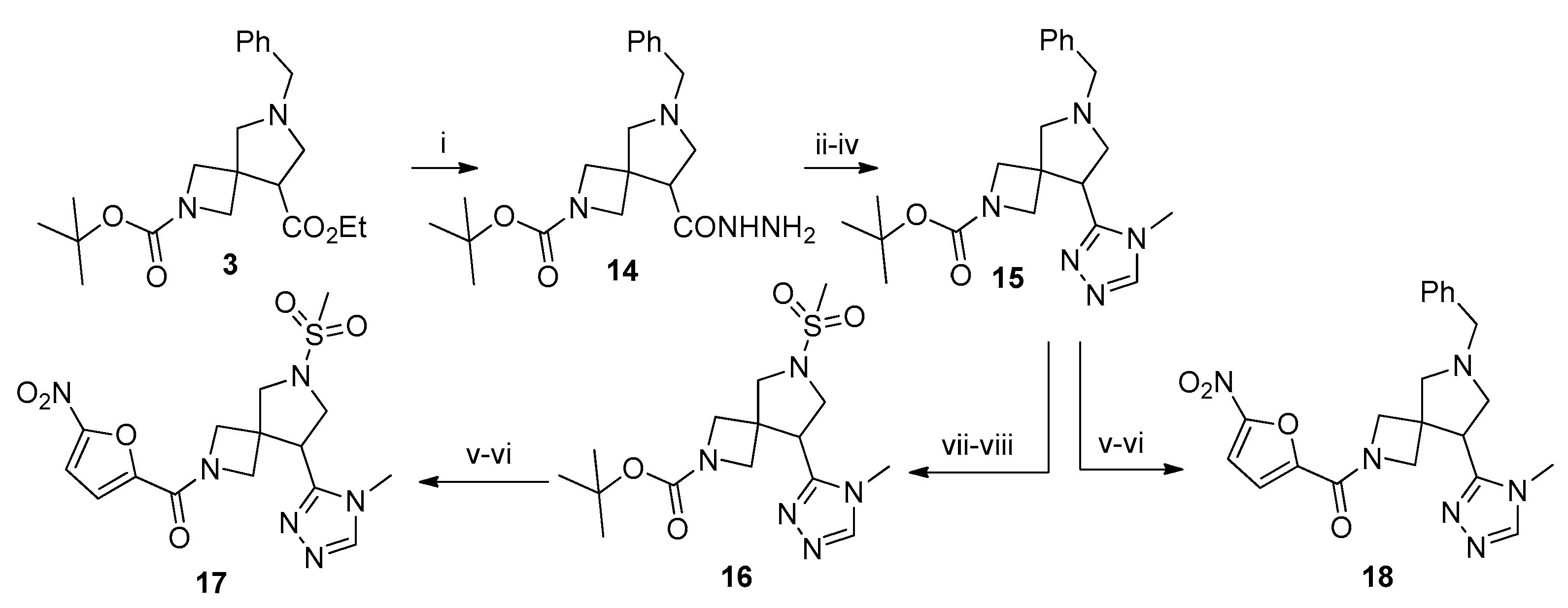
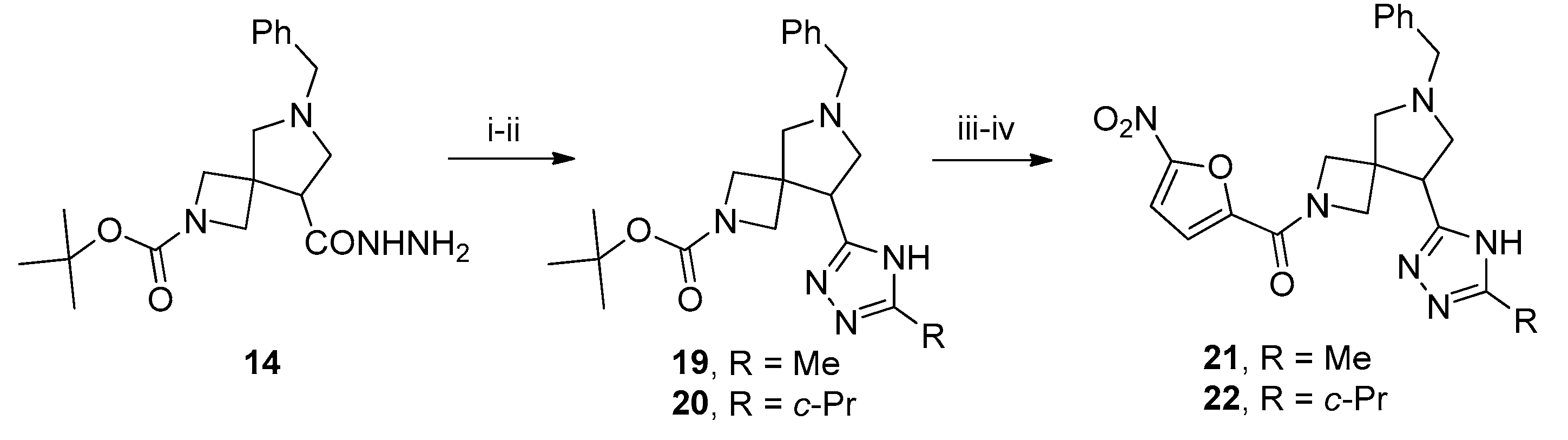
3.9. 2-tert-Butyl 6-benzyl-8-(4-methyl-4H-1,2,4-triazol-3-yl)-2,6-diazaspiro[3.4]octane-2-carboxylate (15)
To a solution of 3 (3 g, 8 mmol) in ethanol (25 mL), N2H4 (64% aq. solution, 1 mL) was added. The reaction mixture was heated under reflux for 8 h. Upon cooling to r. t., the mixture was concentrated in vacuo to give compound 14 (2.8 g, quant., assuming analytical purity), which was used in the next step without further purification.
To a solution of 14 (2 g, 5.5 mmol, 1 equiv.) in ethanol (25 mL), CH3NCS (0.5 g, 6.8 mmol, 1.25 equiv.) was added dropwise and the resulting mixture was heated under reflux for 2 h. Sat. aq. K2CO3 (5 mL) was added and refluxing continued for 8 h. The reaction mixture was concentrated in vacuo, the residue was dissolved in water (25 mL), and the solution was acidified to pH 7 with 5% aq. HCl. The resulting precipitate was filtered off and dissolved in ethanol (25 mL). A suspension of freshly prepared Raney nickel in a minimum amount of ethanol was added and the mixture was heated under reflux with vigorous stirring for 12 h. Upon cooling to r. t., the reaction mixture was filtered through a plug of Celite and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (1.22 g, 58%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.08 (s, 1H), 7.39–7.13 (m, 5H), 4.01–3.84 (m, 2H), 3.74–3.59 (m, 6H), 3.53 (t, J = 8.0 Hz, 1H), 3.35 (d, J = 9.5 Hz, 1H), 3.28 (t, J = 8.8 Hz, 1H), 3.12 (d, J = 9.4 Hz, 1H), 2.97–2.86 (m, 1H), 2.80 (d, J = 9.4 Hz, 1H), 1.37 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 156.25, 154.24, 144.60, 138.35, 128.65, 128.34, 127.19, 79.74, 64.86, 59.57, 58.88, 43.59, 42.08, 30.78, 28.23; HRMS (ESI) m/z calcd for C21H30N5O2 [M + H+] 384.2399, found 384.2405.
3.10. 2-tert-Butoxycarbonyl-6-(methylsulfonyl)-8-(4-methyl-4H-1,2,4-triazol-3-yl)-2,6-diazaspiro[3.4]octane (16)
Compound 15 was hydrogenated as described for the preparation of compound 7 to give 0.76 g of a colorless oil. To a solution of this oil (0.35 g, 1.1 mmol, 1.0 equiv.) in CH2Cl2 (10 mL), Et3N (0.14 g, 1.37 mmol, 1.25 equiv.) was added dropwise. The mixture was cooled to 0 °C and treated with MsCl (0.16 g, 1.37 mmol, 1.25 equiv.). After stirring the mixture overnight, it was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (0.22 g, 53%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.11 (s, 1H), 3.04–3.95 (m, 2H), 3.91–3.75 (m, 4H), 3.64–3.52 (m, 2H), 1.40 (s, 9H).; 13C NMR (75 MHz, CDCl3) δ 156.03, 152.70, 144.60, 80.13, 61.23, 55.73, 53.35, 51.38, 43.74, 41.23, 35.65, 30.95, 28.20; HRMS (ESI) m/z calcd for C15H26N5O4S [M + H+] 372.1705, found 372.1701.
3.11. 2-tert-Butoxycarbonyl-6-benzyl-8-(5-methyl-4H-1,2,4-triazol-3-yl)-2,6-diazaspiro[3.4]octane (19)
To a solution of acetamidine hydrochloride (0.16 g, 1.73 mmol, 1.25 equiv.) in absolute methanol (10 mL), MeONa (0.093 g, 1.73 mmol, 1.25 equiv.) was added. The mixture was stirred for 30 min, whereupon crude 14 (0.5 g, 1.39 mmol, 1.0 equiv.) was added and the stirring continued overnight. The solvent was removed in vacuo and the residue was heated at 170 °C under an argon stream for 15 min. The residue was dissolved in ethyl acetate (25 mL), washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (0.26 g, 48%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.39–7.13 (m, 5H), 3.92 (dd, J = 37.4, 8.6 Hz, 2H), 3.70 (q, J = 12.8 Hz, 2H), 3.59–3.41 (m, 3H), 3.20–2.99 (m, 2H), 2.91 (t, J = 8.0 Hz, 2H), 2.38 (s, 3H), 1.37 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 156.29, 138.23, 128.69, 128.42, 127.27, 79.68, 64.71, 59.87, 53.40, 45.16, 43.19, 28.30, 8.08, −0.05; HRMS (ESI) m/z calcd for C21H30N5O2 [M + H+] 384.2399, found 384.2404.
3.12. 2-tert-Butoxycarbonyl-6-benzyl-8-(3-cyclopropyl-1H-1,2,4-triazol-5-yl)-2,6-diazaspiro[3.4]octane (20)
Synthesized analogously to 19. Yield 0.23 g (42%), colorless oil. 1H NMR 1H NMR (300 MHz, CDCl3) δ 7.39–7.16 (m, 5H), 3.91 (dd, J = 41.5, 8.6 Hz, 2H), 3.69 (q, J = 12.8 Hz, 2H), 3.57–3.41 (m, 3H), 2.04–1.86 (m, 1H), 1.38 (s, 9H), 1.00–0.94 (m, 4H).; 13C NMR (75 MHz, DMSO-d6) δ 156.29, 138.23, 128.69, 128.42, 127.27, 79.68, 64.71, 59.87, 53.40, 45.16, 43.19, 28.30, 8.08, −0.05; HRMS (ESI) m/z calcd for C23H32N5O2 [M + H+] 410.2556, found 410.2561.
3.13. 2-tert-Butoxycarbonyl-6-benzyl-8-(3-methyl-1,2,4-oxadiazol-5-yl)-2,6-diazaspiro[3.4]octane (23)
To a solution of 3 (1 g, 2.29 mmol, 1 equiv.) in 1,4-dioxane, an aqueous solution of LiOH∙H2O (1 mL, 0.175 g, 4.2 mmol,1.25 equiv.) was added and the mixture was stirred for 12 h. HOBt (0.56 g, 4.2 mmol, 1.25 equiv.), EDC∙HCl (0.81 g, 4.2 mmol, 1.25 equiv.), and acetamidoxime (0.35 g, 4.7 mmol, 1.4 equiv.) were added and the resulting mixture was stirred for 12 h. The solvent was removed in vacuo and the residue was dissolved in ethyl acetate (50 mL). The solution was washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was dissolved in toluene (25 mL), TBAF (100 mg) was added, and the resulting mixture was heated under reflux with a Dean-Stark trap for 6 h. The reaction mixture was concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (0.48 g, 41%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.41–7.17 (m, 5H), 3.94 (dd, J = 33.0, 8.6 Hz, 2H), 3.78–3.59 (m, 4H), 3.49 (d, J = 9.3 Hz, 1H), 3.33–3.21 (m, 1H), 3.09 (d, J = 9.4 Hz, 1H), 2.90–2.72 (m, 2H), 2.40 (s, 3H), 1.39 (s, 9H).; 13C NMR (75 MHz, DMSO-d6) δ 178.85, 167.17, 155.96, 138.18, 128.53, 128.37, 127.24, 79.69, 59.41, 57.47, 43.93, 43.22, 28.26, 11.60, −0.05; HRMS (ESI) m/z calcd for C21H29N4O3 [M + H+] 385.2239, found 385.2244.
3.14. 2-tert-Butoxycarbonyl-6-(methylsulfonyl)-2,6-diazaspiro[3.4]octane-8-carboxylic acid (25)
To a solution of crude 7 (0.31 g, 1.1 mmol, 1 equiv.) in CH2Cl2 (10 mL), Et3N (0.14 g, 1.37 mmol, 1.25 equiv.) was added dropwise. The mixture was cooled to 0 °C, treated with dropwise addition of MsCl (0.16 g, 1.37 mmol, 1.25 equiv.), and stirred overnight. It was then washed with 10% aq. K2CO3, brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was dissolved in methanol (10 mL) and treated with dropwise addition of 25% aq. KOH (1 mL). The mixture was stirred for 1 h, concentrated in vacuo, and the residue was dissolved in water. The solution was acidified to pH 6 with 5% aq. HCl. The resulting precipitate was filtered off and dried over NaOH pellets to give the title compound. Yield 0.76 g (100%), white solid, m.p. 102–103 °C. 1H NMR (300 MHz, DMSO-d6) δ 12.94 (s, 1H), 3.99–3.68 (m, 4H), 3.46–3.29 (m, 3H), 3.22 (dd, J = 7.4, 6.0 Hz, 1H), 2.91 (s, 3H), 1.36 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 172.53, 155.92, 79.12, 56.42, 49.37, 48.78, 41.60, 34.30, 28.45; HRMS (ESI) m/z calcd for C13H23N2O6S [M + H+] 335.1276, found 335.1280.
3.15. 2-tert-Butoxycarbonyl-8-(3-cyclopropyl-1,2,4-oxadiazol-5-yl)-6-(methylsulfonyl)-2,6-diazaspiro[3.4]octane (26)
To a solution of 25 (0.25 g, 0.75 mmol, 1 equiv.) in CH2Cl2 (10 mL), CDI (0.15 g, 0.94 mmol, 1.25 equiv.) was added and the mixture was stirred for 1 h, whereupon N′-hydroxycyclopropanecarboximidamide (0.094 g, 0.94 mmol, 1.25 equiv.) was added and the stirring was continued overnight. The reaction mixture was washed with 1% aq. HCl (2 × 15 mL) and concentrated in vacuo. The residue was dissolved in toluene (25 mL), TBAF (100 mg) was added, and the mixture was heated under reflux with a Dean-Stark trap for 6 h. The reaction mixture was evaporated to dryness. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound (0.19 g, 63%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.02–3.91 (m, 2H), 3.85 (t, J = 7.6 Hz, 1H), 3.81–3.65 (m, 5H), 3.56 (d, J = 9.9 Hz, 1H), 2.08 (dt, J = 8.3, 4.9 Hz, 1H), 1.41 (s, 9H), 1.13–0.88 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 156.03, 152.70, 144.60, 80.13, 55.73, 51.38, 43.74, 41.23, 35.65, 30.95, 28.20; HRMS (ESI) m/z calcd for C17H27N4O5S [M + H+] 399.1702, found 399.1697.
3.16. 6-Benzyl-N-methyl-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane-8-carboxamide (5a)
General Procedure B. For the preparation of compounds 5a–c, 6d, 12–3, 17–18, 21–22, 24 and 27. To a solution of 5-nitro-2-furoic acid (75 mg, 0.47 mmol) in DMF (3 mL), CDI (97 mg, 0.6 mmol) was added at 0 °C and the solution was stirred for 1 h.
To a solution of 4a (0.22 g, 0.6 mmol) in CH2Cl2 (5 mL) at 0 °C, trifluoroacetic acid (1 mL) was added and the mixture was stirred for 1 h. The solution was concentrated in vacuo while keeping the bath temperature under 30 °C. The residue was dissolved in DMF (3 mL), triethylamine (0.19 g, 1.9 mmol) was added dropwise, and after 30 min stirring, the mixture was added to the solution of 5-nitro-2-furoic acid imidazolide prepared as described above. The reaction mixture was stirred at r. t. overnight, poured into water (25 mL), and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel using 10% methanol in CH2Cl2 to give the title compound. Yield 133 mg (56%), white solid, m.p. 132–135 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 8.06 (s, 1H), 7.75 (dd, J = 8.8, 3.9 Hz, 1H), 7.40–7.15 (m, 7H), 4.45 (dd, J = 27.0, 9.3 Hz, 1H), 4.31 (s, 1H), 4.10–3.74 (m, 3H), 3.67–3.51 (m, 2H), 3.07–2.82 (m, 4H), 2.75–2.52 (m, 7H), 1.37–1.14 (m, 2H), 0.96–0.73 (m, 1H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers δ 172.28, 156.47, 151.81, 147.95, 147.82, 139.15, 128.88, 128.85, 128.59, 127.27, 117.35, 117.31, 113.49, 113.43, 65.50, 64.72, 61.17, 59.29, 59.21, 56.69, 55.20, 51.47, 51.33, 43.53, 25.91, 25.88; HRMS (ESI) m/z calcd for C20H23N4O5 [M + H+] 399.1668, found 399.1673.
3.17. 6-Benzyl-N-isobutyl-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane-8-carboxamide (5b)
This compound was synthesized according to general procedure B from 4b (0.24 g, 0.6 mmol). Yield 148 mg (56%), white solid, m.p. 166–165 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 8.13 (dd, J = 10.8, 5.4 Hz, 1H), 7.73 (dt, J = 16.3, 8.2 Hz, 1H), 7.42–7.09 (m, 7H), 4.47 (dd, J = 33.9, 9.4 Hz, 1H), 4.37–4.24 (m, 1H), 4.12 (d, J = 10.3 Hz, 1H), 4.00–3.75 (m, 2H), 3.64–3.50 (m, 2H), 3.08–2.68 (m, 6H), 2.67–2.53 (m, 2H), 1.62 (dd, J = 11.3, 6.7 Hz, 1H), 0.89–0.61 (m, 7H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 196.40, 193.60, 174.10, 173.50, 171.66, 168.54, 161.80, 159.89, 156.37, 151.78, 147.97, 147.91, 146.43, 146.33, 145.57, 139.77, 139.14, 134.96, 133.64, 129.86, 129.54, 128.97, 128.85, 128.59, 128.45, 128.31, 127.87, 127.28, 121.61, 121.53, 121.50, 121.45, 120.72, 118.14, 117.31, 113.49, 113.45, 105.49, 65.02, 64.74, 60.90, 59.40, 59.30, 59.23, 56.64, 56.51, 55.38, 55.26, 51.51, 51.44, 46.53, 43.47, 28.38, 28.34, 20.44, 20.35; HRMS (ESI) m/z calcd for C23H29N4O5 [M + H+] 441.2137, found 441.2134.
3.18. 6-Benzyl-N-(cyclopropylmethyl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane-8-carboxamide (5c)
This compound was synthesized according to general procedure B from 4c (0.185 g, 0.6 mmol). Yield 126 mg (48%), white solid, m.p. 180–181 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 8.42–8.08 (m, 1H), 7.76 (dd, J = 8.9, 3.9 Hz, 1H), 7.45–7.11 (m, 8H), 4.62–4.26 (m, 2H), 4.14 (d, J = 10.4 Hz, 1H), 3.94 (d, J = 9.2 Hz, 1H), 3.81 (d, J = 11.1 Hz, 1H), 3.69–3.47 (m, 3H), 3.14–2.77 (m, 6H), 2.62 (dd, J = 16.4, 8.9 Hz, 2H), 1.00–0.70 (m, 1H), 0.33 (dd, J = 18.6, 7.1 Hz, 2H), 0.10 (dd, J = 9.1, 4.1 Hz, 2H).; 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 171.59, 156.38, 151.79, 147.97, 147.91, 139.16, 128.88, 128.85, 128.74, 128.60, 127.27, 117.32, 113.46, 65.22, 64.65, 61.03, 59.30, 59.22, 56.51, 56.35, 55.26, 51.34, 43.54, 43.34, 43.30, 11.33, 11.30, 3.66, 3.55, 3.46; HRMS (ESI) m/z calcd for C23H27N4O5 [M + H+] 439.1981, found 439.1986.
3.19. 1-[6-Benzyl-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]oct-8-yl]-N,N-dimethylmethanamine (6d)
This compound was synthesized according to general procedure B from 5d (0.215 g, 0.6 mmol). Yield 91 mg (38%), white solid, m.p. 154–155 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 7.77 (d, J = 1.9 Hz, 1H), 7.76 (d, J = 1.9 Hz, 1H), 7.43–7.15 (m, 14H), 4.68 (d, J = 9.5 Hz, 1H), 4.39 (s, 2H), 4.31 (d, J = 10.4 Hz, 1H), 4.17 (d, J = 9.6 Hz, 1H), 3.94 (q, J = 10.1 Hz, 2H), 3.70 (d, J = 10.4 Hz, 1H), 3.63–3.48 (m, 5H), 2.98–2.55 (m, 8H), 2.44–2.25 (m, 5H), 2.10 (t, J = 10.9 Hz, 17H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 156.64, 156.48, 151.80, 148.01, 147.96, 139.28, 135.52, 128.84, 128.57, 127.21, 117.20, 113.45, 65.42, 65.36, 64.58, 61.25, 61.13, 60.33, 59.67, 58.65, 58.48, 58.27, 54.17, 45.74, 45.69, 43.23, 43.14, 41.77, 41.47.; HRMS (ESI) m/z calcd for C21H27N4O5 [M + H+] 399.2032, found 399.2028.
3.20. 8-(1-Benzyl-5-methyl-1H-imidazol-2-yl)-6-methyl-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (12)
This compound was synthesized according to general procedure B from 10 (0.237 g, 0.6 mmol). Yield 74 mg (28%), white solid, m.p. 165–167 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 7.74 (dd, J = 13.4, 3.8 Hz, 1H), 7.28–7.06 (m, 3H), 7.02–6.81 (m, 3H), 6.73 (d, J = 7.7 Hz, 1H), 5.37–5.06 (m, 2H), 4.24 (d, J = 9.5 Hz, 1H), 4.15 (d, J = 10.2 Hz, 1H), 3.94–3.53 (m, 4H), 3.38 (d, J = 19.3 Hz, 4H), 3.17–2.94 (m, 2H), 2.76–2.54 (m, 2H), 2.46–2.36 (m, 1H), 2.23 (s, 3H), 2.17–2.00 (m, 4H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 155.99, 151.74, 151.57, 148.01, 147.85, 147.65, 138.48, 138.23, 128.98, 128.91, 127.69, 127.51, 127.38, 127.19, 126.15, 125.97, 125.77, 125.72, 117.17, 117.12, 113.46, 68.02, 67.52, 65.66, 63.11, 62.97, 62.02, 58.47, 55.04, 45.96, 45.87, 45.24, 45.10, 43.51, 43.40, 41.95, 41.87.; HRMS (ESI) m/z calcd for C23H26N5O4 [M + H+] 436.1984, found 436.1988.
3.21. 6-Methyl-8-(5-methyl-1,3-oxazol-2-yl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (13)
This compound was synthesized according to general procedure B from 11 (0.184 g, 0.6 mmol). Yield 99 mg (48%), white solid, m.p. 175–176 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 7.77 (d, J = 3.9 Hz, 1H), 7.72 (d, J = 3.9 Hz, 1H), 7.32 (d, J = 3.9 Hz, 1H), 7.27 (d, J = 3.9 Hz, 1H), 7.02 (s, 1H), 6.80–6.73 (m, 2H), 4.57 (q, J = 9.6 Hz, 2H), 4.31 (d, J = 10.1 Hz, 1H), 4.17 (d, J = 10.4 Hz, 1H), 4.05 (d, J = 10.4 Hz, 1H), 3.97 (d, J = 10.0 Hz, 1H), 3.84–3.64 (m, 3H), 3.55–3.44 (m, 4H), 3.12–2.94 (m, 4H), 2.76 (ddd, J = 17.3, 7.5, 4.9 Hz, 4H), 2.30 (s, 5H), 2.25 (s, 3H), 2.22 (s, 2H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 162.41, 162.31, 156.55, 156.43, 151.85, 149.25, 149.19, 147.67, 147.61, 122.93, 117.41, 113.42, 113.36, 66.17, 64.16, 59.94, 59.25, 58.90, 58.79, 55.23, 45.46, 45.41, 44.98, 41.89, 10.82.; HRMS (ESI) m/z calcd for C16H19N4O5 [M + H+] 347.1355, found 347.1360.
3.22. 6-(Methylsulfonyl)-8-(4-methyl-4H-1,2,4-triazol-3-yl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (17)
This compound was synthesized according to general procedure B from 16 (0.22 g, 0.6 mmol). Yield 138 mg (56%), white solid, m.p. 180–182 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 8.44 (s, 1H), 7.78 (d, J = 3.9 Hz, 1H), 7.73 (d, J = 3.9 Hz, 1H), 7.30 (t, J = 4.1 Hz, 1H), 4.57 (q, J = 9.5 Hz, 2H), 4.21–3.84 (m, 5H), 3.84–3.58 (m, 6H), 3.56–3.37 (m, 1H), 2.85 (d, J = 9.7 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 156.67, 152.31, 151.83, 147.80, 147.70, 145.65, 117.39, 113.50, 113.45, 62.50, 59.36, 57.62, 56.55, 55.27, 53.97, 51.20, 51.05, 44.12, 43.99, 34.59, 34.52, 30.92.; HRMS (ESI) m/z calcd for C15H19N6O6S [M + H+] 411.1086, found 411.1091.
3.23. 6-Benzyl-8-(4-methyl-4H-1,2,4-triazol-3-yl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (18)
This compound was synthesized according to general procedure B from 15 (0.23 g, 0.6 mmol). Yield 157 mg (62%), white solid, m.p. 152–153 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 8.65–8.20 (m, 1H), 7.84–7.59 (m, 2H), 7.57–7.16 (m, 5H), 7.02 (s, 1H), 4.77–3.96 (m, 4H), 3.94–3.06 (m, 13H), 2.85 (dd, J = 21.6, 9.0 Hz, 1H), 2.68 (dd, J = 16.7, 8.9 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers, δ 156.60, 154.43, 154.32, 151.79, 147.75, 145.30, 139.09, 135.51, 129.86, 129.54, 128.91, 128.86, 128.62, 127.32, 122.04, 117.32, 113.43, 65.48, 64.63, 64.49, 61.43, 59.33, 58.92, 58.72, 54.82, 44.51, 30.77; HRMS (ESI) m/z calcd for C21H23N6O4 [M + H+] 423.1780, found 423.1777.
3.24. 6-Benzyl-8-(5-methyl-4H-1,2,4-triazol-3-yl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (21)
This compound was synthesized according to general procedure B from 19 (0.23 g, 0.6 mmol). Yield 108 mg (43%), white solid, m.p. 168–170 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 8.57–8.26 (m, 1H), 7.87–7.60 (m, 2H), 7.52–7.13 (m, 6H), 7.02 (s, 1H), 4.82–3.93 (m, 4H), 3.89–3.52 (m, 7H), 3.27–3.04 (m, 2H), 2.99–2.55 (m, 3H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers δ 158.23, 156.60, 154.43, 154.32, 151.79, 147.91, 147.75, 145.30, 139.09, 135.51, 129.86, 129.54, 128.91, 128.86, 128.62, 127.82, 127.32, 122.04, 117.32, 113.43, 66.85, 65.48, 64.63, 64.49, 61.43, 59.33, 58.92, 58.72, 54.82, 45.93, 44.51, 43.31, 30.77.; HRMS (ESI) m/z calcd for C21H23N6O4 [M + H+] 423.1780, found 423.1784.
3.25. 6-Benzyl-8-(5-cyclopropyl-4H-1,2,4-triazol-3-yl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (22)
This compound was synthesized according to general procedure B from 20 (0.24 g, 0.6 mmol). Yield 102 mg (38%), white solid, m.p. 155–157 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 13.39 (s, 1H), 7.82–7.58 (m, 1H), 7.41–7.10 (m, 5H), 4.59–4.39 (m, 1H), 4.26–3.85 (m, 2H), 3.73–3.57 (m, 2H), 3.14–2.60 (m, 3H), 2.06–1.69 (m, 1H), 1.06–0.49 (m, 4H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers δ 193.60, 167.18, 161.07, 156.43, 156.30, 151.73, 147.85, 147.80, 139.26, 135.51, 134.96, 129.86, 129.54, 128.87, 128.60, 127.26, 121.70, 121.63, 121.57, 121.54, 121.50, 121.48, 118.46, 117.24, 113.44, 113.38, 64.18, 64.10, 63.66, 59.53, 59.38, 57.37, 57.29, 55.38, 55.26, 44.29, 44.20, 9.48, 8.21, 7.60; HRMS (ESI) m/z calcd for C23H25N6O4 [M + H+] 449.1937, found 449.1942.
3.26. 6-Benzyl-8-(3-methyl-1,2,4-oxadiazol-5-yl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (24)
This compound was synthesized according to general procedure B from 23 (0.245 g, 0.6 mmol). Yield 121 mg (48%), white solid, m.p. 180–182 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 7.84–7.63 (m, 1H), 7.41–7.09 (m, 5H), 4.75–4.42 (m, 1H), 4.27 (dd, J = 36.8, 10.2 Hz, 1H), 4.10–3.93 (m, 2H), 3.82 (t, J = 11.6 Hz, 0H), 3.74–3.50 (m, 2H), 3.10 (dt, J = 26.4, 20.0, 9.4 Hz, 2H), 2.92–2.75 (m, 2H), 2.31 (d, J = 8.0 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers δ 179.23, 179.17, 167.29, 167.22, 162.72, 156.61, 151.85, 147.72, 147.58, 138.82, 129.86, 129.54, 129.00, 128.87, 128.66, 127.39, 121.59, 117.42, 113.44, 63.90, 63.78, 59.79, 59.04, 56.78, 56.66, 55.25, 55.16, 44.20, 43.12, 36.17, 31.17, 11.52.; HRMS (ESI) m/z calcd for C21H22N5O5 [M + H+] 424.1620, found 424.1616.
3.27. 8-(3-Cyclopropyl-1,2,4-oxadiazol-5-yl)-6-(methylsulfonyl)-2-(5-nitro-2-furoyl)-2,6-diazaspiro[3.4]octane (27)
This compound was synthesized according to general procedure B from 26 (0.24 g, 0.6 mmol). Yield 165 mg (63%), white solid, m.p. 171–173 °C. 1H NMR (300 MHz, DMSO-d6) mixture of rotamers δ 7.77 (dd, J = 9.8, 3.9 Hz, 1H), 7.31 (dd, J = 8.0, 3.9 Hz, 1H), 4.65 (q, J = 9.9 Hz, 1H), 4.45 (dd, J = 25.5, 10.0 Hz, 1H), 4.26–4.07 (m, 2H), 3.97 (dt, J = 35.2, 9.1 Hz, 1H), 3.82–3.52 (m, 4H), 2.97 (d, J = 1.7 Hz, 3H), 2.18–2.02 (m, 1H), 1.07–0.68 (m, 4H); 13C NMR (75 MHz, DMSO-d6) mixture of rotamers δ 177.00, 176.87, 172.19, 156.67, 151.86, 147.63, 147.52, 122.80, 122.67, 122.56, 117.49, 113.48, 60.70, 57.56, 55.56, 55.26, 54.21, 49.35, 49.15, 43.77, 43.71, 42.72, 42.54, 34.65, 34.59, 7.89, 6.62; HRMS (ESI) m/z calcd for C17H20N5O7S [M + H+] 438.1083, found 438.1088.
3.28. Method to Evaluate Antitubercular Activity of Compounds 5a–c, 6d, 12–13, 17–18, 21–22, 24, and 27
Mycobacterium tuberculosis H37Rv strain (originated from the Institute of Hygiene and Epidemiology in Prague, 1976) was obtained on 7 August 2013 from the Federal Scientific Center for Expertise of Medical Products (RF Ministry of Health Care). The lyophilized strain was seeded in Löwenstein–Jensen growth medium. After 3 weeks, the culture was suspended in physiological solution containing glycerol (15%), transferred into cryotubes, and kept at −80 °C. Three weeks prior to the experiment, the culture was brought to ambient temperature and re-seeded into Löwenstein–Jensen growth medium. Thus, the 2nd generation of the original M. tuberculosis culture was used in the present study.
The minimal inhibitory concentration (MIC) of each compound was determined using the REMA (resazurin microtiter plate assay) [
13]. A 3-week
M. tuberculosis culture was transferred into a dry, sterile tube containing 8–9 3 mm glass beads. The tube was placed on a Vortex shaker for 30–40 s and then 5 mL Middlebrook 7H9 Broth (Becton Dickinson, catalogue No. 271310) was introduced. The turbidity of the bacterial suspension was adjusted to 1.0 McFarland units (corresponding to approximately 3 × 10
8 bacteria/mL) and diluted 20-fold with Middlebrook 7H9 Broth containing OADC enrichment (Becton Dickinson, catalogue No. 245116). The same culture medium was used to prepare the 1:100
M. tuberculosis (1% population) control. The stock solution of the compound in DMSO (10 mg/mL) was diluted with Middlebrook 7H9 Broth (containing OADC enrichment) to a concentration of 100 μg/mL. A 200 μL aliquot of the solution thus obtained was introduced into the 1st row of a 96-well microtiter plate. This row was used to perform 2-fold serial dilutions using an 8-channel pipette to obtain final concentrations of 0.4, 0.8, 1.6, 3.1, 6.2, 12.5, 25, and 50 μg/mL of the compound in rows 2–9 (accounting for 100 μL of bacterial suspension introduced for testing). Similarly, an experiment with 0.004, 0.008, 0.016, 0.031, 0.062, 0.125, 0.25, 0.50 and 1 μg/mL concentrations of the compounds in rows 1–9 was carried out. Row 10 contained the
MTb suspension control while row 11 contained the same culture diluted 10-fold (1% control). Row 12 was used as a blank control for the optical density reading (200 μL of growth medium). The bacterial suspension (100 μL) was introduced into each well, except rows 11 (1% population control) and 12 (blank culture medium), to give a total volume of 200 μL in each well. The plates were incubated at 35 °C for 7 days. At that point, 0.01% aqueous solution (30 μL) of resazurin (Sigma, product No. R7017) was introduced in each well and the incubation continued for 18 h at 35 °C. The fluorescence reading was performed using a FLUOstar Optima plate reader operating at λ
ex = 520 nm and λ
em = 590 nm. The bacterial viability was determined by comparing the mean values (±SD at
p = 0.05) of fluorescence in the control wells (row 12, blank and row 11, 1% control) and the wells containing the compound tested. The MIC was determined as the compound concentration at which the fluorescence reached a plateau or was statistically (t criterion) similar to that of the 1% control (see
Supplementary Materials).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}