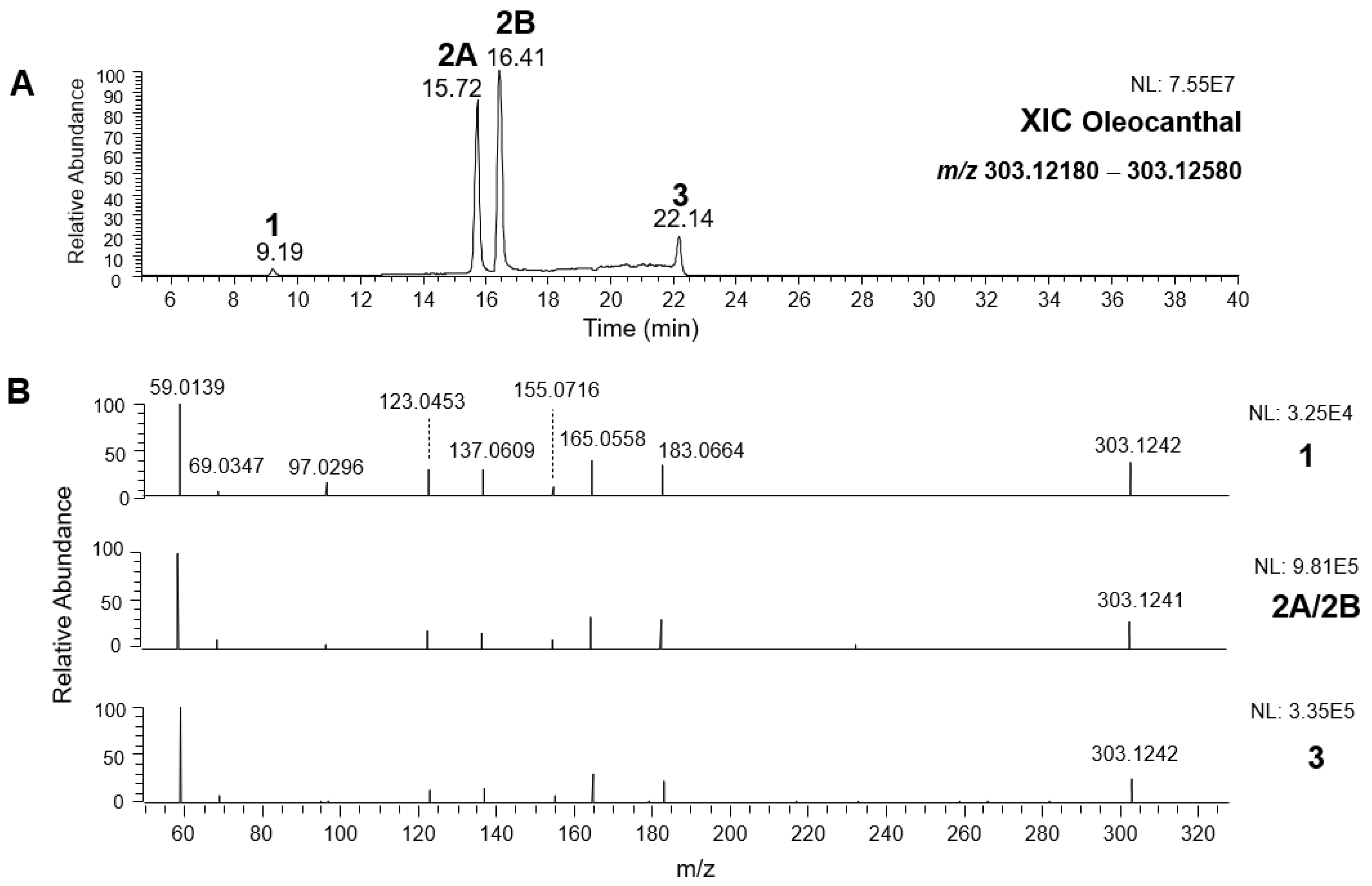
2.1. RPLC-ESI-FTMS Analysis of Oleacin and Oleocanthal in EVOO
As a first step of the structural investigation of OLEA and OLEO in EVOO extracts, eXtracted Ion Current (XIC) chromatograms related to their [M-H]
− ions were obtained by extracting currents using intervals centered on the corresponding exact
m/
z values, 319.1187 and 303.1238, respectively, and having a 0.004
m/
z unit width (see
Figure 2A and
Figure 3A). The chromatographic features were consistent with those previously observed for both secoiridoids in other EVOO extracts [
25,
34,
35], thus showing a qualitative reproducibility of the OLEA and OLEO profiles between different oils. Compared to those of oleuropein and ligstroside aglycones (OA and LA), usually exhibiting more than 10 peaks under the same chromatographic conditions [
8,
9,
25,
34,
35], the profiles of OLEA and OLEO were significantly simpler, due to their inherently more limited structural variability. Indeed, the absence of the carboxymethylic (-CO
2CH
3) moiety on the C4 atom in their molecular structure (see
Figure 1), with the consequent lack of an additional stereogenic center, lowers the number of potential diastereoisomers compared to those of OA and LA. Moreover, the lack of the CO
2CH
3 group prevents the generation of minor, closed-structure isoforms detected for OA and LA and named
Closed Forms II [
8,
9].
As a result, the chromatographic profiles of OLEO and OLEA were dominated by just two almost identical peaks, numbered 2A and 2B in
Figure 2A and
Figure 3A, in apparent analogy with the chromatographic traces reported by Impellizzeri et al. [
31] and Adhami et al. [
32]. However, as emphasized in
Figure 2A and
Figure 3A, those chromatographic peaks were accompanied by some minor peaks (numbered as 1 and 3 in the case of OLEO and 1, 3A/3B and 4A/4B in the case of OLEA), which had not previously been reported. Not surprisingly, peaks related to OLEA were eluted earlier from the C18 column, i.e., between 4 and 17.5 min, than those of OLEO (between 9 and 22.5 min), likely because the additional OH group linked to the phenyl moiety increased the polarity of OLEA, reducing its interaction with the C18 stationary phase. The XIC traces of OLEA and OLEO shared other interesting features. First, the retention time difference between the two major peaks (2A and 2B),
ca. 0.7 min, was almost constant between the two compounds and between different EVOO samples (see the XIC traces reported in Refs. [
25,
34,
35]). Moreover, a careful comparison with the complex XIC profiles obtained for OA and LA [
8,
9] showed a partial similarity in terms of elution patterns, suggesting that some of the isoforms previously discovered for OA and LA were likely present, with the appropriate structural differences, also in the case of OLEA and OLEO. This analogy is highlighted in
Figure 1, in which a key step for the conversion of OA/LA isoforms into OLEA/OLEO ones is represented by the methylesterases activity. Notably, four novel methylesterases were recently identified in
Olea europaea by Volk et al. [
36], exhibiting significant homology to polyneuridine aldehyde esterase (PNAE) and showing variable in vitro activity towards plant methyl esters, such as those of jasmonic acid (MeJA), indole acetic acid (MeIAA), and salicylic acid (MeSA).
Interestingly, none of those methylesterases was found to be active directly on oleuropein and ligstroside, the original secoiridoids occurring in olive drupes. However, two of them, OeEst030 and OeEst228, contributed to the transformation of OA and LA into OLEO and OLEA through demethylation, followed by non-enzymatic decarboxylation [
36], as depicted in
Figure 1.
The predicted structural analogies between the isomeric forms of OA and LA and those of OLEO and OLEA helped in formulating the initial hypotheses on their structures. Indeed, due to the absence of a stereogenic center on C4, two geometric isomers, related to the C=C bond located between C8 and C9, were predicted for
Open Forms I of OLEA/OLEO, whereas a couple of diastereoisomers, arising from the combination of the fixed configuration at the chiral center on C5 with the two possible configurations assumed by the stereogenic center at C9, could be predicted for
Open Forms II (see
Figure 1). As for cyclic isomers, which correspond to
Closed Forms I in
Figure 1, up to four diastereoisomers can be expected in the case of OLEA/OLEO due to the coupling of configurations for the chiral center on C5 and the stereogenic centers on C8 and C9 (see
Figure 1).
By analogy with the chromatographic behavior observed for OA/LA isomeric forms [
8,
9], the retention time order for OLEA/OLEO isomers was expected to be
Open Forms I <
Open Forms II <
Closed Forms I. Consequently, peaks numbered as 1 in XIC traces obtained for the two compounds (see
Figure 2A and
Figure 3A) were initially assigned to
Open Forms I, peaks 2A/2B to
Open Forms II and lately eluting peaks (3, 3A/3B and 4A/4B, according to the case) were associated with
Closed Forms I. High-resolution MS/MS spectrometry, with or without H/D exchange, was subsequently employed to confirm these structural hypotheses and evaluate if stable enolic tautomers, able to provide further structural information, might also be present for one or more isoforms of OLEO and OLEA.
2.2. RPLC-ESI-FTMS/MS Analysis of Oleacin and Oleocanthal Isoforms in EVOO
Tandem MS spectra acquired using the higher energy collisional dissociation (HCD) cell of the
Q-Exactive spectrometer, averaged in the retention time intervals corresponding to the main chromatographic peaks detected for OLEO and OLEA, are shown in panels B of
Figure 2 and
Figure 3, respectively. As is apparent, almost identical fragmentation patterns were obtained for the four main peaks of OLEO (1, 2A/2B, and 3), whereas two main patterns, partially overlapping, were found for OLEA, one related to peaks 1, 2A/2B, and 3B, and the other to peaks 3A, 4A, and 4B. Moreover, some common product ions were detected for isoforms of OLEO and OLEA; notably, such features were found also in the case of OA and LA isoforms [
8,
9], thus strengthening the structural relationship existing between the four secoiridoids.
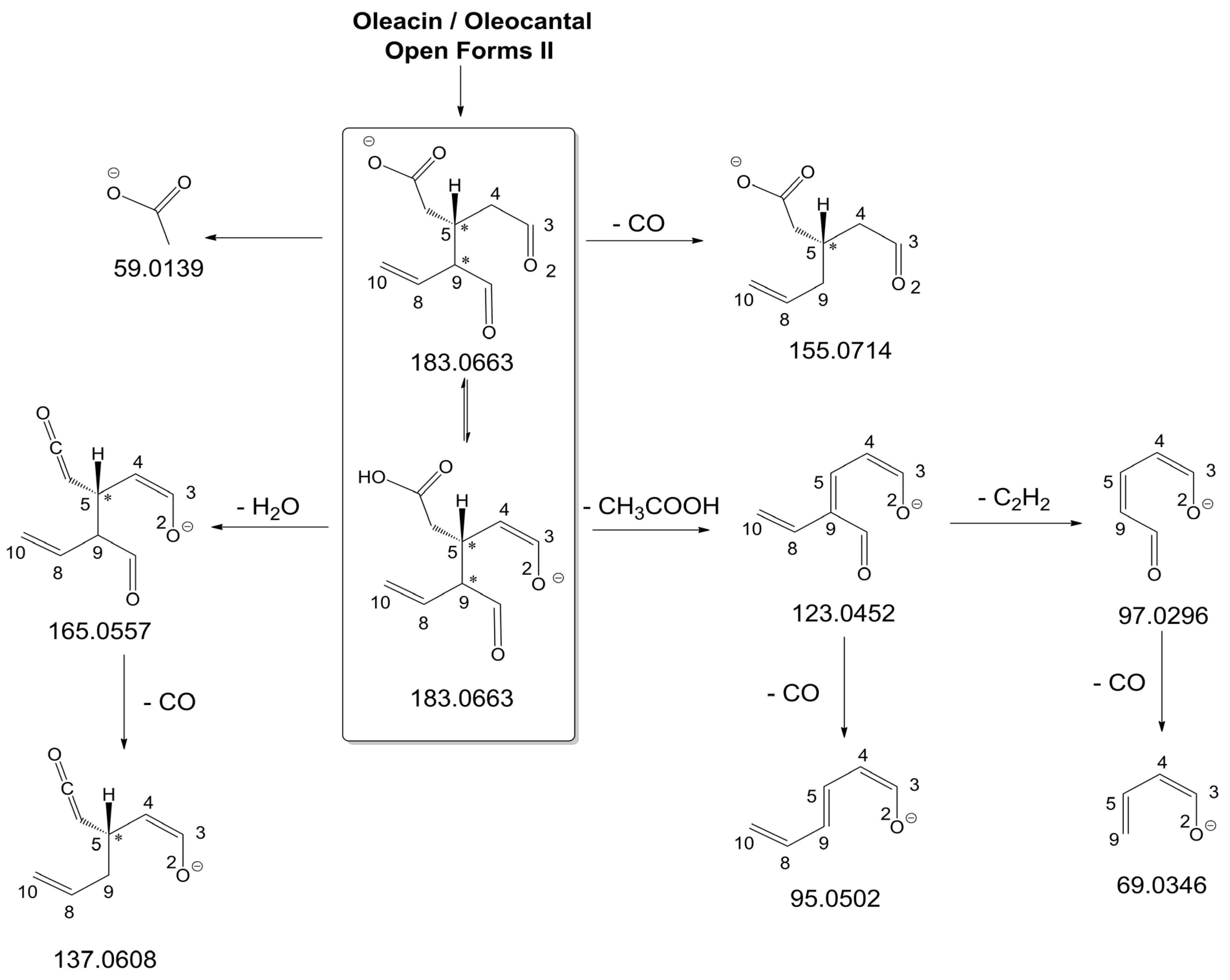
As illustrated in the fragmentation schemes proposed for
Open Forms II in
Figure 4 and for
Open Forms I/
Closed Forms I in
Figure S1 of the Supplementary Material, the key product ion to explain the fragmentation patterns observed for OLEO and OLEA isoforms was the anion of decarboxymethyl-elenolic acid (exact
m/
z 183.0663), which was detected in almost all MS/MS spectra, although its peak signal was more intense in the case of OLEO isoforms (see
Figure 2B and
Figure 3B). By analogy with the generation of deprotonated elenolic acid from the negatively charged precursors of OA and LA isoforms [
8,
9], the decarboxymethyl-elenolic acid anion could be generated upon gas phase collisional dissociation of any of the predicted isoforms of OLEO and OLEA shown in
Figure 1. Indeed, it could be formed through the displacement of the negative charge originally located on a phenolic OH group towards one of the carbonyl groups tautomerized to enol, followed by the neutral loss of the dehydrated forms of tyrosol and hydroxytyrosol, respectively.
Additionally, structures referred to as “decarboxymethyl-elenolic acid isoforms,” arising from the predicted different isoforms for OLEO and OLEA (see
Figure 1) and bearing the negative charge either on an enolic moiety or on the carboxylic one, were reported in
Figure 4 and
Figure S1 as the starting point of the fragmentation cascades. In particular, structures including only carbonyl groups or with one of them tautomerized to a stable enol were drawn in the figure, since only keto-enolic tautomerization was able to explain the generation of some product ions once the negative charge was displaced on the enolic OH group. For example, product ions corresponding to acetate (exact
m/
z 59.0139), which usually corresponds to the prevailing peak signal in almost all MS/MS spectra of OLEO and OLEA, and to decarboxymethyl-elenolic acid after CO loss (exact
m/
z 155.0714) were reasonably generated by the all-aldehydic isoforms of decarboxymethyl-elenolic acid (see
Figure 4), having the negative charge on the COOH group. On the other hand, product ions corresponding to the exact
m/
z ratios 165.0557, 137.0608, 123.0452, 97.0296, 95.0502, and 69.0346 could be accounted for by considering the location of the negative charge on an enolic OH group.
Figure 4 and
Figure S1 emphasize the neutral losses of acetic acid, water, carbon monoxide, and acetylene (C
2H
2) involved in the gas phase formation of those product ions. Notably, a further isoform of the precursor ion, resulting from ring opening and intramolecular proton transfer and bearing the negative charge on an enolic OH functionality, had to be invoked to account for the subsequent generation of those product ions in the case of
Closed Forms I (see
Figure S1).
As discussed so far, each of the different suggested isoforms was potentially able to generate product ions detected in the MS/MS spectra of OLEO, thus fragmentation patterns were not diagnostic for the type of isoform involved. As for OLEA, most of the product ions described so far were also detected in the tandem MS spectra referred to chromatographic peaks 1, 2A/2B, and 3B. In addition, peak signals were detected at
m/
z 275.1612 and 249.0766. For the sake of brevity, their generation from
Open Forms I of OLEA is described in
Figure S2 of the Supplementary Material, involving neutral losses of acetaldehyde or but-2-enal, respectively. However, similar mechanisms can be hypothesized to account for their generation from
Open Forms II and
Closed Forms I of OLEA. In the last case, a preliminary opening of the ring of decarboxymethyl-elenolic acid is required, as described for one of the pathways reported in
Figure S1.
As mentioned before, MS/MS spectra obtained for peaks 3A, 4A, and 4B in the XIC trace of OLEA suggested a partially different fragmentation pattern, as additional product ions were detected at
m/
z 199.0610, 181.0505, 155.0713, 139.0401, 137.0243, 111.0086, and 85.0295, with varying relative abundance (see peaks with underlined
m/
z ratios in
Figure 3B). The interpretation of most of these product ions was already provided in one of our previous studies, concerning the evolution of secoiridoids in EVOO stored for a long time (up to six months) in the presence of oxygen [
25]. Indeed, as described in
Figure S3 of the Supplementary Material, the
m/
z ratio (319.1187) of monoisotopic deprotonated OLEA [C
17H
19O
6]
−, is identical to that of deprotonated oleocanthalic acid, i.e., the anion of oleocanthal in which a carbonyl group has been oxidized to a COOH group. All possible isoforms of oleocanthalic acid are reported in
Figure S3, depending on the OLEO isoform involved in the oxidation process and on the final location of the COOH group (in fact,
Open Forms I and
II of OLEO have two C=O groups that can undergo oxidation to COOH). As shown in the figure, the product ion detected at
m/
z 199.0610, prevailing over others in the MS/MS spectra of peaks 3A, 4A, and 4B shown in
Figure 3B, corresponds to deprotonated oleocanthalic acid after the neutral loss of the dehydrated form of tyrosol (exact
m/
z 199.0612), a typical fragmentation of secoiridoids. Moreover, as shown in
Figure S4 of the Supplementary Material for
Closed Forms I of oleocanthalic acid, the
m/
z 199.0612 ion is the precursor of a fragmentation cascade involving neutral losses of acetic acid, water, acetylene, carbon dioxide, and ethylene, and is, according to the case, able to account for the entire series of additional product ions mentioned above.
Based on these considerations, OLEA peaks labelled 3A, 4A, and 4B in
Figure 3A could be associated with oleocanthalic acid isoforms, formed upon EVOO oxidation during storage before analysis. As recently discussed [
25], their retention times suggest that these peaks are related to diastereoisomers of
Closed Forms I of oleocanthalic acid; due to the presence of two stereogenic centers (at C8 and C9), each with two possible configurations, and to the fixed configuration of the chiral center at C5, four isoforms are expected for them. Actually, other isoforms of oleocanthalic acid, corresponding to
Open Forms II, were detected during our previous investigation, but only after prolonged (i.e., after six months) EVOO storage [
25], and their retention times were much lower than those observed in the present case. It is worth noting that the proximity of peak 3B, related to one of the OLEA isoforms, with peaks 3A and 4A, associated with isoforms of oleocanthalic acid, may explain why the MS/MS spectrum of the latter also included product ions typical of OLEA isoforms (i.e., those detected at
m/
z 183.0660 and 165.0555), due to spectral interference. On the other hand, as evidenced in
Figure S4, product ions with
m/
z ratios compatible with exact values 59.0139 (the acetate ion), 95.0502 and 69.0348, typical of OLEO and OLEA isoforms (see
Figure 4 and
Figure S1), can be generated also by isoforms of oleocanthalic acid, thus they were detected also in the MS/MS spectra related to peaks 3A and 4A of OLEA (see
Figure 3B). Actually, their peak intensity was negligible only in the MS/MS spectrum related to the oleocanthalic acid isoform corresponding to peak 4B, for which the pathway leading to the
m/
z 111.0088 ion was clearly prevailing. Since the key fragmentation towards the generation of this ion was the neutral loss of acetic acid, with the formation of a C=C bond between C5 and C9 (see
Figure S4), peak 4B might correspond to the specific diastereoisomer of oleocanthalic acid bearing the H atom bond to C9 on the same side as the CH
2COOH group bond to C5, thus making the detachment of acetic acid more sterically favorable.
Notably, the same results described so far for OLEO and OLEA extracted from the EVOO sample were obtained by applying the RPLC-ESI-(-)-FTMS method to a mixture of OLEO and OLEA standards available commercially (phyproof
® Reference Substances), each dissolved at a 10 mg/L concentration into the same water/methanol mixture used for the secoiridoid extraction from EVOO. Indeed, as highlighted in
Figure S5 of the Supplementary Material, the profiles inferred from the XIC chromatograms of the two standard compounds were very similar to those obtained from the real EVOO sample, the only relevant exception being the absence of peaks related to oxidized oleocanthal, as expected. Even in this case, the MS/MS spectra obtained for the different detected isoforms of OLEO and OLEA were very similar.
Consequently, despite the ability to provide some structural information on OLEO and OLEA isoforms and also on oleocanthalic acid isoforms, tandem MS analyses were unable to associate each chromatographic peak to specific isoforms of OLEO and OLEA among those depicted in
Figure 1. RPLC-ESI-FTMS analysis in conjunction with H/D exchange was thus undertaken to address this issue.
2.3. RPLC-ESI-FTMS and MS/MS Analysis of Oleacin and Oleocanthal Isoforms in the Presence of H/D Exchange
As previously described [
8,
9], H/D exchange (HDX) proved a powerful approach to detect the existence of stable dienolic tautomers for some isoforms of oleuropein and ligstroside aglycones, namely
Open Forms II, providing fundamental information to recognize them. HDX was performed by using D
2O as the co-solvent of acetonitrile in the RPLC mobile phase. As in the case of OA and LA, labile hydrogen atoms of phenolic and enolic -OH groups of OLEO and OLEA were expected to be involved in the H/D exchange. Thanks to the high mass resolving power available with the Orbitrap analyzer of the
Q-Exactive spectrometer, the
m/
z shift related to the exchange of an H atom with a D one can be recognized distinctly with respect to the one related to the exchange of a
12C atom with a
13C one, the main responsible for the
m/
z differences occurring between the M+0 and M+1 isotopologues of the secoiridoids under study. As an example, the exact
m/
z value for the M+1 isotopologue of deprotonated OLEO, 304.1272, could be distinguished from the one related to the M+0 isotopologue of oleocanthal after one H/D exchange, 304.1301, even though they only differed by 0.0029
m/
z units. Appropriate
m/
z intervals, having a width of 0.004 units and including the exact
m/
z values predicted for the M+0 isotopologues of non-deuterated and of the possible deuterated forms of OLEO and OLEA ions, could thus be adopted to retrieve the respective extracted ion current (XIC) traces from RPLC-ESI(-)-FTMS TIC chromatograms obtained when using D
2O in the mobile phase, as shown in
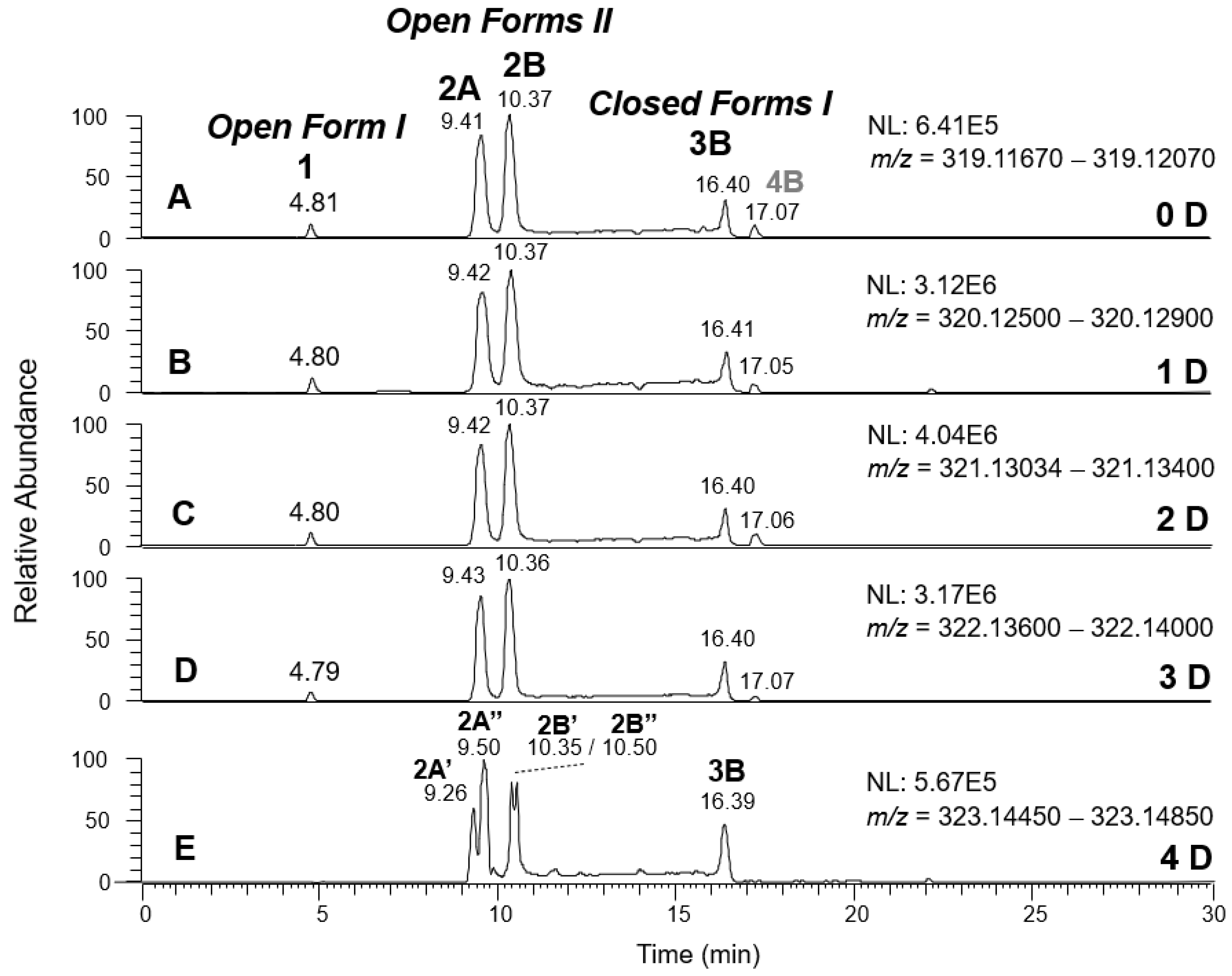
Figure 5 and
Figure 6, respectively. As was apparent, mono-, bis-, or tris-deuterated OLEO and OLEA ions were detected, along with non-deuterated ions, resulting from the total lack of H/D exchange concerning part of the ionic population. Tetra-deuterated ions were also detected for OLEA. As described in detail in
Table 1, all possible isoforms of OLEA and OLEO reported in
Figure 1, along with those arising from them as a result of keto-enolic tautomerism on carbonyl groups, and all the reasonable mechanisms for H/D exchange, including one that was initially not expected (
vide infra), had to be considered to explain the experimental data. As discussed for OA and LA [
8,
9], the H atoms located on the phenolic OH group(s) of OLEO and OLEA, along with those included in enolic OH groups, were considered the first candidates for the HDX process.
Starting from dialdehydic
Open Forms I and
II and mono-aldehydic
Closed Forms I of OLEO (see the first row in the related part of
Table 1), the single phenolic OH group is the only predictable site for H/D exchange, but it is also the site of negative ionization. Consequently, if the phenolic OH group is involved in the H/D exchange, the D atom is subsequently lost as a D
+ ion during the ESI process, and no deuteration can be finally observed for negatively charged OLEO (exact
m/
z value: 303.1238). Conversely, if the enolization of one of the suitable aldehydic groups occurs, the H atom of the resulting enolic OH group can be exchanged with D, and a negative ion with an exact
m/
z of 304.1301 is formed (see the second row in the OLEO section of
Table 1). It is thus not surprising that peaks for all possible OLEO isoforms were detected in the XIC chromatograms referred to exact
m/
z values 303.1238 and 304.1301 (see
Figure 5A,B), since all of them could include none or at least one D atom in the final ion. Notably, since peaks in the two XIC traces were well aligned in terms of retention times, the enolization leading to a D atom introduction in the deprotonated OLEO structure reasonably occurred during the ionization process in the ESI source.
As evidenced in
Figure 5C, isoforms of deprotonated OLEO including two D atoms on their structure (exact
m/
z 305.1364) were also detected, and, once again, the corresponding chromatographic peaks were perfectly aligned with those of non- and mono-deuterated anions. In principle, a double HDX on labile H atoms should have been possible only for
Open Forms II, since only they can undergo a double keto-enolic tautomerization, leading to the presence of two enolic OH groups on their structure. However, the XIC trace related to the
m/
z value 305.1364 (see
Figure 5C) showed all four peaks expected if all OLEO isoforms were able to generate anions with two D atoms on their structures. An interesting possible explanation for this surprising outcome was obtained by considering also the deuteration of specific non-labile H atoms, i.e., those located in the α-position to carbonyl groups. Indeed, Zherebker et al. [
37] disclosed that a relatively high desolvation temperature can facilitate the H/D exchange at non-labile sites of hydroxybenzoic acids and aromatic amino acids during the ESI process. According to this model, schematically described in
Figure 7, for dialdehydic
Open Forms I of OLEO and OLEA, an additional H/D exchange would be induced by the presence of deuterated hydroxide ions in the microdroplets generated during the ESI process in negative polarity. Remarkably, the acceleration of organic reactions in the confined volume of partially solvated microdroplets formed in an ESI source was discussed in a review by Yan et al. [
38]. In the case of OLEO
Open Forms I, an additional deuteration would result from the in-source H/D exchange at C4, along with that occurring on the enolic OH group linked to C3, as shown in the third row of the OLEO-related part of
Table 1. As emphasised in
Figure 7, the deuterated enol with a D also on C4 can be considered in equilibrium with the tautomer bearing a carbonyl group on C3 and two D linked to C4 (only the former structure was reported in
Table 1, for the sake of simplicity). In the case of OLEO
Open Forms II, the described double deuteration mechanism described in
Figure 7 might involve, alternatively, the other C=O group, since this has one H atom on the α-carbon (C9) that can be exchanged with D. As for
Closed Forms I, only a ring opening process occurring during the ESI process, such as the one already described as part of a fragmentation pathway reported in
Figure S1, would finally result in the presence of two enolic groups and, hence, in the detection of OLEO anions including 2 D atoms (see the third row in the upper part of
Table 1), thus explaining why peaks for those isoforms were detected also in the XIC trace referred to the
m/
z value 305.1364 (see
Figure 5C). Notably, all double H/D exchanges described for OLEO isoforms were expected to occur during the ESI process because the corresponding peaks detected in XIC chromatograms of non-, mono-, and bis-deuterated OLEO anions (see
Figure 5A–C) were aligned.
A different scenario was hypothesized to explain the XIC chromatogram obtained for the exact
m/
z value of 306.1426, which corresponds to OLEO anions bearing 3D atoms on their structure. As shown in
Figure 5D, no relevant peak due to
Open Forms I was detected since there was no reasonable way for these forms to undergo three H/D exchanges. However, four peaks were found in the retention time interval related to
Closed Forms I, one of which (3A, at 22.20 min) aligned with the prevailing peak detected in that interval in other XIC traces of deuterated OLEO derivatives. In this case, the already cited ring opening of decarboxymethyl-elenolic acid enabled the third H/D exchange to occur at C4 (see the last row in the upper part of
Table 1), through the mechanism described in
Figure 7. Interestingly, this process seemed to occur with a non-negligible likelihood for all four possible diastereoisomers predicted for
Closed Forms I, since three weak additional chromatographic peaks (labeled 3B, 3C, and 3D in
Figure 5D) were detected along with the prevailing peak 3A.
The detection of four distinct peaks in the XIC chromatogram referred to tris-deuterated OLEO anions instead of the two peaks (2A/2B) putatively associated with
Open Forms II required a specific explanation. In particular, couples of peaks located on both sides of them (thus not aligned with them), labelled as 2A’/2A’’ and 2B’/2B’’ (see
Figure 5D), were detected. As emphasized in the last row of the OLEO-related part of
Table 1, these peaks can be associated only with OLEO isoforms in which a double H/D exchange occurs on the enolic moiety involving C3 and C4, such as the one already described for
Open Forms I, and an additional H/D exchange occurs on the enolic group resulting from the tautomerization on the carbonyl group involving C1. Such an extent of deuteration is possible only for
Open Forms II, since only in their case the C
1=O group can be involved in the keto-enolic tautomerization. The detection of peaks at different retention times compared to those of the two typical peaks observed (2A/2B) suggests that dienolic forms of
Open Forms II can be formed in the mobile phase and be so stable as to be separated from their all-aldehydic counterparts. Those forms likely underwent the first two H/D exchanges on their enolic OH groups, whereas the last H/D exchange, on C4, occurred during the ESI process. This behavior is analogous to the one previously observed for the
Open Forms II of oleuropein and ligstroside aglycones [
8,
9] and can be considered an important confirmation of the structural characteristics of these isoforms (namely, the occurrence of a C=C bond between C8 and C10) that cannot be obtained otherwise using MS. Strikingly, the detection of four distinct peaks for the tris-deuterated derivatives of OLEO
Open Forms II, with no alignment with peaks 2A/2B, is consistent with the occurrence of dienolic forms, in which all the combinations of the geometries (
E or
Z) of the two C=C bonds related to the enolic moieties are possible, as emphasized in
Table 1 by a specific drawing of bonds linking the C3 and C1 atoms to the enolic OH groups.
As evidenced by
Figure 6, OLEA exhibited a behavior similar to that of OLEO when the H/D exchange was performed. In this case, since an additional OH not involved in negative ionization is present on the catechol ring, an additional H/D exchange occurred with respect to OLEO. XIC chromatograms for mono-, bis-, and tris-deuterated isoforms, corresponding to non-, mono-, and bis-deuterated isoforms of OLEO, respectively, showed peaks aligned with those observed for non-deuterated isoforms of OLEA. As shown in the lower part of
Table 1, the first deuteration could be expected to occur on the OH group linked to the phenyl ring. The second deuteration was still possible for all isoforms since all of them had a carbonyl group suitable for the keto-enolic tautomerization in the ESI source, enabling a H/D exchange to occur on an enolic OH group (see the third row in the lower part of
Table 1). The third deuteration required the same mechanism described before for the double deuteration on OLEO isoforms (see
Figure 7) when
Open Forms I and
II of OLEA were involved, whereas a ring opening was essential to enable a further H/D exchange in
Closed Forms I of OLEA (see structures reported in the fourth row of the OLEA-related part of
Table 1). The alignment of corresponding peaks in XIC traces referred to non-, mono-, bis-, and tris-deuterated OLEA isoforms was observed (see
Figure 6A–D), by analogy with OLEO isoforms. Once again, a specific behavior, similar to the one found for tris-deuterated OLEO, was observed when the XIC trace referring to tetra-deuterated OLEA anions was considered (see
Figure 6E). Indeed, while no significant peak was observed for
Open Forms I and an alignment with the corresponding peak in the other traces (peak 3B) was found for
Closed Forms I, two couples of peaks were detected instead of peaks 2A and 2B for
Open Forms II that were not aligned with them. This outcome confirmed the occurrence of stable dienolic forms, with distinct retention times and capability to undergo H/D exchange already in the mobile phase, for
Open Forms II, thus providing an indirect but important proof of the specific structural features of these isoforms, prevailing over others also in the case of oleacin.
A final consideration is due to the peak labeled 4B, detected after
ca. 17 min in four of the five XIC traces reported in
Figure 6. As discussed before, that peak was related to one of the
Closed Forms I of oleocanthalic acid, eluting near the
Closed Forms I of oleacin (see
Figure 3). As it can be inferred by considering the structure of oleocanthalic acid
Closed Forms I (see
Figure S3), these isoforms can undergo one H/D exchange on the OH group linked to the phenyl ring, assuming the carboxylic group as the most reasonable ionization site in this case. One additional H/D exchange can occur on the enolic group linked to C3, resulting from the ring opening occurring during the ESI process and described before for
Closed Forms I of OLEO and OLEA. Finally, a H/D exchange may occur on the C9 atom, which is an α-carbon with respect to the COOH group and thus can participate in the mechanism described in
Figure 7. Accordingly, a peak corresponding to peak 4B in the XIC trace of non-deuterated OLEA was observed in traces referred to as mono-, bis-, and tris-deuterated species (see
Figure 6B–D), whereas there was no reasonable possibility for oleocanthalic acid
Closed Forms I to undergo four H/D exchanges, and thus no significant peak was detected at the retention time of peak 4B in the XIC trace for tetra-deuterated OLEA (see
Figure 6E).




 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























